
Myc beyond Cancer: Regulation of Mammalian Tissue Regeneration

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Overview of Myc-Dependent Transcription
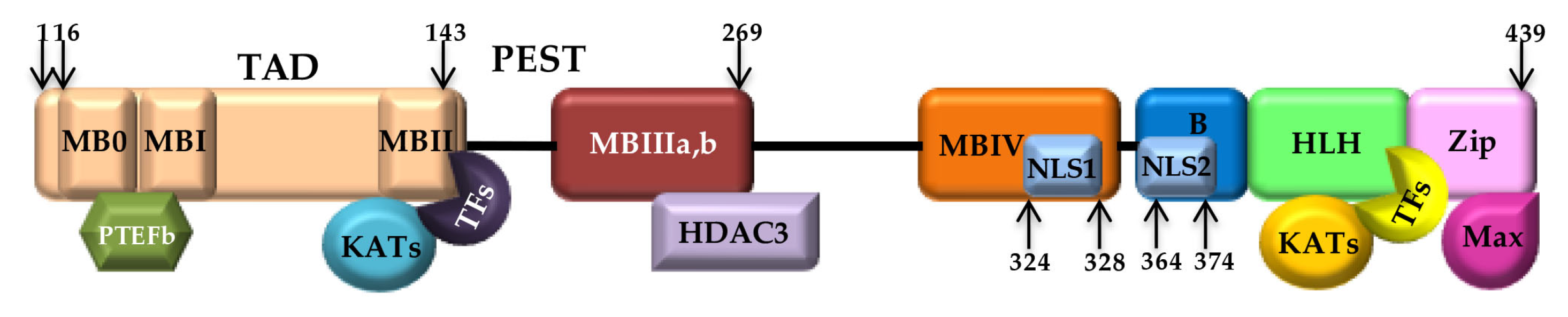
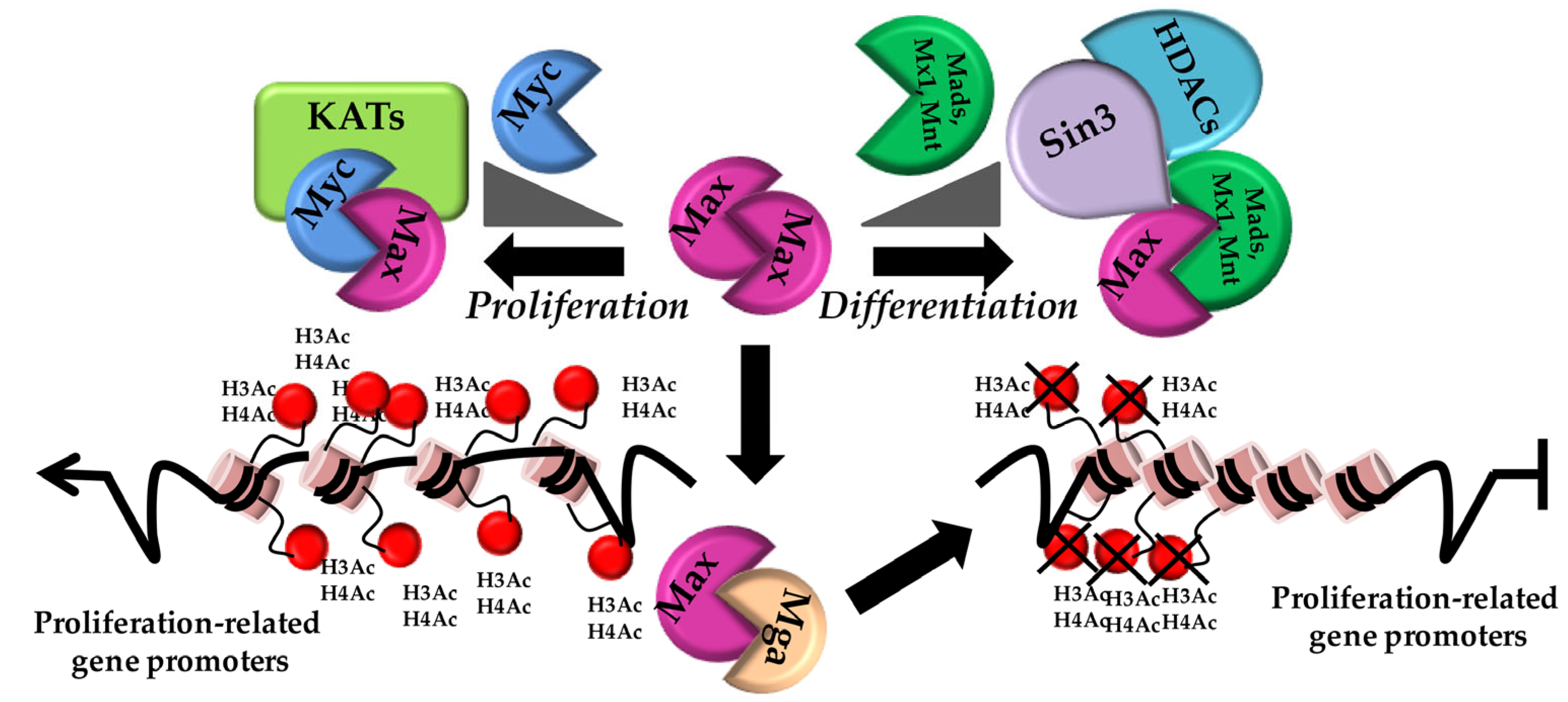
2.1. The Myc (Max) Network
2.2. Myc Transcription beyond DNA Binding
3. The Role of Myc in Pluripotent Stem Cells
3.1. Myc and the Stem Cells Epigenome
3.2. Myc in ES
3.3. Myc and Somatic Cell Reprogramming
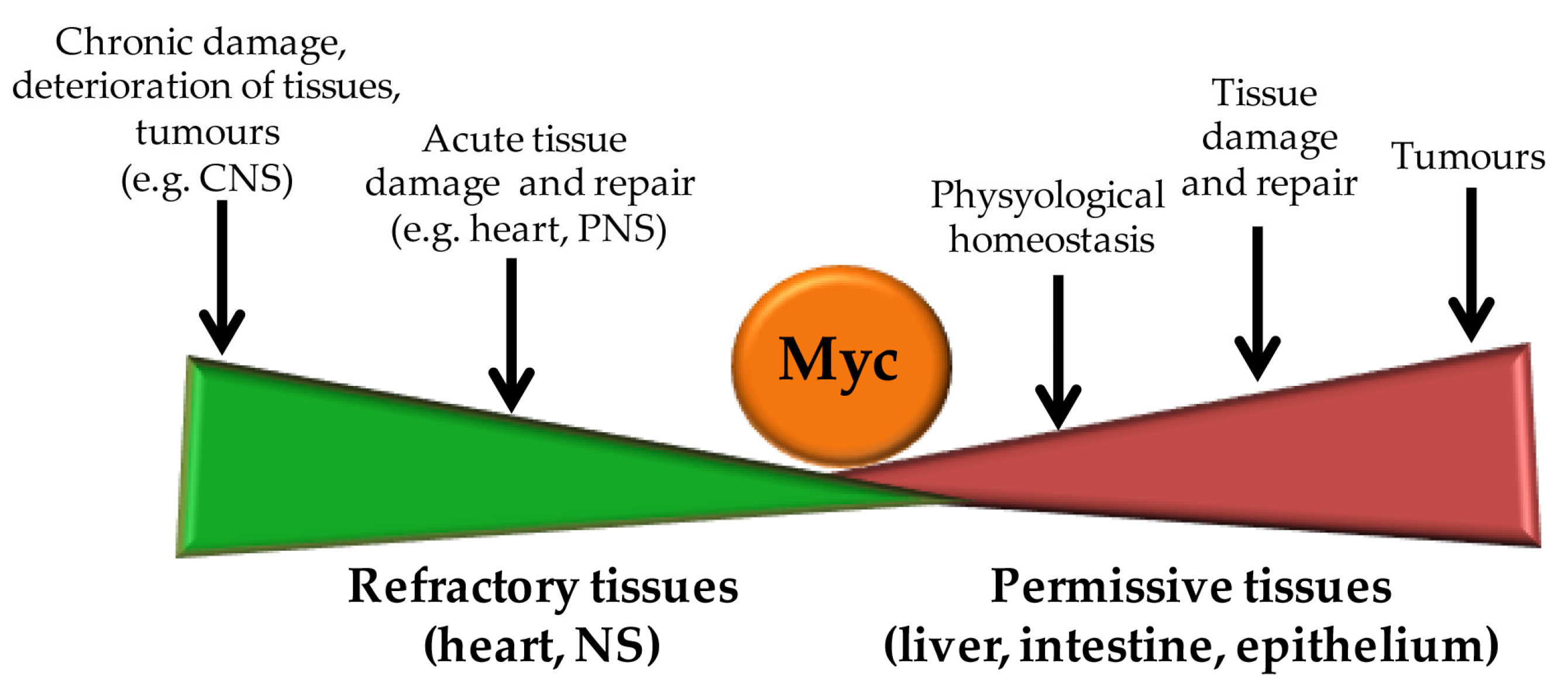
4. Mammalian Tissue Regeneration: A Transcriptional Point of View
5. Myc Action in Tissue Regeneration
5.1. Myc in Liver Regeneration
5.2. Myc Role in Pancreatic β-Cells’ Replacement
5.3. Myc in the Regulation of Intestinal Crypts’ Stemness and Regeneration
5.4. The Role of Myc in Cardiomyocytes’ Proliferation
5.5. Myc and the Regeneration of the Nervous System
6. Conclusions and Translational Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Goldman, J.A.; Poss, K.D. Gene regulatory programmes of tissue regeneration. Nat. Rev. Genet. 2020, 21, 511–525. [Google Scholar] [CrossRef] [PubMed]
- Mokalled, M.H.; Poss, K.D. A Regeneration Toolkit. Dev. Cell 2018, 47, 267–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wells, J.M.; Watt, F.M. Diverse mechanisms for endogenous regeneration and repair in mammalian organs. Nature 2018, 557, 322–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colby, W.W.; Chen, E.Y.; Smith, D.H.; Levinson, A.D. Identification and nucleotide sequence of a human locus homologous to the v-myc oncogene of avian myelocytomatosis virus MC29. Nature 1983, 301, 722–725. [Google Scholar] [CrossRef]
- Blackwood, E.M.; Eisenman, R.N. Max: A helix-loop-helix zipper protein that forms a sequence-specific DNA-binding complex with Myc. Science 1991, 251, 1211–1217. [Google Scholar] [CrossRef]
- Kanazawa, S.; Soucek, L.; Evan, G.; Okamoto, T.; Peterlin, B.M. c-Myc recruits P-TEFb for transcription, cellular proliferation and apoptosis. Oncogene 2003, 22, 5707–5711. [Google Scholar] [CrossRef] [Green Version]
- Evan, G.I.; Wyllie, A.H.; Gilbert, C.S.; Littlewood, T.D.; Land, H.; Brooks, M.; Waters, C.M.; Penn, L.Z.; Hancock, D.C. Induction of apoptosis in fibroblasts by c-myc protein. Cell 1992, 69, 119–128. [Google Scholar] [CrossRef]
- Shi, Y.; Glynn, J.M.; Guilbert, L.J.; Cotter, T.G.; Bissonnette, R.P.; Green, D.R. Role for c-myc in activation-induced apoptotic cell death in T cell hybridomas. Science 1992, 257, 212–214. [Google Scholar] [CrossRef]
- Soucie, E.L.; Annis, M.G.; Sedivy, J.; Filmus, J.; Leber, B.; Andrews, D.W.; Penn, L.Z. Myc potentiates apoptosis by stimulating Bax activity at the mitochondria. Mol. Cell Biol. 2001, 21, 4725–4736. [Google Scholar] [CrossRef] [Green Version]
- Shim, H.; Dolde, C.; Lewis, B.C.; Wu, C.S.; Dang, G.; Jungmann, R.A.; Dalla-Favera, R.; Dang, C.V. c-Myc transactivation of LDH-A: Implications for tumor metabolism and growth. Proc. Natl. Acad. Sci. USA 1997, 94, 6658–6663. [Google Scholar] [CrossRef]
- Gao, P.; Tchernyshyov, I.; Chang, T.C.; Lee, Y.S.; Kita, K.; Ochi, T.; Zeller, K.I.; De Marzo, A.M.; Van Eyk, J.E.; Mendell, J.T.; et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 2009, 458, 762–765. [Google Scholar] [CrossRef] [Green Version]
- Bello-Fernandez, C.; Packham, G.; Cleveland, J.L. The ornithine decarboxylase gene is a transcriptional target of c-Myc. Proc. Natl. Acad. Sci. USA 1993, 90, 7804–7808. [Google Scholar] [CrossRef]
- Hydbring, P.; Larsson, L.G. Cdk2: A key regulator of the senescence control function of Myc. Aging 2010, 2, 244–250. [Google Scholar] [CrossRef] [Green Version]
- Singh, V.P.; Hassan, H.; Deng, F.; Tsuchiya, D.; McKinney, S.; Ferro, K.; Gerton, J.L. c-Myc promotes polyploidy in murine trophoblast cells and suppresses senescence. Development 2023, 150, 11. [Google Scholar] [CrossRef]
- Cartwright, P.; McLean, C.; Sheppard, A.; Rivett, D.; Jones, K.; Dalton, S. LIF/STAT3 controls ES cell self-renewal and pluripotency by a Myc-dependent mechanism. Development 2005, 132, 885–896. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, K.; Yamanaka, S. A decade of transcription factor-mediated reprogramming to pluripotency. Nat. Rev. Mol. Cell Biol. 2016, 17, 183–193. [Google Scholar] [CrossRef]
- Rahl, P.B.; Lin, C.Y.; Seila, A.C.; Flynn, R.A.; McCuine, S.; Burge, C.B.; Sharp, P.A.; Young, R.A. c-Myc regulates transcriptional pause release. Cell 2010, 141, 432–445. [Google Scholar] [CrossRef] [Green Version]
- Baluapuri, A.; Hofstetter, J.; Dudvarski Stankovic, N.; Endres, T.; Bhandare, P.; Vos, S.M.; Adhikari, B.; Schwarz, J.D.; Narain, A.; Vogt, M.; et al. MYC Recruits SPT5 to RNA Polymerase II to Promote Processive Transcription Elongation. Mol. Cell 2019, 74, 674–687.e611. [Google Scholar] [CrossRef] [Green Version]
- Scagnoli, F.; Palma, A.; Favia, A.; Scuoppo, C.; Illi, B.; Nasi, S. A New Insight into MYC Action: Control of RNA Polymerase II Methylation and Transcription Termination. Biomedicines 2023, 11, 412. [Google Scholar] [CrossRef]
- Koh, C.M.; Bezzi, M.; Low, D.H.; Ang, W.X.; Teo, S.X.; Gay, F.P.; Al-Haddawi, M.; Tan, S.Y.; Osato, M.; Sabo, A.; et al. MYC regulates the core pre-mRNA splicing machinery as an essential step in lymphomagenesis. Nature 2015, 523, 96–100. [Google Scholar] [CrossRef] [PubMed]
- See, Y.X.; Chen, K.; Fullwood, M.J. MYC overexpression leads to increased chromatin interactions at super-enhancers and MYC binding sites. Genome Res. 2022, 32, 629–642. [Google Scholar] [CrossRef] [PubMed]
- Psathas, J.N.; Thomas-Tikhonenko, A. MYC and the art of microRNA maintenance. Cold Spring Harb. Perspect. Med. 2014, 4, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dang, C.V. MYC on the path to cancer. Cell 2012, 149, 22–35. [Google Scholar] [CrossRef] [Green Version]
- Kohl, N.E.; Kanda, N.; Schreck, R.R.; Bruns, G.; Latt, S.A.; Gilbert, F.; Alt, F.W. Transposition and amplification of oncogene-related sequences in human neuroblastomas. Cell 1983, 35, 359–367. [Google Scholar] [CrossRef]
- Nau, M.M.; Brooks, B.J.; Battey, J.; Sausville, E.; Gazdar, A.F.; Kirsch, I.R.; McBride, O.W.; Bertness, V.; Hollis, G.F.; Minna, J.D. L-myc, a new myc-related gene amplified and expressed in human small cell lung cancer. Nature 1985, 318, 69–73. [Google Scholar] [CrossRef]
- Kalkat, M.; Resetca, D.; Lourenco, C.; Chan, P.K.; Wei, Y.; Shiah, Y.J.; Vitkin, N.; Tong, Y.; Sunnerhagen, M.; Done, S.J.; et al. MYC Protein Interactome Profiling Reveals Functionally Distinct Regions that Cooperate to Drive Tumorigenesis. Mol. Cell 2018, 72, 836–848.e837. [Google Scholar] [CrossRef] [Green Version]
- Beaulieu, M.E.; Castillo, F.; Soucek, L. Structural and Biophysical Insights into the Function of the Intrinsically Disordered Myc Oncoprotein. Cells 2020, 9, 1038. [Google Scholar] [CrossRef]
- Cole, M.D. Myc meets its Max. Cell 1991, 65, 715–716. [Google Scholar] [CrossRef]
