1. Introduction
Geum rivale L. (water avens, purple avens) is an herbaceous plant of Rosaceae family that is widespread in the regions of temperate climate of northern hemisphere [
1]. Together with closely related
G. urbanum L., it has been used in traditional medicine as astringent and antiseptic agent in the treatment of diarrhoea, digestive disorders, and haemorrhoids [
2]. Isolation studies concerning the species have so far been limited to the aerial parts and led to the isolation and identification of triterpenoid, flavonoid, and phenolic acid derivatives [
3,
4]. The plant is also known to accumulate large amounts of tannins, especially in underground organs [
5]. In previous studies, the authors of this paper demonstrated that the rhizome of
G. rivale is particularly rich source of ellagic acid (60.6 mg/g dry weight (dw) of the plant material), found mainly in a bound form [
6].
Ellagic acid is a bi-lactone compound occurring widely in plant kingdom. It has been broadly studied due to its strong antioxidant and chemopreventive potential and was demonstrated to have a variety of other biological effects, e.g., cytotoxic, antimicrobial, and anti-inflammatory activities [
7]. It is formed in plants from hexahydroxydiphenic acid—a product of oxidative dimerization of two vicinal galloyl moieties in gallotannin molecules—and accumulated in a free form or transformed further into various derivatives, usually by methylation and glycosylation or very rarely by sulphation [
7,
8]. Because of the activity of the parent compound, these derivatives are often investigated to find out the impact of different substituents on its pharmacological properties.
Sulphate derivatives of ellagic acid are especially rare in nature. So far, they have been found in only eight species including
Potentilla candicans Humb. and Bonpl. ex Nestl.,
Tamarix tetragyna Ehrenb.,
Tamarix amplexicaulis Ehrenb.,
Frankenia laevis L.,
Euphorbia sororia A. Schrenk,
Langerstroemia speciosa (L.) Pers., and
Reaumuria vermiculata L. [
9,
10,
11,
12,
13,
14,
15]. Of particular interest and best recognized is their inhibitory activity towards aldose reductase—an enzyme taking part in the pathogenesis of diabetes complications. In fact, 3,3′,4-trimethoxy-4-sulphoxyellagic acid potassium salt isolated from
Potentilla candicans is among the strongest aldose reductase inhibitors of natural origin (IC
50 = 0.08 μM), and the presence of sulphate moiety was demonstrated to have crucial impact on its effectiveness, enhancing it over twice in comparison to ellagic acid (IC
50 = 0.20 μM) [
9]. However, the influence of sulphation on other activity parameters of ellagic acid such as cytotoxicity has not been investigated to date, despite the known direct apoptotic and anti-proliferative effects (IC
50 = 37–72 μM) of the basic structure towards a number of cancer cell lines (Caco-2 colon, HT-29 colon, HTC 116 colon, MCF-7 breast, Hs 578T breast, DU 145 prostatic) [
16,
17].
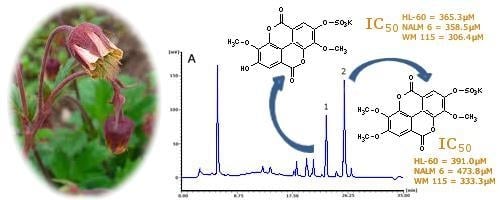
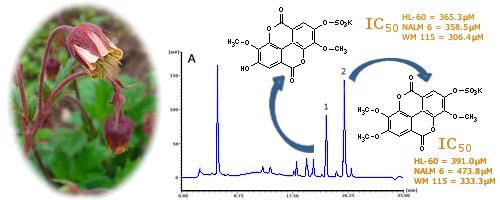
The research on this interesting class of compounds is undoubtedly hindered by their scant occurrence, and lack of the analytical methodology allowing their standardisation in plant tissues and selection of potential new natural sources. This work describes the isolation and identification of two of these rare derivatives—3,3′-dimethoxy-4-sulphoxyellagic acid potassium salt (1) and 3,3′,4′-trimethoxy-4-sulphoxyellagic acid potassium salt (2)—from the rhizome of a common European species G. rivale. Furthermore, HPLC-PDA methodology was optimized and validated to evaluate G. rivale rhizome as a source of sulphate derivatives of ellagic acid in comparison to related plant materials. The cytotoxicity of both isolates was also tested and compared with that of ellagic acid to assess the influence of the sulphate moiety on the anti-cancer activity of the parent compound.
