
COVID-19-Induced Diabetes Mellitus: Comprehensive Cellular and Molecular Mechanistic Insights


Abstract
1. Introduction
2. SARS-CoV-2 Infection
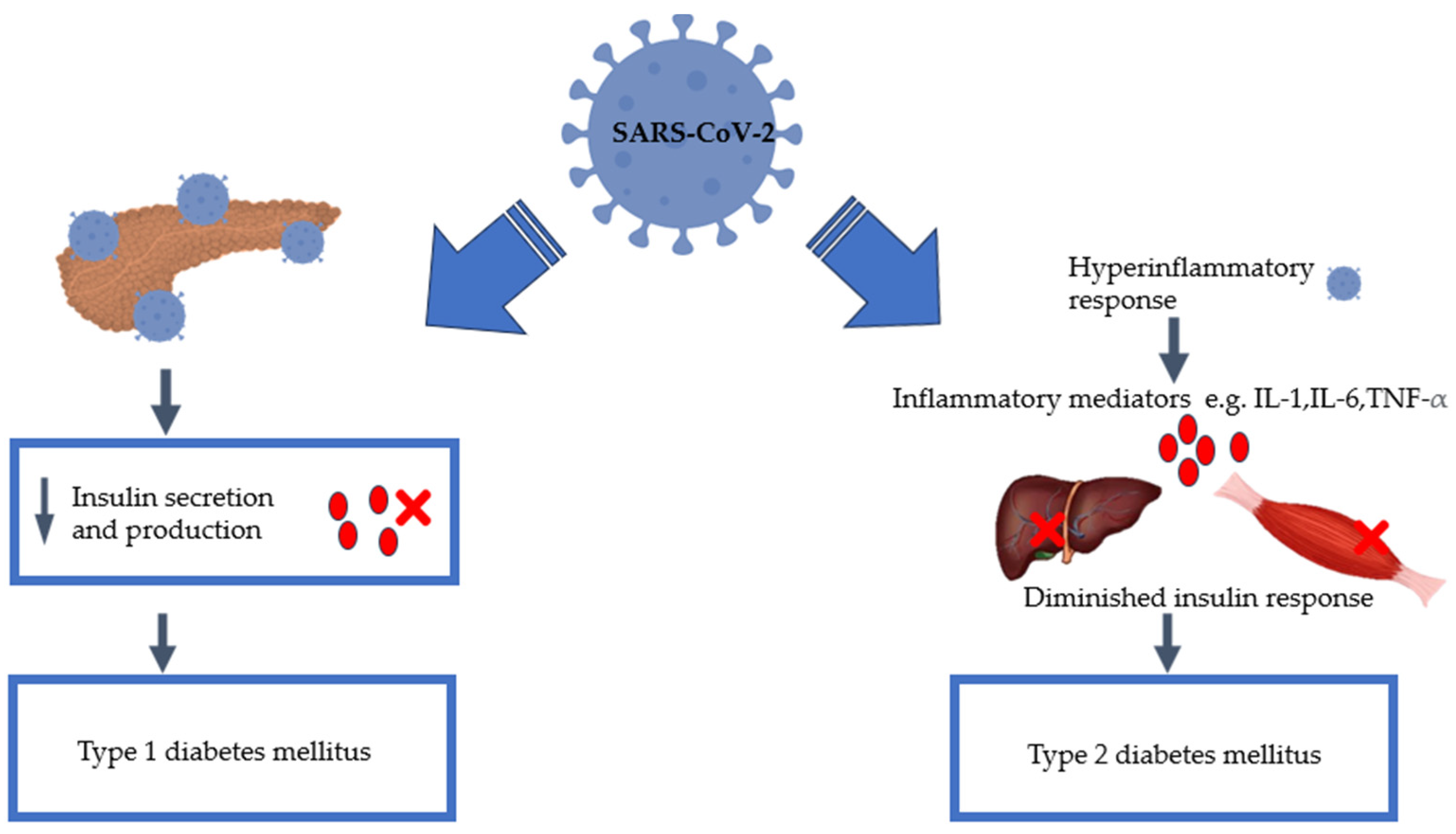
3. SARS-CoV-2 Infection and Diabetes Mellitus
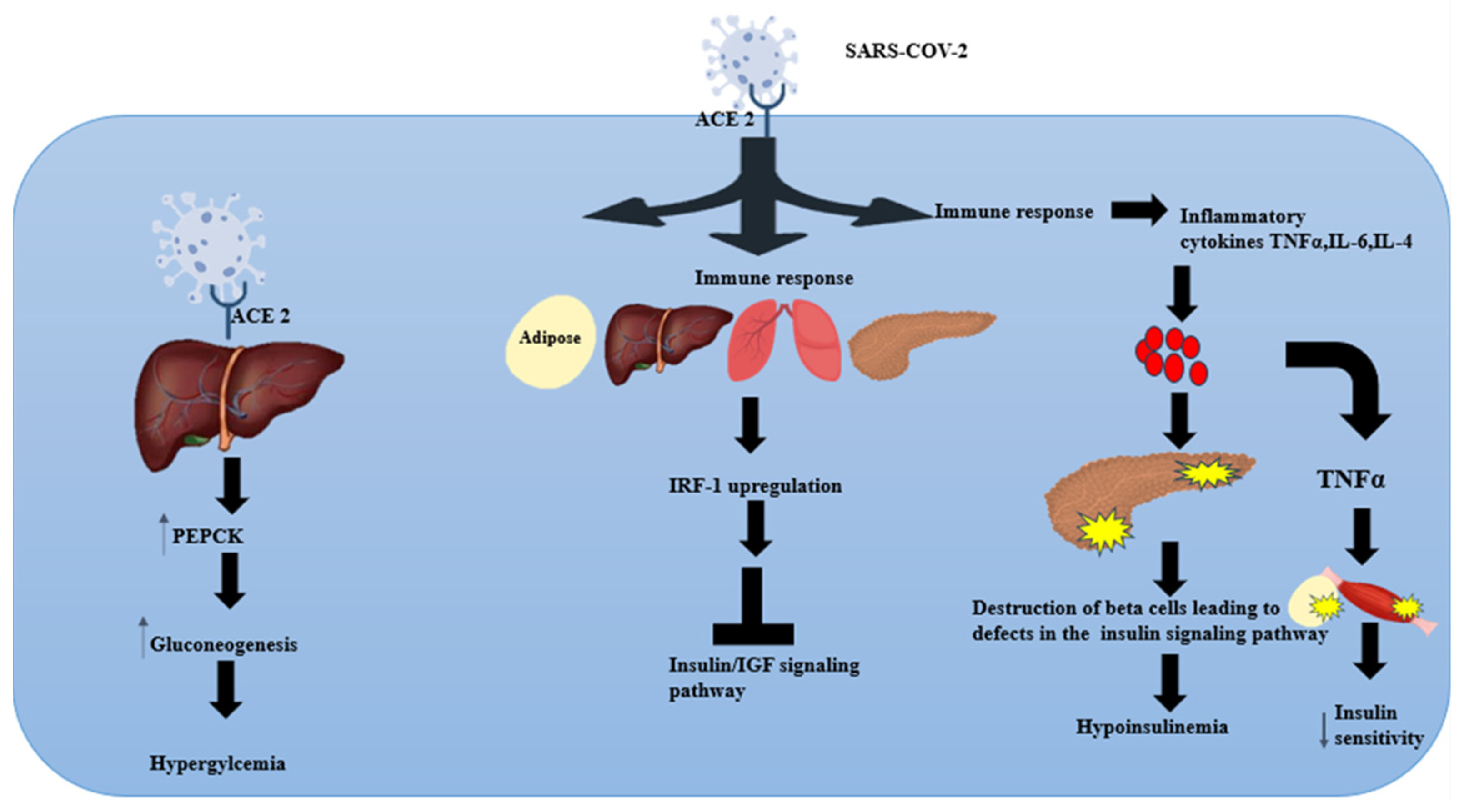
4. Insulin Signalling Pathway: Effect of SARS-CoV-2
5. Virus-Induced Glucose Homeostasis Alteration
6. Pancreatic Damage: Effect of SARS-CoV-2
7. SARS-CoV-2-Induced Glycolytic and Hepatic Gluconeogenic Pathways
8. SARS-CoV-2 and Inflammation
9. COVID-19 and New-Onset Diabetes: Outcomes with New-Onset Hyperglycaemia with or without Diabetes in People Who Have Suffered from COVID-19
10. Authors’ Perspectives and Recommendations
Author Contributions
Funding
Conflicts of Interest
References
- Abegunde, D.O.; Mathers, C.D.; Adam, T.; Ortegon, M.; Strong, K. The burden and costs of chronic diseases in low-income and middle-income countries. Lancet 2007, 370, 1929–1938. [Google Scholar] [CrossRef] [PubMed]
- Khetan, A.K.; Rajagopalan, S. Prediabetes. Can. J. Cardiol. 2018, 34, 615–623. [Google Scholar] [CrossRef] [PubMed]
- Kiernan, K.; MacIver, N.J. Viral Infection “Interferes” with Glucose Tolerance. Immunity 2018, 49, 6–8. [Google Scholar] [CrossRef] [PubMed]
- American Diabetes Association. 2. Classification and Diagnosis of Diabetes: Standards of Medical Care in Diabetes—2018. Diabetes Care 2017, 41, S13–S27. [Google Scholar] [CrossRef]