- Grandori, C.; Cowley, S.M.; James, L.P.; Eisenman, R.N. The Myc/Max/Mad network and the transcriptional control of cell behavior. Annu. Rev. Cell Dev. Biol. 2000, 16, 653–699. [Google Scholar] [CrossRef]
- Forman-Kay, J.D.; Mittag, T. From sequence and forces to structure, function, and evolution of intrinsically disordered proteins. Structure 2013, 21, 1492–1499. [Google Scholar] [CrossRef] [Green Version]
- McDuff, F.O.; Naud, J.F.; Montagne, M.; Sauve, S.; Lavigne, P. The Max homodimeric b-HLH-LZ significantly interferes with the specific heterodimerization between the c-Myc and Max b-HLH-LZ in absence of DNA: A quantitative analysis. J. Mol. Recognit. 2009, 22, 261–269. [Google Scholar] [CrossRef]
- Sammak, S.; Hamdani, N.; Gorrec, F.; Allen, M.D.; Freund, S.M.V.; Bycroft, M.; Zinzalla, G. Crystal Structures and Nuclear Magnetic Resonance Studies of the Apo Form of the c-MYC:MAX bHLHZip Complex Reveal a Helical Basic Region in the Absence of DNA. Biochemistry 2019, 58, 3144–3154. [Google Scholar] [CrossRef] [Green Version]
- Nair, S.K.; Burley, S.K. X-ray structures of Myc-Max and Mad-Max recognizing DNA. Molecular bases of regulation by proto-oncogenic transcription factors. Cell 2003, 112, 193–205. [Google Scholar] [CrossRef] [Green Version]
- Whitfield, J.R.; Beaulieu, M.E.; Soucek, L. Strategies to Inhibit Myc and Their Clinical Applicability. Front. Cell Dev. Biol. 2017, 5, 10. [Google Scholar] [CrossRef] [Green Version]
- Berg, T.; Cohen, S.B.; Desharnais, J.; Sonderegger, C.; Maslyar, D.J.; Goldberg, J.; Boger, D.L.; Vogt, P.K. Small-molecule antagonists of Myc/Max dimerization inhibit Myc-induced transformation of chicken embryo fibroblasts. Proc. Natl. Acad. Sci. USA 2002, 99, 3830–3835. [Google Scholar] [CrossRef]
- Yin, X.; Giap, C.; Lazo, J.S.; Prochownik, E.V. Low molecular weight inhibitors of Myc-Max interaction and function. Oncogene 2003, 22, 6151–6159. [Google Scholar] [CrossRef] [Green Version]
- Han, H.; Jain, A.D.; Truica, M.I.; Izquierdo-Ferrer, J.; Anker, J.F.; Lysy, B.; Sagar, V.; Luan, Y.; Chalmers, Z.R.; Unno, K.; et al. Small-Molecule MYC Inhibitors Suppress Tumor Growth and Enhance Immunotherapy. Cancer Cell 2019, 36, 483–497.e415. [Google Scholar] [CrossRef]
- Soucek, L.; Helmer-Citterich, M.; Sacco, A.; Jucker, R.; Cesareni, G.; Nasi, S. Design and properties of a Myc derivative that efficiently homodimerizes. Oncogene 1998, 17, 2463–2472. [Google Scholar] [CrossRef] [Green Version]
- Savino, M.; Annibali, D.; Carucci, N.; Favuzzi, E.; Cole, M.D.; Evan, G.I.; Soucek, L.; Nasi, S. The action mechanism of the Myc inhibitor termed Omomyc may give clues on how to target Myc for cancer therapy. PLoS ONE 2011, 6, e22284. [Google Scholar] [CrossRef] [Green Version]
- Delmore, J.E.; Issa, G.C.; Lemieux, M.E.; Rahl, P.B.; Shi, J.; Jacobs, H.M.; Kastritis, E.; Gilpatrick, T.; Paranal, R.M.; Qi, J.; et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 2011, 146, 904–917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dauch, D.; Rudalska, R.; Cossa, G.; Nault, J.C.; Kang, T.W.; Wuestefeld, T.; Hohmeyer, A.; Imbeaud, S.; Yevsa, T.; Hoenicke, L.; et al. A MYC-aurora kinase A protein complex represents an actionable drug target in p53-altered liver cancer. Nat. Med. 2016, 22, 744–753. [Google Scholar] [CrossRef] [PubMed]
- Grandori, C.; Eisenman, R.N. Myc target genes. Trends Biochem. Sci. 1997, 22, 177–181. [Google Scholar] [CrossRef] [PubMed]
- Farhana, L.; Dawson, M.I.; Fontana, J.A. Down regulation of miR-202 modulates Mxd1 and Sin3A repressor complexes to induce apoptosis of pancreatic cancer cells. Cancer Biol. Ther. 2015, 16, 115–124. [Google Scholar] [CrossRef]
- Moroy, T.; Saba, I.; Kosan, C. The role of the transcription factor Miz-1 in lymphocyte development and lymphomagenesis-Binding Myc makes the difference. Semin. Immunol. 2011, 23, 379–387. [Google Scholar] [CrossRef]
- Nie, Z.; Hu, G.; Wei, G.; Cui, K.; Yamane, A.; Resch, W.; Wang, R.; Green, D.R.; Tessarollo, L.; Casellas, R.; et al. c-Myc is a universal amplifier of expressed genes in lymphocytes and embryonic stem cells. Cell 2012, 151, 68–79. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.Y.; Loven, J.; Rahl, P.B.; Paranal, R.M.; Burge, C.B.; Bradner, J.E.; Lee, T.I.; Young, R.A. Transcriptional amplification in tumor cells with elevated c-Myc. Cell 2012, 151, 56–67. [Google Scholar] [CrossRef] [Green Version]
- Nie, Z.; Guo, C.; Das, S.K.; Chow, C.C.; Batchelor, E.; Simons, S.S.J.; Levens, D. Dissecting transcriptional amplification by MYC. eLife 2020, 9, e52483. [Google Scholar] [CrossRef]
- Walz, S.; Lorenzin, F.; Morton, J.; Wiese, K.E.; von Eyss, B.; Herold, S.; Rycak, L.; Dumay-Odelot, H.; Karim, S.; Bartkuhn, M.; et al. Activation and repression by oncogenic MYC shape tumour-specific gene expression profiles. Nature 2014, 511, 483–487. [Google Scholar] [CrossRef]
- Guccione, E.; Martinato, F.; Finocchiaro, G.; Luzi, L.; Tizzoni, L.; Dall’ Olio, V.; Zardo, G.; Nervi, C.; Bernard, L.; Amati, B. Myc-binding-site recognition in the human genome is determined by chromatin context. Nat. Cell Biol. 2006, 8, 764–770. [Google Scholar] [CrossRef]
- Das, S.K.; Kuzin, V.; Cameron, D.P.; Sanford, S.; Jha, R.K.; Nie, Z.; Rosello, M.T.; Holewinski, R.; Andresson, T.; Wisniewski, J.; et al. MYC assembles and stimulates topoisomerases 1 and 2 in a “topoisome”. Mol. Cell 2022, 82, 140–158.e112. [Google Scholar] [CrossRef]
- Cowling, V.H.; Cole, M.D. The Myc transactivation domain promotes global phosphorylation of the RNA polymerase II carboxy-terminal domain independently of direct DNA binding. Mol. Cell Biol. 2007, 27, 2059–2073. [Google Scholar] [CrossRef] [Green Version]
- Galardi, S.; Savino, M.; Scagnoli, F.; Pellegatta, S.; Pisati, F.; Zambelli, F.; Illi, B.; Annibali, D.; Beji, S.; Orecchini, E.; et al. Resetting cancer stem cell regulatory nodes upon MYC inhibition. EMBO Rep. 2016, 17, 1872–1889. [Google Scholar] [CrossRef] [Green Version]
- Davis, A.C.; Wims, M.; Spotts, G.D.; Hann, S.R.; Bradley, A. A null c-myc mutation causes lethality before 10.5 days of gestation in homozygotes and reduced fertility in heterozygous female mice. Genes Dev. 1993, 7, 671–682. [Google Scholar] [CrossRef] [Green Version]
- Zimmerman, K.A.; Yancopoulos, G.D.; Collum, R.G.; Smith, R.K.; Kohl, N.E.; Denis, K.A.; Nau, M.M.; Witte, O.N.; Toran-Allerand, D.; Gee, C.E.; et al. Differential expression of myc family genes during murine development. Nature 1986, 319, 780–783. [Google Scholar] [CrossRef]
- Malynn, B.A.; de Alboran, I.M.; O’Hagan, R.C.; Bronson, R.; Davidson, L.; DePinho, R.A.; Alt, F.W. N-myc can functionally replace c-myc in murine development, cellular growth, and differentiation. Genes Dev. 2000, 14, 1390–1399. [Google Scholar] [CrossRef]