2. Materials and Methods
2.1. General
UV-Vis spectra of the isolates 1 and 2 and the standard ellagic acid (Sigma-Aldrich, Seelze, Germany/St. Louis, MO, USA) were recorded in water at 25 °C on a Perkin-Elmer Lambda 25 spectrophotometer (Waltham, MA, USA). HR-ESI-TOF-MS spectra were measured on a MaldiSYNAPT G2-S HDMS spectrometer (Waters, Milford, MA, USA) with ESI as an ion source (operating in the negative-ion mode) and a TOF detector. 1H NMR, 13C NMR, 1H-1H COSY, HMQC, and HMBC spectra were recorded at 25 °C on a Bruker Avance III 600 spectrometer (Bruker BioSpin Co., Billerica, MA, USA) in D2O (600 MHz for 1 H and 150.9 MHz for 13C), with TMS as the internal standard. The TLC was performed on silica gel 60 G precoated plates (Merck, Darmstadt, Germany) using horizontal DC chambers (Chromdes, Lublin, Poland). Chromatograms were visualized by UV-Vis at 366 nm before and after spraying with diphenylboryloxyethylamine (Sigma-Aldrich). Sephadex LH-20 (50 g, Sigma-Aldrich, Germany/USA) was used for column chromatography. All solvents and chemicals of analytical grade used in the preparative studies were obtained from Chempur (Piekary Slaskie, Poland).
2.2. Plant Material
The rhizome of G. rivale as well as the aerial parts of G. rivale and the aerial parts and rhizome of G. urbanum were collected in Lodz in May 2012 from plants growing in natural habitats. The material was identified by Prof. Jan Gudej from the Department of Pharmacognosy, Medical University of Lodz, Poland. A voucher specimens (KFG/HB/05012-GRV, KFG/HB/05012-GUR) was deposited in the Department of Pharmacognosy, Medical University of Lodz, Poland. Prior to the analysis, the plant samples were powdered using an electric grinder, sieved through a 0.315-mm sieve, and stored in airtight containers at ambient temperature and in darkness until used.