- Jeong, S.U.; Kang, D.G.; Lee, D.H.; Lee, K.W.; Lim, D.-M.; Kim, B.J.; Park, K.-Y.; Chin, H.-J.; Koh, G. Clinical Characteristics of Type 2 Diabetes Patients according to Family History of Diabetes. Korean Diabetes J. 2010, 34, 222–228. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Roep, B.O.; Thomaidou, S.; van Tienhoven, R.; Zaldumbide, A. Type 1 diabetes mellitus as a disease of the β-cell (do not blame the immune system?). Nat. Rev. Endocrinol. 2021, 17, 150–161. [Google Scholar] [CrossRef]
- Cavaghan, M.K.; Ehrmann, D.A.; Polonsky, K.S. Interactions between insulin resistance and insulin secretion in the development of glucose intolerance. J. Clin. Investig. 2000, 106, 329–333. [Google Scholar] [CrossRef]
- Fazakerley, D.J.; Krycer, J.R.; Kearney, A.L.; Hocking, S.L.; James, D.E. Muscle and adipose tissue insulin resistance: Malady without mechanism? J. Lipid Res. 2019, 60, 1720–1732. [Google Scholar] [CrossRef]
- Metwally, A.A.; Mehta, P.; Johnson, B.S.; Nagarjuna, A.; Snyder, M.P. COVID-19–Induced New-Onset Diabetes: Trends and Technologies. Diabetes 2021, 70, 2733–2744. [Google Scholar] [CrossRef]
- Pal, R.; Banerjee, M. COVID-19 and the endocrine system: Exploring the unexplored. J. Endocrinol. Investig. 2020, 43, 1027–1031. [Google Scholar] [CrossRef]
- Iacobellis, G.; Penaherrera, C.A.; Bermudez, L.E.; Bernal Mizrachi, E. Admission hyperglycemia and radiological findings of SARS-CoV2 in patients with and without diabetes. Diabetes Res. Clin. Pract. 2020, 164, 108185. [Google Scholar] [CrossRef] [PubMed]
- Coppelli, A.; Giannarelli, R.; Aragona, M.; Penno, G.; Falcone, M.; Tiseo, G.; Ghiadoni, L.; Barbieri, G.; Monzani, F.; Virdis, A.; et al. Hyperglycemia at Hospital Admission Is Associated with Severity of the Prognosis in Patients Hospitalized for COVID-19: The Pisa COVID-19 Study. Diabetes Care 2020, 43, 2345–2348. [Google Scholar] [CrossRef] [PubMed]