- Smith, K.N.; Singh, A.M.; Dalton, S. Myc represses primitive endoderm differentiation in pluripotent stem cells. Cell Stem Cell 2010, 7, 343–354. [Google Scholar] [CrossRef] [Green Version]
- Varlakhanova, N.V.; Cotterman, R.F.; deVries, W.N.; Morgan, J.; Donahue, L.R.; Murray, S.; Knowles, B.B.; Knoepfler, P.S. myc maintains embryonic stem cell pluripotency and self-renewal. Differentiation 2010, 80, 9–19. [Google Scholar] [CrossRef] [Green Version]
- Gaspar-Maia, A.; Alajem, A.; Meshorer, E.; Ramalho-Santos, M. Open chromatin in pluripotency and reprogramming. Nat. Rev. Mol. Cell Biol. 2011, 12, 36–47. [Google Scholar] [CrossRef] [Green Version]
- Tee, W.W.; Reinberg, D. Chromatin features and the epigenetic regulation of pluripotency states in ESCs. Development 2014, 141, 2376–2390. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Adli, M.; Zou, J.Y.; Verstappen, G.; Coyne, M.; Zhang, X.; Durham, T.; Miri, M.; Deshpande, V.; De Jager, P.L.; et al. Genome-wide chromatin state transitions associated with developmental and environmental cues. Cell 2013, 152, 642–654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernstein, B.E.; Mikkelsen, T.S.; Xie, X.; Kamal, M.; Huebert, D.J.; Cuff, J.; Fry, B.; Meissner, A.; Wernig, M.; Plath, K.; et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 2006, 125, 315–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, G.; Tian, S.; Nie, J.; Yang, C.; Ruotti, V.; Wei, H.; Jonsdottir, G.A.; Stewart, R.; Thomson, J.A. Whole-genome analysis of histone H3 lysine 4 and lysine 27 methylation in human embryonic stem cells. Cell Stem Cell 2007, 1, 299–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frye, M.; Fisher, A.G.; Watt, F.M. Epidermal stem cells are defined by global histone modifications that are altered by Myc-induced differentiation. PLoS ONE 2007, 2, e763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cotterman, R.; Jin, V.X.; Krig, S.R.; Lemen, J.M.; Wey, A.; Farnham, P.J.; Knoepfler, P.S. N-Myc regulates a widespread euchromatic program in the human genome partially independent of its role as a classical transcription factor. Cancer Res. 2008, 68, 9654–9662. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.H.; Lin, C.; Tanaka, H.; Fero, M.L.; Eisenman, R.N. Gene regulation and epigenetic remodeling in murine embryonic stem cells by c-Myc. PLoS ONE 2009, 4, e7839. [Google Scholar] [CrossRef]
- Krepelova, A.; Neri, F.; Maldotti, M.; Rapelli, S.; Oliviero, S. Myc and max genome-wide binding sites analysis links the Myc regulatory network with the polycomb and the core pluripotency networks in mouse embryonic stem cells. PLoS ONE 2014, 9, e88933. [Google Scholar] [CrossRef]
- Kim, J.; Chu, J.; Shen, X.; Wang, J.; Orkin, S.H. An extended transcriptional network for pluripotency of embryonic stem cells. Cell 2008, 132, 1049–1061. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Woo, A.J.; Chu, J.; Snow, J.W.; Fujiwara, Y.; Kim, C.G.; Cantor, A.B.; Orkin, S.H. A Myc network accounts for similarities between embryonic stem and cancer cell transcription programs. Cell 2010, 143, 313–324. [Google Scholar] [CrossRef] [Green Version]
- Das, P.P.; Shao, Z.; Beyaz, S.; Apostolou, E.; Pinello, L.; De Los Angeles, A.; O’Brien, K.; Atsma, J.M.; Fujiwara, Y.; Nguyen, M.; et al. Distinct and combinatorial functions of Jmjd2b/Kdm4b and Jmjd2c/Kdm4c in mouse embryonic stem cell identity. Mol. Cell 2014, 53, 32–48. [Google Scholar] [CrossRef] [Green Version]
- Amente, S.; Lania, L.; Majello, B. The histone LSD1 demethylase in stemness and cancer transcription programs. Biochim. Biophys. Acta 2013, 1829, 981–986. [Google Scholar] [CrossRef] [Green Version]
- Smith, K.N.; Lim, J.M.; Wells, L.; Dalton, S. Myc orchestrates a regulatory network required for the establishment and maintenance of pluripotency. Cell Cycle 2011, 10, 592–597. [Google Scholar] [CrossRef]
- Hu, G.; Wade, P.A. NuRD and pluripotency: A complex balancing act. Cell Stem Cell 2012, 10, 497–503. [Google Scholar] [CrossRef] [Green Version]
- Fagnocchi, L.; Mazzoleni, S.; Zippo, A. Integration of Signaling Pathways with the Epigenetic Machinery in the Maintenance of Stem Cells. Stem Cells Int. 2016, 2016, 8652748. [Google Scholar] [CrossRef] [Green Version]
- Bonasio, R.; Tu, S.; Reinberg, D. Molecular signals of epigenetic states. Science 2010, 330, 612–616. [Google Scholar] [CrossRef] [Green Version]
- Laurenti, E.; Varnum-Finney, B.; Wilson, A.; Ferrero, I.; Blanco-Bose, W.E.; Ehninger, A.; Knoepfler, P.S.; Cheng, P.F.; MacDonald, H.R.; Eisenman, R.N.; et al. Hematopoietic stem cell function and survival depend on c-Myc and N-Myc activity. Cell Stem Cell 2008, 3, 611–624. [Google Scholar] [CrossRef] [Green Version]
- Smith, K.; Dalton, S. Myc transcription factors: Key regulators behind establishment and maintenance of pluripotency. Regen. Med. 2010, 5, 947–959. [Google Scholar] [CrossRef] [Green Version]
- Gibson, J.D.; Jakuba, C.M.; Boucher, N.; Holbrook, K.A.; Carter, M.G.; Nelson, C.E. Single-cell transcript analysis of human embryonic stem cells. Integr. Biol. 2009, 1, 540–551. [Google Scholar] [CrossRef]
- Doyon, Y.; Cote, J. The highly conserved and multifunctional NuA4 HAT complex. Curr. Opin. Genet. Dev. 2004, 14, 147–154. [Google Scholar] [CrossRef]
- Faast, R.; White, J.; Cartwright, P.; Crocker, L.; Sarcevic, B.; Dalton, S. Cdk6-cyclin D3 activity in murine ES cells is resistant to inhibition by p16(INK4a). Oncogene 2004, 23, 491–502. [Google Scholar] [CrossRef]
- Stead, E.; White, J.; Faast, R.; Conn, S.; Goldstone, S.; Rathjen, J.; Dhingra, U.; Rathjen, P.; Walker, D.; Dalton, S. Pluripotent cell division cycles are driven by ectopic Cdk2, cyclin A/E and E2F activities. Oncogene 2002, 21, 8320–8333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savatier, P.; Huang, S.; Szekely, L.; Wiman, K.G.; Samarut, J. Contrasting patterns of retinoblastoma protein expression in mouse embryonic stem cells and embryonic fibroblasts. Oncogene 1994, 9, 809–818. [Google Scholar] [PubMed]
- White, J.; Stead, E.; Faast, R.; Conn, S.; Cartwright, P.; Dalton, S. Developmental activation of the Rb-E2F pathway and establishment of cell cycle-regulated cyclin-dependent kinase activity during embryonic stem cell differentiation. Mol. Biol. Cell 2005, 16, 2018–2027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bretones, G.; Delgado, M.D.; Leon, J. Myc and cell cycle control. Biochim. Biophys. Acta 2015, 1849, 506–516. [Google Scholar] [CrossRef]
- Brenner, C.; Deplus, R.; Didelot, C.; Loriot, A.; Vire, E.; De Smet, C.; Gutierrez, A.; Danovi, D.; Bernard, D.; Boon, T.; et al. Myc represses transcription through recruitment of DNA methyltransferase corepressor. EMBO J. 2005, 24, 336–346. [Google Scholar] [CrossRef]
- Sela, Y.; Molotski, N.; Golan, S.; Itskovitz-Eldor, J.; Soen, Y. Human embryonic stem cells exhibit increased propensity to differentiate during the G1 phase prior to phosphorylation of retinoblastoma protein. Stem Cells 2012, 30, 1097–1108. [Google Scholar] [CrossRef]