2.3. Preparative Extraction and Isolation
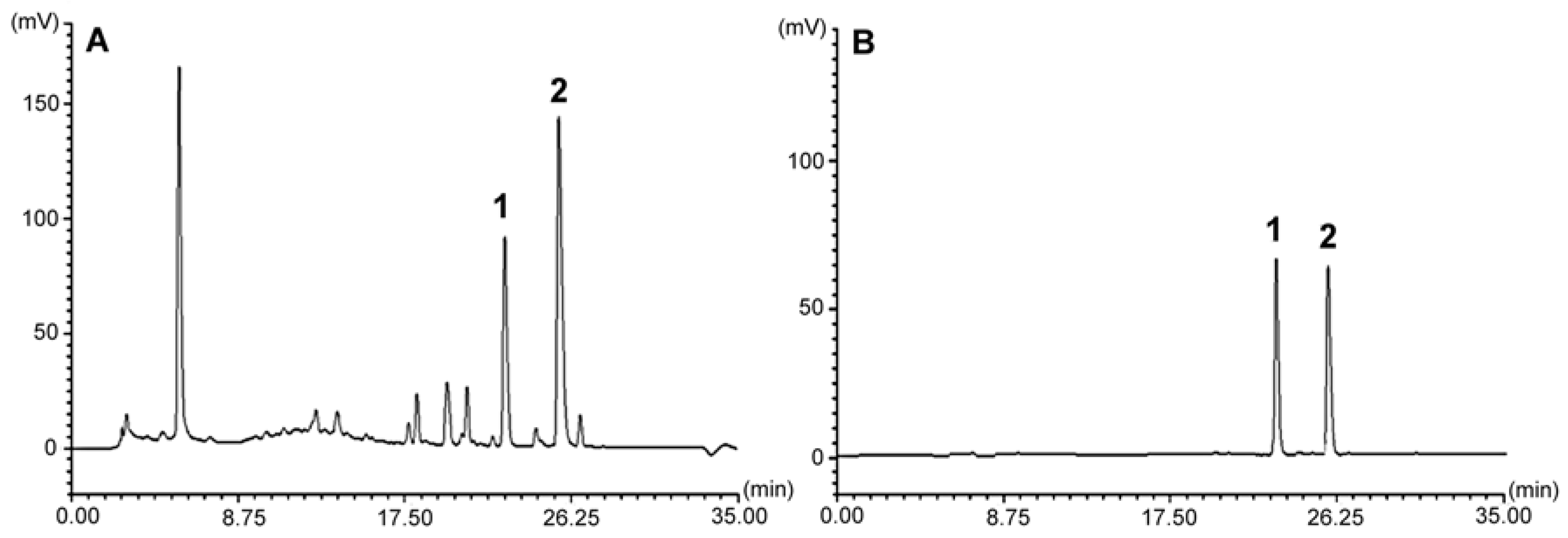
A preparative sample (300 g) of the dried and grinded plant material was exhaustively extracted with acetone-water 70:30 (v/v) at room temperature (22–25 °C) by mechanical shaking (6 × 3 L × 8 h). The combined acetone-water extracts were concentrated in vacuum until complete removal of acetone and then extracted with ethyl acetate (6 × 100 mL) to remove less polar constituents. The water residue containing the compounds of interest was evaporated to dryness and resolved in boiling methanol (500 mL). After 24 h, the crystallised residue (12 g) containing compounds 1 and 2 was obtained. A portion of the precipitate (0.5 g) was subjected to open column gel permeation chromatography over Sephadex LH-20 and eluted with water. The eluates were monitored by TLC (ethyl acetate : methanol : formic acid, 18:1:1, v/v/v) on silica gel 60 G precoated plates. The fractions containing the separated analytes were combined and independently re-chromatographed over Sephadex LH-20 to obtain 35 mg of compound 1 and 40 mg of compound 2, respectively. The replicate rounds of chromatography of two further portions (2 × 0.5 g) of the precipitate confirmed the isolation yield of 2.80 mg/g and 3.20 mg/g dry weight (dw) of the plant material, respectively. The purity of the isolates assessed by HPLC-PDA was 97.3% and 98.1%, respectively.
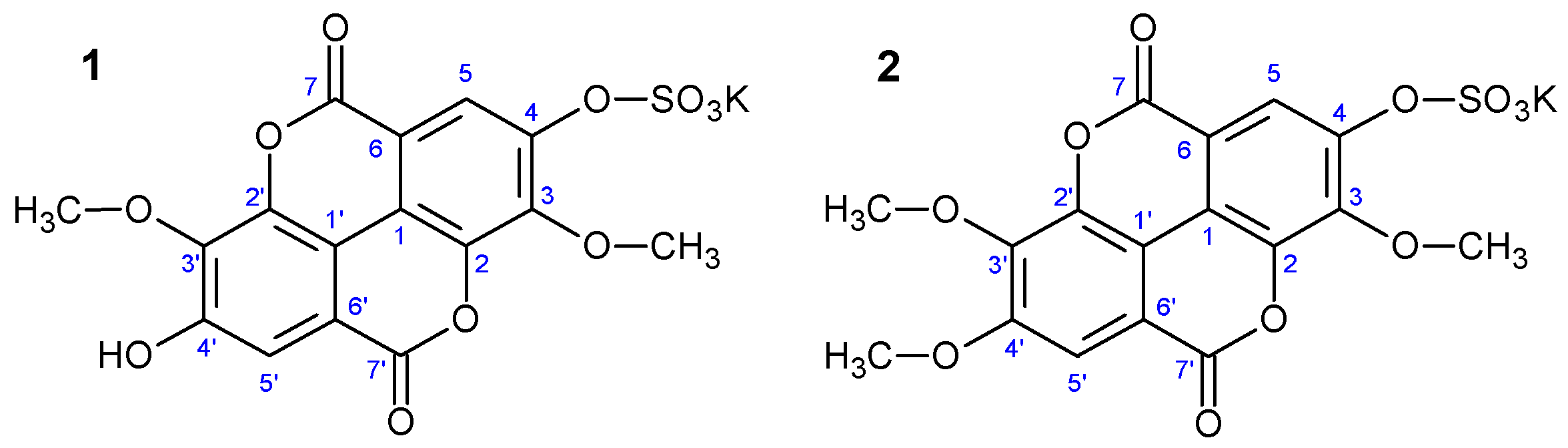
3,3′-dimethoxy-4-sulphoxyellagic acid potassium salt (
1)—whitish amorphous powder; UV (H
2O) λ
max: 245, 350sh, 362; HR-ESI-TOF-MS: 408.9869 [M–K]
− (calcd. for C
16H
9O
11S, 408.9877), 329.0244 [M–KSO
3]
−;
1H and
13C NMR data (D
2O): see
Table 1.