- Baidya, A.; Singh, S.K.; Bajaj, S.; Zargar, A.H.; Singh, P.; Das, S.; Shankar, A. Diabetes and COVID-19: A Review. J. ASEAN Fed. Endocr. Soc. 2020, 35, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Kaul, D. An overview of coronaviruses including the SARS-2 coronavirus—Molecular biology, epidemiology and clinical implications. Curr. Med. Res. Pract. 2020, 10, 54–64. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Deep, S. In-silico drug repurposing for targeting SARS-CoV-2 main protease (Mpro). J. Biomol. Struct. Dyn. 2022, 40, 3003–3010. [Google Scholar] [CrossRef] [PubMed]
- Dallavalasa, S.; Tulimilli, S.V.; Prakash, J.; Ramachandra, R.; Madhunapantula, S.V.; Veeranna, R.P. COVID-19: Diabetes Perspective—Pathophysiology and Management. Pathogens 2023, 12, 184. [Google Scholar] [CrossRef] [PubMed]
- Yuki, K.; Fujiogi, M.; Koutsogiannaki, S. COVID-19 pathophysiology: A review. Clin. Immunol. 2020, 215, 108427. [Google Scholar] [CrossRef] [PubMed]
- Gangadharan, C.; Ahluwalia, R.; Sigamani, A. Diabetes and COVID-19: Role of insulin resistance as a risk factor for COVID-19 severity. World J. Diabetes 2021, 12, 1550–1562. [Google Scholar] [CrossRef] [PubMed]
- Malkova, A.; Kudlay, D.; Kudryavtsev, I.; Starshinova, A.; Yablonskiy, P.; Shoenfeld, Y. Immunogenetic Predictors of Severe COVID-19. Vaccines 2021, 9, 211. [Google Scholar] [CrossRef]
- WHO Coronavirus (COVID-19) Dashboard. Available online: https://covid19.who.int (accessed on 27 June 2023).