- Pauklin, S.; Vallier, L. The cell-cycle state of stem cells determines cell fate propensity. Cell 2013, 155, 135–147. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.M.; Dalton, S. The cell cycle and Myc intersect with mechanisms that regulate pluripotency and reprogramming. Cell Stem Cell 2009, 5, 141–149. [Google Scholar] [CrossRef] [Green Version]
- Gu, W.; Gaeta, X.; Sahakyan, A.; Chan, A.B.; Hong, C.S.; Kim, R.; Braas, D.; Plath, K.; Lowry, W.E.; Christofk, H.R. Glycolytic Metabolism Plays a Functional Role in Regulating Human Pluripotent Stem Cell State. Cell Stem Cell 2016, 19, 476–490. [Google Scholar] [CrossRef] [Green Version]
- Epstein, T.; Gatenby, R.A.; Brown, J.S. The Warburg effect as an adaptation of cancer cells to rapid fluctuations in energy demand. PLoS ONE 2017, 12, e0185085. [Google Scholar] [CrossRef] [Green Version]
- Stine, Z.E.; Walton, Z.E.; Altman, B.J.; Hsieh, A.L.; Dang, C.V. MYC, Metabolism, and Cancer. Cancer Discov. 2015, 5, 1024–1039. [Google Scholar] [CrossRef] [Green Version]
- Cencioni, C.; Scagnoli, F.; Spallotta, F.; Nasi, S.; Illi, B. The “Superoncogene” Myc at the Crossroad between Metabolism and Gene Expression in Glioblastoma Multiforme. Int. J. Mol. Sci. 2023, 24, 4217. [Google Scholar] [CrossRef]
- Cao, Y.; Guo, W.T.; Tian, S.; He, X.; Wang, X.W.; Liu, X.; Gu, K.L.; Ma, X.; Huang, D.; Hu, L.; et al. miR-290/371-Mbd2-Myc circuit regulates glycolytic metabolism to promote pluripotency. EMBO J. 2015, 34, 609–623. [Google Scholar] [CrossRef] [Green Version]
- Edmunds, L.R.; Sharma, L.; Kang, A.; Lu, J.; Vockley, J.; Basu, S.; Uppala, R.; Goetzman, E.S.; Beck, M.E.; Scott, D.; et al. c-Myc programs fatty acid metabolism and dictates acetyl-CoA abundance and fate. J. Biol. Chem. 2014, 289, 25382–25392. [Google Scholar] [CrossRef] [Green Version]
- Shyh-Chang, N.; Locasale, J.W.; Lyssiotis, C.A.; Zheng, Y.; Teo, R.Y.; Ratanasirintrawoot, S.; Zhang, J.; Onder, T.; Unternaehrer, J.J.; Zhu, H.; et al. Influence of threonine metabolism on S-adenosylmethionine and histone methylation. Science 2013, 339, 222–226. [Google Scholar] [CrossRef] [Green Version]
- Moussaieff, A.; Rouleau, M.; Kitsberg, D.; Cohen, M.; Levy, G.; Barasch, D.; Nemirovski, A.; Shen-Orr, S.; Laevsky, I.; Amit, M.; et al. Glycolysis-mediated changes in acetyl-CoA and histone acetylation control the early differentiation of embryonic stem cells. Cell Metab. 2015, 21, 392–402. [Google Scholar] [CrossRef] [Green Version]
- Shiraki, N.; Shiraki, Y.; Tsuyama, T.; Obata, F.; Miura, M.; Nagae, G.; Aburatani, H.; Kume, K.; Endo, F.; Kume, S. Methionine metabolism regulates maintenance and differentiation of human pluripotent stem cells. Cell Metab. 2014, 19, 780–794. [Google Scholar] [CrossRef] [Green Version]
- Carey, B.W.; Finley, L.W.; Cross, J.R.; Allis, C.D.; Thompson, C.B. Intracellular alpha-ketoglutarate maintains the pluripotency of embryonic stem cells. Nature 2015, 518, 413–416. [Google Scholar] [CrossRef] [Green Version]
- Spitalieri, P.; Talarico, V.R.; Murdocca, M.; Novelli, G.; Sangiuolo, F. Human induced pluripotent stem cells for monogenic disease modelling and therapy. World J. Stem Cells 2016, 8, 118–135. [Google Scholar] [CrossRef]
- Streckfuss-Bomeke, K.; Wolf, F.; Azizian, A.; Stauske, M.; Tiburcy, M.; Wagner, S.; Hubscher, D.; Dressel, R.; Chen, S.; Jende, J.; et al. Comparative study of human-induced pluripotent stem cells derived from bone marrow cells, hair keratinocytes, and skin fibroblasts. Eur. Heart J. 2013, 34, 2618–2629. [Google Scholar] [CrossRef] [Green Version]
- Rao, L.; Qian, Y.; Khodabukus, A.; Ribar, T.; Bursac, N. Engineering human pluripotent stem cells into a functional skeletal muscle tissue. Nat. Commun. 2018, 9, 126. [Google Scholar] [CrossRef] [Green Version]
- Cyganek, L.; Tiburcy, M.; Sekeres, K.; Gerstenberg, K.; Bohnenberger, H.; Lenz, C.; Henze, S.; Stauske, M.; Salinas, G.; Zimmermann, W.H.; et al. Deep phenotyping of human induced pluripotent stem cell-derived atrial and ventricular cardiomyocytes. JCI Insight 2018, 3, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Böscke, R.; Tang, P.C.; Hartman, B.H.; Heller, S.; Koehler, K.R. Hair Follicle Development in Mouse Pluripotent Stem Cell-Derived Skin Organoids. Cell Rep. 2018, 22, 242–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, Y.; Gotoh, S.; Korogi, Y.; Seki, M.; Konishi, S.; Ikeo, S.; Sone, N.; Nagasaki, T.; Matsumoto, H.; Muro, S.; et al. Long-term expansion of alveolar stem cells derived from human iPS cells in organoids. Nat. Methods 2017, 14, 1097–1106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ardeshirylajimi, A. Applied Induced Pluripotent Stem Cells in Combination with Biomaterials in Bone Tissue Engineering. J. Cell. Biochem. 2017, 118, 3034–3042. [Google Scholar] [CrossRef]
- Wang, A.; Tang, Z.; Park, I.H.; Zhu, Y.; Patel, S.; Daley, G.Q.; Li, S. Induced pluripotent stem cells for neural tissue engineering. Biomaterials 2011, 32, 5023–5032. [Google Scholar] [CrossRef] [Green Version]
- Brambrink, T.; Foreman, R.; Welstead, G.G.; Lengner, C.J.; Wernig, M.; Suh, H.; Jaenisch, R. Sequential expression of pluripotency markers during direct reprogramming of mouse somatic cells. Cell Stem Cell 2008, 2, 151–159. [Google Scholar] [CrossRef] [Green Version]
- Sridharan, R.; Tchieu, J.; Mason, M.J.; Yachechko, R.; Kuoy, E.; Horvath, S.; Zhou, Q.; Plath, K. Role of the murine reprogramming factors in the induction of pluripotency. Cell 2009, 136, 364–377. [Google Scholar] [CrossRef] [Green Version]
- Soufi, A.; Donahue, G.; Zaret, K.S. Facilitators and impediments of the pluripotency reprogramming factors’ initial engagement with the genome. Cell 2012, 151, 994–1004. [Google Scholar] [CrossRef] [Green Version]
- Okita, K.; Ichisaka, T.; Yamanaka, S. Generation of germline-competent induced pluripotent stem cells. Nature 2007, 448, 313–317. [Google Scholar] [CrossRef]
- Nakagawa, M.; Koyanagi, M.; Tanabe, K.; Takahashi, K.; Ichisaka, T.; Aoi, T.; Okita, K.; Mochiduki, Y.; Takizawa, N.; Yamanaka, S. Generation of induced pluripotent stem cells without Myc from mouse and human fibroblasts. Nat. Biotechnol. 2008, 26, 101–106. [Google Scholar] [CrossRef]
- Wernig, M.; Meissner, A.; Cassady, J.P.; Jaenisch, R. c-Myc is dispensable for direct reprogramming of mouse fibroblasts. Cell Stem Cell 2008, 2, 10–12. [Google Scholar] [CrossRef] [Green Version]
- Nakagawa, M.; Takizawa, N.; Narita, M.; Ichisaka, T.; Yamanaka, S. Promotion of direct reprogramming by transformation-deficient Myc. Proc. Natl. Acad. Sci. USA 2010, 107, 14152–14157. [Google Scholar] [CrossRef]
- Torchy, M.P.; Hamiche, A.; Klaholz, B.P. Structure and function insights into the NuRD chromatin remodeling complex. Cell. Mol. Life Sci. 2015, 72, 2491–2507. [Google Scholar] [CrossRef]