- Yang, X.; Yu, Y.; Xu, J.; Shu, H.; Xia, J.; Liu, H.; Wu, Y.; Zhang, L.; Yu, Z.; Fang, M.; et al. Clinical course and outcomes of critically ill patients with SARS-CoV-2 pneumonia in Wuhan, China: A single-centered, retrospective, observational study. Lancet Respir. Med. 2020, 8, 475–481. [Google Scholar] [CrossRef]
- Wu, Z.; McGoogan, J.M. Characteristics of and Important Lessons From the Coronavirus Disease 2019 (COVID-19) Outbreak in China: Summary of a Report of 72 314 Cases From the Chinese Center for Disease Control and Prevention. JAMA 2020, 323, 1239–1242. [Google Scholar] [CrossRef]
- Yang, J.-K.; Lin, S.-S.; Ji, X.-J.; Guo, L.-M. Binding of SARS coronavirus to its receptor damages islets and causes acute diabetes. Acta Diabetol. 2010, 47, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.; Bae, J.H.; Kwon, H.-S.; Nauck, M.A. COVID-19 and diabetes mellitus: From pathophysiology to clinical management. Nat. Rev. Endocrinol. 2021, 17, 11–30. [Google Scholar] [CrossRef]
- Lui, G.C.-Y.; Yip, T.C.-F.; Wong, V.W.-S.; Chow, V.C.-Y.; Ho, T.H.-Y.; Li, T.C.-M.; Tse, Y.-K.; Chan, H.L.-Y.; Hui, D.S.-C.; Wong, G.L.-H. Significantly Lower Case-fatality Ratio of Coronavirus Disease 2019 (COVID-19) than Severe Acute Respiratory Syndrome (SARS) in Hong Kong—A Territory-Wide Cohort Study. Clin. Infect. Dis. 2021, 72, e466–e475. [Google Scholar] [CrossRef]
- Yu, B.; Li, C.; Sun, Y.; Wang, D.W. Insulin Treatment Is Associated with Increased Mortality in Patients with COVID-19 and Type 2 Diabetes. Cell Metab. 2021, 33, 65–77.e2. [Google Scholar] [CrossRef]
- Riahi, S.; Sombra, L.R.S.; Lo, K.B.; Chacko, S.R.; Neto, A.G.M.; Azmaiparashvili, Z.; Patarroyo-Aponte, G.; Rangaswami, J.; Anastasopoulou, C. Insulin Use, Diabetes Control, and Outcomes in Patients with COVID-19. Endocr. Res. 2021, 46, 45–50. [Google Scholar] [CrossRef]
- Wang, W.; Sun, Y.; Wang, S.; Sun, Y. The Relationship between Insulin Use and Increased Mortality in Patients with COVID-19 and Diabetes: A Meta-Analysis. Endocr. Res. 2022, 47, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Cai, Z.; Zhang, J. Insulin Treatment May Increase Adverse Outcomes in Patients with COVID-19 and Diabetes: A Systematic Review and Meta-Analysis. Front. Endocrinol. 2021, 12, 696087. [Google Scholar] [CrossRef] [PubMed]
- Sardu, C.; D’Onofrio, N.; Balestrieri, M.L.; Barbieri, M.; Rizzo, M.R.; Messina, V.; Maggi, P.; Coppola, N.; Paolisso, G.; Marfella, R. Outcomes in Patients with Hyperglycemia Affected by COVID-19: Can We Do More on Glycemic Control? Diabetes Care 2020, 43, 1408–1415. [Google Scholar] [CrossRef]
- Unsworth, R.; Wallace, S.; Oliver, N.S.; Yeung, S.; Kshirsagar, A.; Naidu, H.; Kwong, R.M.W.; Kumar, P.; Logan, K.M. New-Onset Type 1 Diabetes in Children During COVID-19: Multicenter Regional Findings in the U.K. Diabetes Care 2020, 43, e170–e171. [Google Scholar] [CrossRef]
- Müller, J.A.; Groß, R.; Conzelmann, C.; Krüger, J.; Merle, U.; Steinhart, J.; Weil, T.; Koepke, L.; Bozzo, C.P.; Read, C.; et al. SARS-CoV-2 infects and replicates in cells of the human endocrine and exocrine pancreas. Nat. Metab. 2021, 3, 149–165. [Google Scholar] [CrossRef]
- Fadini, G.P.; Morieri, M.L.; Boscari, F.; Fioretto, P.; Maran, A.; Busetto, L.; Bonora, B.M.; Selmin, E.; Arcidiacono, G.; Pinelli, S.; et al. Newly-diagnosed diabetes and admission hyperglycemia predict COVID-19 severity by aggravating respiratory deterioration. Diabetes Res. Clin. Pract. 2020, 168, 108374. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.K.; Khunti, K. COVID-19 and Diabetes. Annu. Rev. Med. 2022, 73, 129–147. [Google Scholar] [CrossRef]
- Bode, B.; Garrett, V.; Messler, J.; McFarland, R.; Crowe, J.; Booth, R.; Klonoff, D.C. Glycemic Characteristics and Clinical Outcomes of COVID-19 Patients Hospitalized in the United States. J. Diabetes Sci. Technol. 2020, 14, 813–821. [Google Scholar] [CrossRef] [PubMed]
- Sibiya, N. The Effects of Oxidovanadium Complexes on Glucose Metabolism in Liver and Skeletal Muscle Cell Lines. Ph.D. Thesis, University of KwaZulu-Natal, Durban, South Africa, 2014. Available online: https://www.semanticscholar.org/paper/The-effects-of-oxidovanadium-complexes-on-glucose-Sibiya/1d2701fa785e4c499132c38c31fdaa959bddbc5c (accessed on 21 December 2023).
- Lizcano, J.M.; Alessi, D.R. The insulin signalling pathway. Curr. Biol. 2002, 12, R236–R238. [Google Scholar] [CrossRef]
- Saltiel, A.R.; Kahn, C.R. Insulin signalling and the regulation of glucose and lipid metabolism. Nature 2001, 414, 799–806. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Chiang, S.-H.; Saltiel, A.R. Insulin Signaling and the Regulation of Glucose Transport. Mol. Med. 2004, 10, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Sechi, L.A.; Griffin, C.A.; Giacchetti, G.; Zingaro, L.; Catena, C.; Bartoli, E.; Schambelan, M. Abnormalities of Insulin Receptors in Spontaneously Hypertensive Rats. Hypertension 1996, 27, 955–961. [Google Scholar] [CrossRef]
- Suryawanshi, R.K.; Koganti, R.; Agelidis, A.; Patil, C.D.; Shukla, D. Dysregulation of Cell Signaling by SARS-CoV-2. Trends Microbiol. 2021, 29, 224–237. [Google Scholar] [CrossRef]
- Shin, J.; Toyoda, S.; Nishitani, S.; Onodera, T.; Fukuda, S.; Kita, S.; Fukuhara, A.; Shimomura, I. SARS-CoV-2 infection impairs the insulin/IGF signaling pathway in the lung, liver, adipose tissue, and pancreatic cells via IRF1. Metabolism 2022, 133, 155236. [Google Scholar] [CrossRef]
- Kelesidis, T.; Mantzoros, C.S. Cross-talk between SARS-CoV-2 infection and the insulin/IGF signaling pathway: Implications for metabolic diseases in COVID-19 and for post-acute sequelae of SARS-CoV-2 infection. Metabolism 2022, 134, 155267. [Google Scholar] [CrossRef] [PubMed]
- Šestan, M.; Marinović, S.; Kavazović, I.; Cekinović, Đ.; Wueest, S.; Turk Wensveen, T.; Brizić, I.; Jonjić, S.; Konrad, D.; Wensveen, F.M.; et al. Virus-Induced Interferon-γ Causes Insulin Resistance in Skeletal Muscle and Derails Glycemic Control in Obesity. Immunity 2018, 49, 164–177.e6. [Google Scholar] [CrossRef] [PubMed]
- Girdhar, K.; Powis, A.; Raisingani, A.; Chrudinová, M.; Huang, R.; Tran, T.; Sevgi, K.; Dogru, Y.D.; Altindis, E. Viruses and Metabolism: The Effects of Viral Infections and Viral Insulins on Host Metabolism. Annu. Rev. Virol. 2021, 8, 373–391. [Google Scholar] [CrossRef] [PubMed]
- Diehl, N.; Schaal, H. Make Yourself at Home: Viral Hijacking of the PI3K/Akt Signaling Pathway. Viruses 2013, 5, 3192–3212. [Google Scholar] [CrossRef] [PubMed]
- Cheng, M.-L.; Chien, K.-Y.; Lai, C.-H.; Li, G.-J.; Lin, J.-F.; Ho, H.-Y. Metabolic Reprogramming of Host Cells in Response to Enteroviral Infection. Cells 2020, 9, 473. [Google Scholar] [CrossRef] [PubMed]
- Fontaine, K.A.; Sanchez, E.L.; Camarda, R.; Lagunoff, M. Dengue Virus Induces and Requires Glycolysis for Optimal Replication. J. Virol. 2015, 89, 2358–2366. [Google Scholar] [CrossRef] [PubMed]
- Van Belle, T.L.; Coppieters, K.T.; Von Herrath, M.G. Type 1 Diabetes: Etiology, Immunology, and Therapeutic Strategies. Physiol. Rev. 2011, 91, 79–118. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.W.; Onodera, T.; Notkins, A.L. Virus-induced diabetes mellitus. XV. Beta cell damage and insulin-dependent hyperglycemia in mice infected with coxsackie virus B4. J. Exp. Med. 1978, 148, 1068–1080. [Google Scholar] [CrossRef] [PubMed]
- Dotta, F.; Censini, S.; van Halteren, A.G.S.; Marselli, L.; Masini, M.; Dionisi, S.; Mosca, F.; Boggi, U.; Muda, A.O.; Prato, S.D.; et al. Coxsackie B4 virus infection of β cells and natural killer cell insulitis in recent-onset type 1 diabetic patients. Proc. Natl. Acad. Sci. USA 2007, 104, 5115–5120. [Google Scholar] [CrossRef]