- Gingold, H.; Tehler, D.; Christoffersen, N.R.; Nielsen, M.M.; Asmar, F.; Kooistra, S.M.; Christophersen, N.S.; Christensen, L.L.; Borre, M.; Sorensen, K.D.; et al. A dual program for translation regulation in cellular proliferation and differentiation. Cell 2014, 158, 1281–1292. [Google Scholar] [CrossRef] [Green Version]
- Zviran, A.; Mor, N.; Rais, Y.; Gingold, H.; Peles, S.; Chomsky, E.; Viukov, S.; Buenrostro, J.D.; Scognamiglio, R.; Weinberger, L.; et al. Deterministic Somatic Cell Reprogramming Involves Continuous Transcriptional Changes Governed by Myc and Epigenetic-Driven Modules. Cell Stem Cell 2019, 24, 328–341.e329. [Google Scholar] [CrossRef] [Green Version]
- Felton-Edkins, Z.A.; Kenneth, N.S.; Brown, T.R.; Daly, N.L.; Gomez-Roman, N.; Grandori, C.; Eisenman, R.N.; White, R.J. Direct regulation of RNA polymerase III transcription by RB, p53 and c-Myc. Cell Cycle 2003, 2, 181–184. [Google Scholar] [CrossRef] [Green Version]
- Campbell, K.J.; White, R.J. MYC regulation of cell growth through control of transcription by RNA polymerases I and III. Cold Spring Harb. Perspect. Med. 2014, 4, 5. [Google Scholar] [CrossRef]
- Lepilina, A.; Coon, A.N.; Kikuchi, K.; Holdway, J.E.; Roberts, R.W.; Burns, C.G.; Poss, K.D. A dynamic epicardial injury response supports progenitor cell activity during zebrafish heart regeneration. Cell 2006, 127, 607–619. [Google Scholar] [CrossRef] [Green Version]
- Fausett, B.V.; Goldman, D. A role for alpha1 tubulin-expressing Muller glia in regeneration of the injured zebrafish retina. J. Neurosci. 2006, 26, 6303–6313. [Google Scholar] [CrossRef] [Green Version]
- Ransom, R.C.; Carter, A.C.; Salhotra, A.; Leavitt, T.; Marecic, O.; Murphy, M.P.; Lopez, M.L.; Wei, Y.; Marshall, C.D.; Shen, E.Z.; et al. Mechanoresponsive stem cells acquire neural crest fate in jaw regeneration. Nature 2018, 563, 514–521. [Google Scholar] [CrossRef] [PubMed]
- Gupta, V.; Gemberling, M.; Karra, R.; Rosenfeld, G.E.; Evans, T.; Poss, K.D. An injury-responsive gata4 program shapes the zebrafish cardiac ventricle. Curr. Biol. 2013, 23, 1221–1227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schindler, Y.L.; Garske, K.M.; Wang, J.; Firulli, B.A.; Firulli, A.B.; Poss, K.D.; Yelon, D. Hand2 elevates cardiomyocyte production during zebrafish heart development and regeneration. Development 2014, 141, 3112–3122. [Google Scholar] [CrossRef] [Green Version]
- Oka, T.; Xu, J.; Molkentin, J.D. Re-employment of developmental transcription factors in adult heart disease. Semin. Cell Dev. Biol. 2007, 18, 117–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bisping, E.; Ikeda, S.; Kong, S.W.; Tarnavski, O.; Bodyak, N.; McMullen, J.R.; Rajagopal, S.; Son, J.K.; Ma, Q.; Springer, Z.; et al. Gata4 is required for maintenance of postnatal cardiac function and protection from pressure overload-induced heart failure. Proc. Natl. Acad. Sci. USA 2006, 103, 14471–14476. [Google Scholar] [CrossRef]
- Eming, S.A.; Wynn, T.A.; Martin, P. Inflammation and metabolism in tissue repair and regeneration. Science 2017, 356, 1026–1030. [Google Scholar] [CrossRef] [Green Version]
- Matias Santos, D.; Rita, A.M.; Casanellas, I.; Brito Ova, A.; Araujo, I.M.; Power, D.; Tiscornia, G. Ear wound regeneration in the African spiny mouse Acomys cahirinus. Regeneration 2016, 3, 52–61. [Google Scholar] [CrossRef]
- Simkin, J.; Gawriluk, T.R.; Gensel, J.C.; Seifert, A.W. Macrophages are necessary for epimorphic regeneration in African spiny mice. eLife 2017, 6, e24623. [Google Scholar] [CrossRef]
- Yang, K.; Kang, J. Tissue Regeneration Enhancer Elements: A Way to Unlock Endogenous Healing Power. Dev. Dyn. 2019, 248, 34–42. [Google Scholar] [CrossRef] [Green Version]
- Kang, J.; Hu, J.; Karra, R.; Dickson, A.L.; Tornini, V.A.; Nachtrab, G.; Gemberling, M.; Goldman, J.A.; Black, B.L.; Poss, K.D. Modulation of tissue repair by regeneration enhancer elements. Nature 2016, 532, 201–206. [Google Scholar] [CrossRef] [Green Version]
- Da Silva, S.M.; Gates, P.B.; Brockes, J.P. The newt ortholog of CD59 is implicated in proximodistal identity during amphibian limb regeneration. Dev. Cell 2002, 3, 547–555. [Google Scholar] [CrossRef]
- Vizcaya-Molina, E.; Klein, C.C.; Serras, F.; Mishra, R.K.; Guigo, R.; Corominas, M. Damage-responsive elements in Drosophila regeneration. Genome Res. 2018, 28, 1852–1866. [Google Scholar] [CrossRef] [Green Version]
- Yan, R.; Cigliola, V.; Oonk, K.A.; Petrover, Z.; DeLuca, S.; Wolfson, D.W.; Vekstein, A.; Mendiola, M.A.; Devlin, G.; Bishawi, M.; et al. An enhancer-based gene-therapy strategy for spatiotemporal control of cargoes during tissue repair. Cell Stem Cell 2023, 30, 96–111.e116. [Google Scholar] [CrossRef]
- Soukup, A.A.; Zheng, Y.; Mehta, C.; Wu, J.; Liu, P.; Cao, M.; Hofmann, I.; Zhou, Y.; Zhang, J.; Johnson, K.D.; et al. Single-nucleotide human disease mutation inactivates a blood-regenerative GATA2 enhancer. J. Clin. Investig. 2019, 129, 1180–1192. [Google Scholar] [CrossRef] [Green Version]
- Hewitt, K.J.; Katsumura, K.R.; Matson, D.R.; Devadas, P.; Tanimura, N.; Hebert, A.S.; Coon, J.J.; Kim, J.S.; Dewey, C.N.; Keles, S.; et al. GATA Factor-Regulated Samd14 Enhancer Confers Red Blood Cell Regeneration and Survival in Severe Anemia. Dev. Cell 2017, 42, 213–225.e214. [Google Scholar] [CrossRef] [Green Version]
- Gadye, L.; Das, D.; Sanchez, M.A.; Street, K.; Baudhuin, A.; Wagner, A.; Cole, M.B.; Choi, Y.G.; Yosef, N.; Purdom, E.; et al. Injury Activates Transient Olfactory Stem Cell States with Diverse Lineage Capacities. Cell Stem Cell 2017, 21, 775–790.e779. [Google Scholar] [CrossRef] [Green Version]
- Rocha-Martins, M.; de Toledo, B.C.; Santos-Franca, P.L.; Oliveira-Valenca, V.M.; Vieira-Vieira, C.H.; Matos-Rodrigues, G.E.; Linden, R.; Norden, C.; Martins, R.A.P.; Silveira, M.S. De novo genesis of retinal ganglion cells by targeted expression of Klf4 in vivo. Development 2019, 146, 176586. [Google Scholar] [CrossRef] [Green Version]
- Mahmoud, A.I.; Kocabas, F.; Muralidhar, S.A.; Kimura, W.; Koura, A.S.; Thet, S.; Porrello, E.R.; Sadek, H.A. Meis1 regulates postnatal cardiomyocyte cell cycle arrest. Nature 2013, 497, 249–253. [Google Scholar] [CrossRef] [Green Version]
- Xin, M.; Kim, Y.; Sutherland, L.B.; Murakami, M.; Qi, X.; McAnally, J.; Porrello, E.R.; Mahmoud, A.I.; Tan, W.; Shelton, J.M.; et al. Hippo pathway effector Yap promotes cardiac regeneration. Proc. Natl. Acad. Sci. USA 2013, 110, 13839–13844. [Google Scholar] [CrossRef]
- Patterson, M.; Barske, L.; Van Handel, B.; Rau, C.D.; Gan, P.; Sharma, A.; Parikh, S.; Denholtz, M.; Huang, Y.; Yamaguchi, Y.; et al. Frequency of mononuclear diploid cardiomyocytes underlies natural variation in heart regeneration. Nat. Genet. 2017, 49, 1346–1353. [Google Scholar] [CrossRef]
- Guzman-Ayala, M.; Sachs, M.; Koh, F.M.; Onodera, C.; Bulut-Karslioglu, A.; Lin, C.J.; Wong, P.; Nitta, R.; Song, J.S.; Ramalho-Santos, M. Chd1 is essential for the high transcriptional output and rapid growth of the mouse epiblast. Development 2015, 142, 118–127. [Google Scholar] [CrossRef] [Green Version]
- Mattout, A.; Meshorer, E. Chromatin plasticity and genome organization in pluripotent embryonic stem cells. Curr. Opin. Cell Biol. 2010, 22, 334–341. [Google Scholar] [CrossRef] [PubMed]
- Percharde, M.; Bulut-Karslioglu, A.; Ramalho-Santos, M. Hypertranscription in Development, Stem Cells, and Regeneration. Dev. Cell 2017, 40, 9–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jao, C.Y.; Salic, A. Exploring RNA transcription and turnover in vivo by using click chemistry. Proc. Natl. Acad. Sci. USA 2008, 105, 15779–15784. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.S.; Cserjesi, P.; Markham, B.E.; Molkentin, J.D. The transcription factors GATA4 and dHAND physically interact to synergistically activate cardiac gene expression through a p300-dependent mechanism. J. Biol. Chem. 2002, 277, 24390–24398. [Google Scholar] [CrossRef] [Green Version]
- Goldman, J.A.; Kuzu, G.; Lee, N.; Karasik, J.; Gemberling, M.; Foglia, M.J.; Karra, R.; Dickson, A.L.; Sun, F.; Tolstorukov, M.Y.; et al. Resolving Heart Regeneration by Replacement Histone Profiling. Dev. Cell 2017, 40, 392–404.e395. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Long, Q.; Wu, H.; Zhou, Y.; Duan, L.; Yuan, H.; Ding, Y.; Huang, Y.; Wu, Y.; Huang, J.; et al. Nuclear localization of mitochondrial TCA cycle enzymes modulates pluripotency via histone acetylation. Nat. Commun. 2022, 13, 7414. [Google Scholar] [CrossRef]
- Li, L.; Chen, K.; Wang, T.; Wu, Y.; Xing, G.; Chen, M.; Hao, Z.; Zhang, C.; Zhang, J.; Ma, B.; et al. Glis1 facilitates induction of pluripotency via an epigenome-metabolome-epigenome signalling cascade. Nat. Metab. 2020, 2, 882–892. [Google Scholar] [CrossRef]
- Yucel, N.; Wang, Y.X.; Mai, T.; Porpiglia, E.; Lund, P.J.; Markov, G.; Garcia, B.A.; Bendall, S.C.; Angelo, M.; Blau, H.M. Glucose Metabolism Drives Histone Acetylation Landscape Transitions that Dictate Muscle Stem Cell Function. Cell Rep. 2019, 27, 3939–3955.e3936. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Wang, S.; Liu, S.; Qin, D.; Liu, Z.; Wang, L.; Chen, X.; Zhang, L. MSL1 Promotes Liver Regeneration by Driving Phase Separation of STAT3 and Histone H4 and Enhancing Their Acetylation. Adv. Sci. 2023, e2301094. [Google Scholar] [CrossRef]
- Das, S.; Morvan, F.; Morozzi, G.; Jourde, B.; Minetti, G.C.; Kahle, P.; Rivet, H.; Brebbia, P.; Toussaint, G.; Glass, D.J.; et al. ATP Citrate Lyase Regulates Myofiber Differentiation and Increases Regeneration by Altering Histone Acetylation. Cell Rep. 2017, 21, 3003–3011. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Ji, Y.; Liang, Q.; Ming, M.; Chen, Y.; Zhang, Q.; Zhou, S.; Shen, M.; Ding, F. Expression of Protein Acetylation Regulators During Peripheral Nerve Development, Injury, and Regeneration. Front. Mol. Neurosci. 2022, 15, 888523. [Google Scholar] [CrossRef]
- Huynh, N.C.; Everts, V.; Ampornaramveth, R.S. Histone deacetylases and their roles in mineralized tissue regeneration. Bone Rep. 2017, 7, 33–40. [Google Scholar] [CrossRef]
- Yamauchi, Y.; Cooper, P.R.; Shimizu, E.; Kobayashi, Y.; Smith, A.J.; Duncan, H.F. Histone Acetylation as a Regenerative Target in the Dentine-Pulp Complex. Front. Genet. 2020, 11, 1. [Google Scholar] [CrossRef]
- Yadav, A.; Huang, T.C.; Chen, S.H.; Ramasamy, T.S.; Hsueh, Y.Y.; Lin, S.P.; Lu, F.I.; Liu, Y.H.; Wu, C.C. Sodium phenylbutyrate inhibits Schwann cell inflammation via HDAC and NFkappaB to promote axonal regeneration and remyelination. J. Neuroinflammation 2021, 18, 238. [Google Scholar] [CrossRef]
- Cho, Y.; Cavalli, V. HDAC signaling in neuronal development and axon regeneration. Curr. Opin. Neurobiol. 2014, 27, 118–126. [Google Scholar] [CrossRef] [Green Version]
- Ganai, S.A.; Ramadoss, M.; Mahadevan, V. Histone Deacetylase (HDAC) Inhibitors–emerging roles in neuronal memory, learning, synaptic plasticity and neural regeneration. Curr. Neuropharmacol. 2016, 14, 55–71. [Google Scholar] [CrossRef] [Green Version]
- Fessler, E.B.; Chibane, F.L.; Wang, Z.; Chuang, D.M. Potential roles of HDAC inhibitors in mitigating ischemia-induced brain damage and facilitating endogenous regeneration and recovery. Curr. Pharm. Des. 2013, 19, 5105–5120. [Google Scholar] [CrossRef]
- Stewart, S.; Tsun, Z.Y.; Izpisua Belmonte, J.C. A histone demethylase is necessary for regeneration in zebrafish. Proc. Natl. Acad. Sci. USA 2009, 106, 19889–19894. [Google Scholar] [CrossRef]
- Ma, K.H.; Duong, P.; Moran, J.J.; Junaidi, N.; Svaren, J. Polycomb repression regulates Schwann cell proliferation and axon regeneration after nerve injury. Glia 2018, 66, 2487–2502. [Google Scholar] [CrossRef]
- Duan, R.S.; Tang, G.B.; Du, H.Z.; Hu, Y.W.; Liu, P.P.; Xu, Y.J.; Zeng, Y.Q.; Zhang, S.F.; Wang, R.Y.; Teng, Z.Q.; et al. Polycomb protein family member CBX7 regulates intrinsic axon growth and regeneration. Cell Death Differ. 2018, 25, 1598–1611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, C.; Balsara, Z.R.; Hill, W.G.; Li, X. Stage- and subunit-specific functions of polycomb repressive complex 2 in bladder urothelial formation and regeneration. Development 2017, 144, 400–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ai, S.; Yu, X.; Li, Y.; Peng, Y.; Li, C.; Yue, Y.; Tao, G.; Li, C.; Pu, W.T.; He, A. Divergent Requirements for EZH1 in Heart Development Versus Regeneration. Circ. Res. 2017, 121, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Palacios, D.; Mozzetta, C.; Consalvi, S.; Caretti, G.; Saccone, V.; Proserpio, V.; Marquez, V.E.; Valente, S.; Mai, A.; Forcales, S.V.; et al. TNF/p38alpha/polycomb signaling to Pax7 locus in satellite cells links inflammation to the epigenetic control of muscle regeneration. Cell Stem Cell 2010, 7, 455–469. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Yang, L.; He, Q.; Hu, C.; Zhu, L.; Ma, X.; Ma, X.; Bao, S.; Li, L.; Chen, Y.; et al. A Homeostatic Arid1a-Dependent Permissive Chromatin State Licenses Hepatocyte Responsiveness to Liver-Injury-Associated YAP Signaling. Cell Stem Cell 2019, 25, 54–68.e55. [Google Scholar] [CrossRef]
- Wang, J.; Liu, S.; Heallen, T.; Martin, J.F. The Hippo pathway in the heart: Pivotal roles in development, disease, and regeneration. Nat. Rev. Cardiol. 2018, 15, 672–684. [Google Scholar] [CrossRef]
- Moya, I.M.; Halder, G. Hippo-YAP/TAZ signalling in organ regeneration and regenerative medicine. Nat. Rev. Mol. Cell Biol. 2019, 20, 211–226. [Google Scholar] [CrossRef]
- Benham-Pyle, B.W.; Pruitt, B.L.; Nelson, W.J. Cell adhesion. Mechanical strain induces E-cadherin-dependent Yap1 and beta-catenin activation to drive cell cycle entry. Science 2015, 348, 1024–1027. [Google Scholar] [CrossRef] [Green Version]