- Honeyman, M.C.; Coulson, B.S.; Stone, N.L.; Gellert, S.A.; Goldwater, P.N.; Steele, C.E.; Couper, J.J.; Tait, B.D.; Colman, P.G.; Harrison, L.C. Association between rotavirus infection and pancreatic islet autoimmunity in children at risk of developing type 1 diabetes. Diabetes 2000, 49, 1319–1324. [Google Scholar] [CrossRef]
- Sibiya, N.; Mzimela, N.; Mbatha, B.; Ngubane, P.; Khathi, A. The Insights on Why Diabetes Prevalence May Increase Amid or Post COVID-19 Pandemic. Curr. Diabetes Rev. 2023, 19, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Gęca, T.; Wojtowicz, K.; Guzik, P.; Góra, T. Increased Risk of COVID-19 in Patients with Diabetes Mellitus—Current Challenges in Pathophysiology, Treatment and Prevention. Int. J. Environ. Res. Public Health 2022, 19, 6555. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Long, X.; Zhang, B.; Zhang, W.; Chen, X.; Zhang, Z. ACE2 Expression in Pancreas May Cause Pancreatic Damage After SARS-CoV-2 Infection. Clin. Gastroenterol. Hepatol. 2020, 18, 2128–2130.e2. [Google Scholar] [CrossRef] [PubMed]
- Morris, A. Effects of pancreatic SARS-CoV-2 infection identified. Nat. Rev. Endocrinol. 2021, 17, 192. [Google Scholar] [CrossRef] [PubMed]
- Qadir, M.M.F.; Bhondeley, M.; Beatty, W.; Gaupp, D.D.; Doyle-Meyers, L.A.; Fischer, T.; Bandyopadhyay, I.; Blair, R.V.; Bohm, R.; Rappaport, J.; et al. SARS-CoV-2 infection of the pancreas promotes thrombofibrosis and is associated with new-onset diabetes. JCI Insight 2021, 6, e151551. [Google Scholar] [CrossRef] [PubMed]
- Wagenknecht, L.E.; Roseman, J.M.; Herman, W.H. Increased Incidence of Insulin-Dependent Diabetes Mellitus Following an Epidemic of Coxsackievirus B5. Am. J. Epidemiol. 1991, 133, 1024–1031. [Google Scholar] [CrossRef] [PubMed]
- Sioofy-Khojine, A.-B.; Lehtonen, J.; Nurminen, N.; Laitinen, O.H.; Oikarinen, S.; Huhtala, H.; Pakkanen, O.; Ruokoranta, T.; Hankaniemi, M.M.; Toppari, J.; et al. Coxsackievirus B1 infections are associated with the initiation of insulin-driven autoimmunity that progresses to type 1 diabetes. Diabetologia 2018, 61, 1193–1202. [Google Scholar] [CrossRef] [PubMed]
- Hanley, B.; Naresh, K.N.; Roufosse, C.; Nicholson, A.G.; Weir, J.; Cooke, G.S.; Thursz, M.; Manousou, P.; Corbett, R.; Goldin, R.; et al. Histopathological findings and viral tropism in UK patients with severe fatal COVID-19: A post-mortem study. Lancet Microbe 2020, 1, e245–e253. [Google Scholar] [CrossRef] [PubMed]
- Barreto, E.A.; Cruz, A.S.; Veras, F.P.; Martins, R.; Bernardelli, R.S.; Paiva, I.M.; Lima, T.M.; Singh, Y.; Guimarães, R.C.; Damasceno, S.; et al. COVID-19-related hyperglycemia is associated with infection of hepatocytes and stimulation of gluconeogenesis. Proc. Natl. Acad. Sci. USA 2023, 120, e2217119120. [Google Scholar] [CrossRef]
- Mercado-Gómez, M.; Prieto-Fernández, E.; Goikoetxea-Usandizaga, N.; Vila-Vecilla, L.; Azkargorta, M.; Bravo, M.; Serrano-Maciá, M.; Egia-Mendikute, L.; Rodríguez-Agudo, R.; Lachiondo-Ortega, S.; et al. The spike of SARS-CoV-2 promotes metabolic rewiring in hepatocytes. Commun. Biol. 2022, 5, 827. [Google Scholar] [CrossRef]