- Chang, L.; Azzolin, L.; Di Biagio, D.; Zanconato, F.; Battilana, G.; Lucon Xiccato, R.; Aragona, M.; Giulitti, S.; Panciera, T.; Gandin, A.; et al. The SWI/SNF complex is a mechanoregulated inhibitor of YAP and TAZ. Nature 2018, 563, 265–269. [Google Scholar] [CrossRef]
- Baena, E.; Gandarillas, A.; Vallespinos, M.; Zanet, J.; Bachs, O.; Redondo, C.; Fabregat, I.; Martinez, A.C.; de Alboran, I.M. c-Myc regulates cell size and ploidy but is not essential for postnatal proliferation in liver. Proc. Natl. Acad. Sci. USA 2005, 102, 7286–7291. [Google Scholar] [CrossRef]
- Wang, H.; Lu, J.; Alencastro, F.; Roberts, A.; Fiedor, J.; Carroll, P.; Eisenman, R.N.; Ranganathan, S.; Torbenson, M.; Duncan, A.W.; et al. Coordinated Cross-Talk Between the Myc and Mlx Networks in Liver Regeneration and Neoplasia. Cell Mol. Gastroenterol. Hepatol. 2022, 13, 1785–1804. [Google Scholar] [CrossRef]
- Lasorella, A.; Noseda, M.; Beyna, M.; Yokota, Y.; Iavarone, A. Id2 is a retinoblastoma protein target and mediates signalling by Myc oncoproteins. Nature 2000, 407, 592–598. [Google Scholar] [CrossRef]
- Lasorella, A.; Boldrini, R.; Dominici, C.; Donfrancesco, A.; Yokota, Y.; Inserra, A.; Iavarone, A. Id2 is critical for cellular proliferation and is the oncogenic effector of N-myc in human neuroblastoma. Cancer Res. 2002, 62, 301–306. [Google Scholar] [PubMed]
- Cotta, C.V.; Leventaki, V.; Atsaves, V.; Vidaki, A.; Schlette, E.; Jones, D.; Medeiros, L.J.; Rassidakis, G.Z. The helix-loop-helix protein Id2 is expressed differentially and induced by myc in T-cell lymphomas. Cancer 2008, 112, 552–561. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, J.L.; Sandoval, J.; Serviddio, G.; Sastre, J.; Morante, M.; Perrelli, M.G.; Martinez-Chantar, M.L.; Vina, J.; Vina, J.R.; Mato, J.M.; et al. Id2 leaves the chromatin of the E2F4-p130-controlled c-myc promoter during hepatocyte priming for liver regeneration. Biochem. J. 2006, 398, 431–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Chang, C.; Gao, H.; Wang, Q.; Zhang, F.; Xu, C. MiR-429 regulates rat liver regeneration and hepatocyte proliferation by targeting JUN/MYC/BCL2/CCND1 signaling pathway. Cell. Signal. 2018, 50, 80–89. [Google Scholar] [CrossRef]
- Winkler, M.; Staniczek, T.; Kurschner, S.W.; Schmid, C.D.; Schonhaber, H.; Cordero, J.; Kessler, L.; Mathes, A.; Sticht, C.; Nessling, M.; et al. Endothelial GATA4 controls liver fibrosis and regeneration by preventing a pathogenic switch in angiocrine signaling. J. Hepatol. 2021, 74, 380–393. [Google Scholar] [CrossRef]
- Jonas, J.C.; Laybutt, D.R.; Steil, G.M.; Trivedi, N.; Pertusa, J.G.; Van de Casteele, M.; Weir, G.C.; Henquin, J.C. High glucose stimulates early response gene c-Myc expression in rat pancreatic beta cells. J. Biol. Chem. 2001, 276, 35375–35381. [Google Scholar] [CrossRef] [Green Version]
- Puri, S.; Roy, N.; Russ, H.A.; Leonhardt, L.; French, E.K.; Roy, R.; Bengtsson, H.; Scott, D.K.; Stewart, A.F.; Hebrok, M. Replication confers beta cell immaturity. Nat. Commun. 2018, 9, 485. [Google Scholar] [CrossRef] [Green Version]
- Laybutt, D.R.; Weir, G.C.; Kaneto, H.; Lebet, J.; Palmiter, R.D.; Sharma, A.; Bonner-Weir, S. Overexpression of c-Myc in beta-cells of transgenic mice causes proliferation and apoptosis, downregulation of insulin gene expression, and diabetes. Diabetes 2002, 51, 1793–1804. [Google Scholar] [CrossRef] [Green Version]
- Cheung, L.; Zervou, S.; Mattsson, G.; Abouna, S.; Zhou, L.; Ifandi, V.; Pelengaris, S.; Khan, M. c-Myc directly induces both impaired insulin secretion and loss of beta-cell mass, independently of hyperglycemia in vivo. Islets 2010, 2, 37–45. [Google Scholar] [CrossRef] [Green Version]
- Pelengaris, S.; Khan, M.; Evan, G.I. Suppression of Myc-induced apoptosis in beta cells exposes multiple oncogenic properties of Myc and triggers carcinogenic progression. Cell 2002, 109, 321–334. [Google Scholar] [CrossRef] [Green Version]
- Rosselot, C.; Kumar, A.; Lakshmipathi, J.; Zhang, P.; Lu, G.; Katz, L.S.; Prochownik, E.V.; Stewart, A.F.; Lambertini, L.; Scott, D.K.; et al. Myc Is Required for Adaptive beta-Cell Replication in Young Mice but Is Not Sufficient in One-Year-Old Mice Fed with a High-Fat Diet. Diabetes 2019, 68, 1934–1949. [Google Scholar] [CrossRef]
- Rosselot, C.; Baumel-Alterzon, S.; Li, Y.; Brill, G.; Lambertini, L.; Katz, L.S.; Lu, G.; Garcia-Ocana, A.; Scott, D.K. The many lives of Myc in the pancreatic beta-cell. J. Biol. Chem. 2021, 296, 100122. [Google Scholar] [CrossRef]
- Bettess, M.D.; Dubois, N.; Murphy, M.J.; Dubey, C.; Roger, C.; Robine, S.; Trumpp, A. c-Myc is required for the formation of intestinal crypts but dispensable for homeostasis of the adult intestinal epithelium. Mol. Cell. Biol. 2005, 25, 7868–7878. [Google Scholar] [CrossRef] [Green Version]
- Konsavage, W.M., Jr.; Jin, G.; Yochum, G.S. The Myc 3’ Wnt-responsive element regulates homeostasis and regeneration in the mouse intestinal tract. Mol. Cell. Biol. 2012, 32, 3891–3902. [Google Scholar] [CrossRef] [Green Version]
- Muncan, V.; Sansom, O.J.; Tertoolen, L.; Phesse, T.J.; Begthel, H.; Sancho, E.; Cole, A.M.; Gregorieff, A.; de Alboran, I.M.; Clevers, H.; et al. Rapid loss of intestinal crypts upon conditional deletion of the Wnt/Tcf-4 target gene c-Myc. Mol. Cell. Biol. 2006, 26, 8418–8426. [Google Scholar] [CrossRef] [Green Version]
- Ashton, G.H.; Morton, J.P.; Myant, K.; Phesse, T.J.; Ridgway, R.A.; Marsh, V.; Wilkins, J.A.; Athineos, D.; Muncan, V.; Kemp, R.; et al. Focal adhesion kinase is required for intestinal regeneration and tumorigenesis downstream of Wnt/c-Myc signaling. Dev. Cell 2010, 19, 259–269. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.J.; Xia, B.; Suh, H.N.; Lee, S.H.; Jun, S.; Lien, E.M.; Zhang, J.; Chen, K.; Park, J.I. PAF-Myc-Controlled Cell Stemness Is Required for Intestinal Regeneration and Tumorigenesis. Dev. Cell 2018, 44, 582–596.e584. [Google Scholar] [CrossRef]
- De Pretis, S.; Kress, T.R.; Morelli, M.J.; Sabo, A.; Locarno, C.; Verrecchia, A.; Doni, M.; Campaner, S.; Amati, B.; Pelizzola, M. Integrative analysis of RNA polymerase II and transcriptional dynamics upon MYC activation. Genome Res. 2017, 27, 1658–1664. [Google Scholar] [CrossRef] [Green Version]
- Bywater, M.J.; Burkhart, D.L.; Straube, J.; Sabo, A.; Pendino, V.; Hudson, J.E.; Quaife-Ryan, G.A.; Porrello, E.R.; Rae, J.; Parton, R.G.; et al. Reactivation of Myc transcription in the mouse heart unlocks its proliferative capacity. Nat. Commun. 2020, 11, 1827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Zheng, H.; Han, Y.; Chen, Y.; Li, B.; Chen, G.; Chen, X.; Huang, S.; He, X.; Wei, G.; et al. LncRNA Snhg1-driven self-reinforcing regulatory network promoted cardiac regeneration and repair after myocardial infarction. Theranostics 2021, 11, 9397–9414. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Luttmann, F.F.; Schoger, E.; Scholer, H.R.; Zelarayan, L.C.; Kim, K.P.; Haigh, J.J.; Kim, J.; Braun, T. Reversible reprogramming of cardiomyocytes to a fetal state drives heart regeneration in mice. Science 2021, 373, 1537–1540. [Google Scholar] [CrossRef] [PubMed]