- Icard, P.; Lincet, H.; Wu, Z.; Coquerel, A.; Forgez, P.; Alifano, M.; Fournel, L. The key role of Warburg effect in SARS-CoV-2 replication and associated inflammatory response. Biochimie 2021, 180, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Codo, A.C.; Davanzo, G.G.; de Brito Monteiro, L.; de Souza, G.F.; Muraro, S.P.; Virgilio-da-Silva, J.V.; Prodonoff, J.S.; Carregari, V.C.; de Biagi Junior, C.A.O.; Crunfli, F.; et al. Elevated Glucose Levels Favor SARS-CoV-2 Infection and Monocyte Response through a HIF-1α/Glycolysis-Dependent Axis. Cell Metab. 2020, 32, 437–446.e5. [Google Scholar] [CrossRef]
- Kumar, V. How could we forget immunometabolism in SARS-CoV2 infection or COVID-19? Int. Rev. Immunol. 2021, 40, 72–107. [Google Scholar] [CrossRef] [PubMed]
- Santos, A.F.; Póvoa, P.; Paixão, P.; Mendonça, A.; Taborda-Barata, L. Changes in Glycolytic Pathway in SARS-COV-2 Infection and Their Importance in Understanding the Severity of COVID-19. Front. Chem. 2021, 9, 685196. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.-Y.; Huang, M.-Y.; Xiao, Z.; Yang, S.; Chen, X.-Q. Lactate dehydrogenase elevations is associated with severity of COVID-19: A meta-analysis. Crit. Care 2020, 24, 459. [Google Scholar] [CrossRef] [PubMed]
- Choi, C.S.; Kim, Y.-B.; Lee, F.N.; Zabolotny, J.M.; Kahn, B.B.; Youn, J.H. Lactate induces insulin resistance in skeletal muscle by suppressing glycolysis and impairing insulin signaling. Am. J. Physiol. Endocrinol. Metab. 2002, 283, E233–E240. [Google Scholar] [CrossRef] [PubMed]
- Alfano, G.; Fontana, F.; Mori, G.; Giaroni, F.; Ferrari, A.; Giovanella, S.; Ligabue, G.; Ascione, E.; Cazzato, S.; Ballestri, M.; et al. Acid base disorders in patients with COVID-19. Int. Urol. Nephrol. 2022, 54, 405–410. [Google Scholar] [CrossRef] [PubMed]
- Tay, M.Z.; Poh, C.M.; Rénia, L.; MacAry, P.A.; Ng, L.F.P. The trinity of COVID-19: Immunity, inflammation and intervention. Nat. Rev. Immunol. 2020, 20, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Pickup, J.C. Inflammation and Activated Innate Immunity in the Pathogenesis of Type 2 Diabetes. Diabetes Care 2004, 27, 813–823. [Google Scholar] [CrossRef]
- Chen, T.; Wu, D.; Chen, H.; Yan, W.; Yang, D.; Chen, G.; Ma, K.; Xu, D.; Yu, H.; Wang, H.; et al. Clinical characteristics of 113 deceased patients with coronavirus disease 2019: Retrospective study. BMJ 2020, 368, m1091. [Google Scholar] [CrossRef]
- Queiroz, M.A.F.; das Neves, P.F.M.; Lima, S.S.; da Costa Lopes, J.; da Silva Torres, M.K.; Vallinoto, I.M.V.C.; Bichara, C.D.A.; dos Santos, E.F.; de Brito, M.T.F.M.; da Silva, A.L.S.; et al. Cytokine Profiles Associated With Acute COVID-19 and Long COVID-19 Syndrome. Front. Cell. Infect. Microbiol. 2022, 12, 922422. [Google Scholar] [CrossRef] [PubMed]
- Bhaskar, S.; Sinha, A.; Banach, M.; Mittoo, S.; Weissert, R.; Kass, J.S.; Rajagopal, S.; Pai, A.R.; Kutty, S. Cytokine Storm in COVID-19—Immunopathological Mechanisms, Clinical Considerations, and Therapeutic Approaches: The REPROGRAM Consortium Position Paper. Front. Immunol. 2020, 11, 1648. [Google Scholar] [CrossRef] [PubMed]
- Wellen, K.E.; Hotamisligil, G.S. Inflammation, Stress, and Diabetes. Available online: https://www.jci.org/articles/view/25102/cite (accessed on 7 April 2023).
- Plomgaard, P.; Bouzakri, K.; Krogh-Madsen, R.; Mittendorfer, B.; Zierath, J.R.; Pedersen, B.K. Tumor Necrosis Factor-α Induces Skeletal Muscle Insulin Resistance in Healthy Human Subjects via Inhibition of Akt Substrate 160 Phosphorylation. Diabetes 2005, 54, 2939–2945. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, G.R.; Michell, B.J.; van Denderen, B.J.W.; Watt, M.J.; Carey, A.L.; Fam, B.C.; Andrikopoulos, S.; Proietto, J.; Görgün, C.Z.; Carling, D.; et al. Tumor necrosis factor α-induced skeletal muscle insulin resistance involves suppression of AMP-kinase signaling. Cell Metab. 2006, 4, 465–474. [Google Scholar] [CrossRef] [PubMed]
- Govender, N.; Khaliq, O.P.; Moodley, J.; Naicker, T. Insulin resistance in COVID-19 and diabetes. Prim. Care Diabetes 2021, 15, 629–634. [Google Scholar] [CrossRef] [PubMed]
- Pickup, J.C.; Mattock, M.B.; Chusney, G.D.; Burt, D. NIDDM as a disease of the innate immune system: Association of acute-phase reactants and interleukin-6 with metabolic syndrome X. Diabetologia 1997, 40, 1286–1292. [Google Scholar] [CrossRef] [PubMed]
- Santos, A.; Magro, D.O.; Evangelista-Poderoso, R.; Saad, M.J.A. Diabetes, obesity, and insulin resistance in COVID-19: Molecular interrelationship and therapeutic implications. Diabetol. Metab. Syndr. 2021, 13, 23. [Google Scholar] [CrossRef]
- Seino, S.; Shibasaki, T.; Minami, K. Dynamics of insulin secretion and the clinical implications for obesity and diabetes. J. Clin. Investig. 2011, 121, 2118–2125. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Study Reference | Study Design and Data | Key Findings, Associated Effects, and Remarks |
|---|---|---|
| Unsworth et al., 2020 [31] | Multi-centre regional study: data on new-onset type 1 diabetes for 30 children aged 23 months to 16.8 years that developed new-onset type 1 diabetes | The study supports the bidirectional relationship between COVID-19 and new-onset diabetes. This is the first report to outline a noticeable rise in the occurrence of new-onset T1D among children during the COVID-19 pandemic, with indications of SARS-CoV-2 infection or exposure identified in a portion of the tested individuals. |
| Müller et al., 2021 [32] | ACE2 pancreatic islet cells | SARS-CoV-2 infects and replicates in cultured human islets. This supports the bidirectional relationship between COVID-19 and new-onset diabetes. |
| Coppelli et al. [12] | Cohort study of 271 hospitalised COVID-19 patients with diabetes and no diabetes | COVID-19 patients who developed hyperglycaemia without having pre-existing diabetes had a higher mortality rate when compared to those with normoglycemia. Hyperglycaemia is indicative of a link between COVID-19 and diabetes. |
| Fadini et al. [33] | Retrospective study: 21 COVID-19 patients with new-onset diabetes | A higher rate of ICU admission and mortality among COVID-19 patients with new-onset diabetes than among those with pre-existing diabetes and normoglycemia was observed. New-onset diabetes had the worst prognosis out of the three. This paper supports the bidirectional relationship between COVID-19 and new-onset diabetes. |
| Singh and Khunti et al. [34] | Literature review | New-onset hyperglycaemia and new-onset diabetes have been increasingly recognised in the context of COVID-19. |
| Bode et al. [35] | Retrospective observational study | Individuals with new-onset hyperglycaemia have poorer outcomes than those with pre-existing diabetes. New-onset hyperglycaemia without diabetes had poorer outcomes than pre-existing diabetes. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nhau, P.T.; Gamede, M.; Sibiya, N. COVID-19-Induced Diabetes Mellitus: Comprehensive Cellular and Molecular Mechanistic Insights. Pathophysiology 2024, 31, 197-209. https://doi.org/10.3390/pathophysiology31020016
Nhau PT, Gamede M, Sibiya N. COVID-19-Induced Diabetes Mellitus: Comprehensive Cellular and Molecular Mechanistic Insights. Pathophysiology. 2024; 31(2):197-209. https://doi.org/10.3390/pathophysiology31020016
Chicago/Turabian StyleNhau, Praise Tatenda, Mlindeli Gamede, and Ntethelelo Sibiya. 2024. "COVID-19-Induced Diabetes Mellitus: Comprehensive Cellular and Molecular Mechanistic Insights" Pathophysiology 31, no. 2: 197-209. https://doi.org/10.3390/pathophysiology31020016
APA StyleNhau, P. T., Gamede, M., & Sibiya, N. (2024). COVID-19-Induced Diabetes Mellitus: Comprehensive Cellular and Molecular Mechanistic Insights. Pathophysiology, 31(2), 197-209. https://doi.org/10.3390/pathophysiology31020016