- Klein, J.A.; Ackerman, S.L. Oxidative stress, cell cycle, and neurodegeneration. J. Clin. Investig. 2003, 111, 785–793. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.G.; Wang, X.W.; Qian, C.; Zhou, F.Q. Reprogramming neurons for regeneration: The fountain of youth. Prog. Neurobiol. 2022, 214, 102284. [Google Scholar] [CrossRef]
- Ferrer, I.; Blanco, R.; Carmona, M.; Puig, B. Phosphorylated c-MYC expression in Alzheimer disease, Pick’s disease, progressive supranuclear palsy and corticobasal degeneration. Neuropathol. Appl. Neurobiol. 2001, 27, 343–351. [Google Scholar] [CrossRef]
- Ferrer, I.; Blanco, R. N-myc and c-myc expression in Alzheimer disease, Huntington disease and Parkinson disease. Brain Res. Mol. Brain Res. 2000, 77, 270–276. [Google Scholar] [CrossRef]
- Porta, C.; Paglino, C.; Mosca, A. Targeting PI3K/Akt/mTOR Signaling in Cancer. Front. Oncol. 2014, 4, 64. [Google Scholar] [CrossRef] [Green Version]
- Pei, J.J.; Hugon, J. mTOR-dependent signalling in Alzheimer’s disease. J. Cell. Mol. Med. 2008, 12, 2525–2532. [Google Scholar] [CrossRef] [Green Version]
- Arendt, T. Synaptic plasticity and cell cycle activation in neurons are alternative effector pathways: The ‘Dr. Jekyll and Mr. Hyde concept’ of Alzheimer’s disease or the yin and yang of neuroplasticity. Prog. Neurobiol. 2003, 71, 83–248. [Google Scholar] [CrossRef]
- Hay, N. Interplay between FOXO, TOR, and Akt. Biochim. Biophys. Acta 2011, 1813, 1965–1970. [Google Scholar] [CrossRef] [Green Version]
- Gomez-Crisostomo, N.P.; Rodriguez Martinez, E.; Rivas-Arancibia, S. Oxidative stress activates the transcription factors FoxO 1a and FoxO 3a in the hippocampus of rats exposed to low doses of ozone. Oxidative Med. Cell. Longev. 2014, 2014, 805764. [Google Scholar] [CrossRef] [Green Version]
- Chandramohan, V.; Jeay, S.; Pianetti, S.; Sonenshein, G.E. Reciprocal control of Forkhead box O 3a and c-Myc via the phosphatidylinositol 3-kinase pathway coordinately regulates p27Kip1 levels. J. Immunol. 2004, 172, 5522–5527. [Google Scholar] [CrossRef] [Green Version]
- Riddell, M.; Nakayama, A.; Hikita, T.; Mirzapourshafiyi, F.; Kawamura, T.; Pasha, A.; Li, M.; Masuzawa, M.; Looso, M.; Steinbacher, T.; et al. aPKC controls endothelial growth by modulating c-Myc via FoxO1 DNA-binding ability. Nat. Commun. 2018, 9, 5357. [Google Scholar] [CrossRef] [Green Version]
- Majd, S.; Power, J.; Majd, Z. Alzheimer’s Disease and Cancer: When Two Monsters Cannot Be Together. Front. Neurosci. 2019, 13, 155. [Google Scholar] [CrossRef] [Green Version]
- Qin, Z.H.; Chen, R.W.; Wang, Y.; Nakai, M.; Chuang, D.M.; Chase, T.N. Nuclear factor kappaB nuclear translocation upregulates c-Myc and p53 expression during NMDA receptor-mediated apoptosis in rat striatum. J. Neurosci. 1999, 19, 4023–4033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belin, S.; Nawabi, H.; Wang, C.; Tang, S.; Latremoliere, A.; Warren, P.; Schorle, H.; Uncu, C.; Woolf, C.J.; He, Z.; et al. Injury-induced decline of intrinsic regenerative ability revealed by quantitative proteomics. Neuron 2015, 86, 1000–1014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, J.J.; Ju, X.; Xu, R.J.; Wang, W.H.; Luo, Z.P.; Liu, C.M.; Yang, L.; Li, B.; Chen, J.Q.; Meng, B.; et al. Telomerase Reverse Transcriptase and p53 Regulate Mammalian Peripheral Nervous System and CNS Axon Regeneration Downstream of c-Myc. J. Neurosci. 2019, 39, 9107–9118. [Google Scholar] [CrossRef]
- Peck, B.; Ferber, E.C.; Schulze, A. Antagonism between FOXO and MYC Regulates Cellular Powerhouse. Front. Oncol. 2013, 3, 96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oli, V.; Gupta, R.; Kumar, P. FOXO and related transcription factors binding elements in the regulation of neurodegenerative disorders. J. Chem. Neuroanat. 2021, 116, 102012. [Google Scholar] [CrossRef] [PubMed]
- Hishida, T.; Vazquez-Ferrer, E.; Hishida-Nozaki, Y.; Takemoto, Y.; Hatanaka, F.; Yoshida, K.; Prieto, J.; Sahu, S.K.; Takahashi, Y.; Reddy, P.; et al. Myc Supports Self-Renewal of Basal Cells in the Esophageal Epithelium. Front. Cell Dev. Biol. 2022, 10, 786031. [Google Scholar] [CrossRef]
- Huang, X.; Sun, J.; Chen, G.; Niu, C.; Wang, Y.; Zhao, C.; Sun, J.; Huang, H.; Huang, S.; Liang, Y.; et al. Resveratrol Promotes Diabetic Wound Healing via SIRT1-FOXO1-c-Myc Signaling Pathway-Mediated Angiogenesis. Front. Pharmacol. 2019, 10, 421. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Rabadan Ros, R.; Martinez-Redondo, P.; Ma, Z.; Shi, L.; Xue, Y.; Guillen-Guillen, I.; Huang, L.; Hishida, T.; Liao, H.K.; et al. In vivo partial reprogramming of myofibers promotes muscle regeneration by remodeling the stem cell niche. Nat. Commun. 2021, 12, 3094. [Google Scholar] [CrossRef]
- Shu, Y.; Li, W.; Huang, M.; Quan, Y.Z.; Scheffer, D.; Tian, C.; Tao, Y.; Liu, X.; Hochedlinger, K.; Indzhykulian, A.A.; et al. Renewed proliferation in adult mouse cochlea and regeneration of hair cells. Nat. Commun. 2019, 10, 5530. [Google Scholar] [CrossRef] [Green Version]
- Ocampo, A.; Reddy, P.; Martinez-Redondo, P.; Platero-Luengo, A.; Hatanaka, F.; Hishida, T.; Li, M.; Lam, D.; Kurita, M.; Beyret, E.; et al. In Vivo Amelioration of Age-Associated Hallmarks by Partial Reprogramming. Cell 2016, 167, 1719–1733.e1712. [Google Scholar] [CrossRef] [Green Version]
- Kaufmann, K.B.; Buning, H.; Galy, A.; Schambach, A.; Grez, M. Gene therapy on the move. EMBO Mol. Med. 2013, 5, 1642–1661. [Google Scholar] [CrossRef]
- Deng, X.Y.; Wang, H.; Wang, T.; Fang, X.T.; Zou, L.L.; Li, Z.Y.; Liu, C.B. Non-viral methods for generating integration-free, induced pluripotent stem cells. Curr. Stem. Cell. Res. Ther. 2015, 10, 153–158. [Google Scholar] [CrossRef] [Green Version]
- Martin, I.; Simmons, P.J.; Williams, D.F. Manufacturing challenges in regenerative medicine. Sci. Transl. Med. 2014, 6, 232fs216. [Google Scholar] [CrossRef]
- Cossu, G.; Fears, R.; Griffin, G.; Ter Meulen, V. Regenerative medicine: Challenges and opportunities. Lancet 2020, 395, 1746–1747. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Illi, B.; Nasi, S. Myc beyond Cancer: Regulation of Mammalian Tissue Regeneration. Pathophysiology 2023, 30, 346-365. https://doi.org/10.3390/pathophysiology30030027
Illi B, Nasi S. Myc beyond Cancer: Regulation of Mammalian Tissue Regeneration. Pathophysiology. 2023; 30(3):346-365. https://doi.org/10.3390/pathophysiology30030027
Chicago/Turabian StyleIlli, Barbara, and Sergio Nasi. 2023. "Myc beyond Cancer: Regulation of Mammalian Tissue Regeneration" Pathophysiology 30, no. 3: 346-365. https://doi.org/10.3390/pathophysiology30030027