3,3′,4′-trimethoxy-4-sulphoxyellagic acid potassium salt (
2)—whitish amorphous powder; UV (H
2O) λ
max: 246, 347sh, 361; HR-ESI-TOF-MS: 423.0009 [M–K]
− (calcd. for C
17H
11O
11S, 423.0022), 343.0397 [M–KSO
3]
−;
1H and
13C NMR data (D
2O): see
Table 1.
2.4. Quantitative HPLC-PDA Assay
2.4.1. HPLC-PDA Equipment and Methodology
Qualitative HPLC analyses were carried out using Waters 600E Multisolvent Delivery System (Waters, Milford, MA, USA) with a PDA detector (Waters 2998) scanning in the wavelength range of 220–450 nm, a model 7725 sample injection valve (Rheodyne, Pittsburgh, PA, USA) with a 20 μL injection loop, and an LC workstation equipped with Waters Empower 2 software for data collection and acquisition. A Nucleodur C18 HPLC column (250 × 4.6 mm i.d., 5 μm; Macherey-Nagel, Bethlehem, PA, USA) guarded by a C18 Hypersil ODS pre-column (5 mm, 4 mm × 4 mm, i.d.; Agilent Technologies, Palo Alto, CA, USA) was used. Constant temperature of the column was maintained at 25 °C using a Jetstream Plus 5480 thermostat (Peltier, Langenzersdorf, Austria). The phenolic compounds were separated using the mobile phase consisting of solvent A (0.5% water solution of orthophosphoric acid, w/v) and solvent B (methanol), with the elution profile as follows: 0–1 min, 20% (v/v) B; 1–25 min, 20–70% (v/v) B; 25–28 min, 70% (v/v) B; 28–29 min, 70–20% (v/v) B; 29–35 min, 20% (v/v) B (equilibration). All gradients were linear. Flow rate: 1.0 mL/min. Detection wavelength: 360 nm. All chemicals used in HPLC analyses including methanol (Avantor Performance Materials, Gliwice, Poland), water, and 85% (w/w) orthophosphoric acid (Merck, Darmstadt, Germany) were of HPLC grade.
2.4.2. Sample Preparation
An accurately weighted sample of the plant material (1000 mg) was extracted three times with deionised water by mechanical shaking (15 min for each run) using sequential volumes of 20 mL, 10 mL, and another 10 mL of the solvent. The obtained extracts were combined, diluted with water to 50 mL, filtered through the syringe filter (25 mm, 0.2 µm, Vitrum, Praha, Czech Republic) and directly subjected to the HPLC system (20 µL). Determinations were performed after three separate extractions of each sample, and each extract was injected in triplicate.
2.4.3. Method Validation
The analytical method was validated by determination of the selectivity, linearity, precision, accuracy, and stability of each analyte, according to the International Council for Harmonisation (ICH) Guidance for Industry [
18] and some previous literature reports [
19].
The standard stock solution of compounds 1 and 2 was prepared in triplicate in deionised water and serially diluted (in two replicates) with the same solvent to six concentration levels (2%, 10%, 25%, 50%, 75%, and 100% of the stock concentration) within the ranges of 2.98–149.30 μg/mL and 3.14–157.20 μg/mL, respectively. The replicate solutions were injected three times into the HPLC system. Calibration tests were run at the beginning, midpoint, and at the end of the analytical tests. A calibration graph was constructed by plotting the mean peak area versus concentration. The linearity was tested by the regression method of least squares and the statistical F- and t-tests at the 99% confidence level. The residuals from linear regression models were tested ex post to check the validity of the assumptions of normality, independence and homoscedasticity of the response variables. The possible matrix effects were evaluated by constructing the sensitivity plots for the standards dissolved in the real sample of the rhizome of G. rivale. Concentration ranges of the added standards were the same as those used for calibration. Statistical differences between the slopes of the matrix-matched linear regression equations and the calibration curves were tested by the Tukey’s honest significant difference (HSD) test at the 95% confidence level.
The limit of detection (LOD) and limit of quantitation (LOQ) values were estimated from the standard solution diluted with water to provide serial solutions with their concentrations decreasing to the smallest detectable peaks. The LODs were accepted with the 3-signal-to-noise (S/N) ratio, while the LOQs were accepted if the relative standard deviation (RSD) values for peak area were less than 15% for both intraday and inter-day variability with S/N ratio greater than 10, according to [
20].
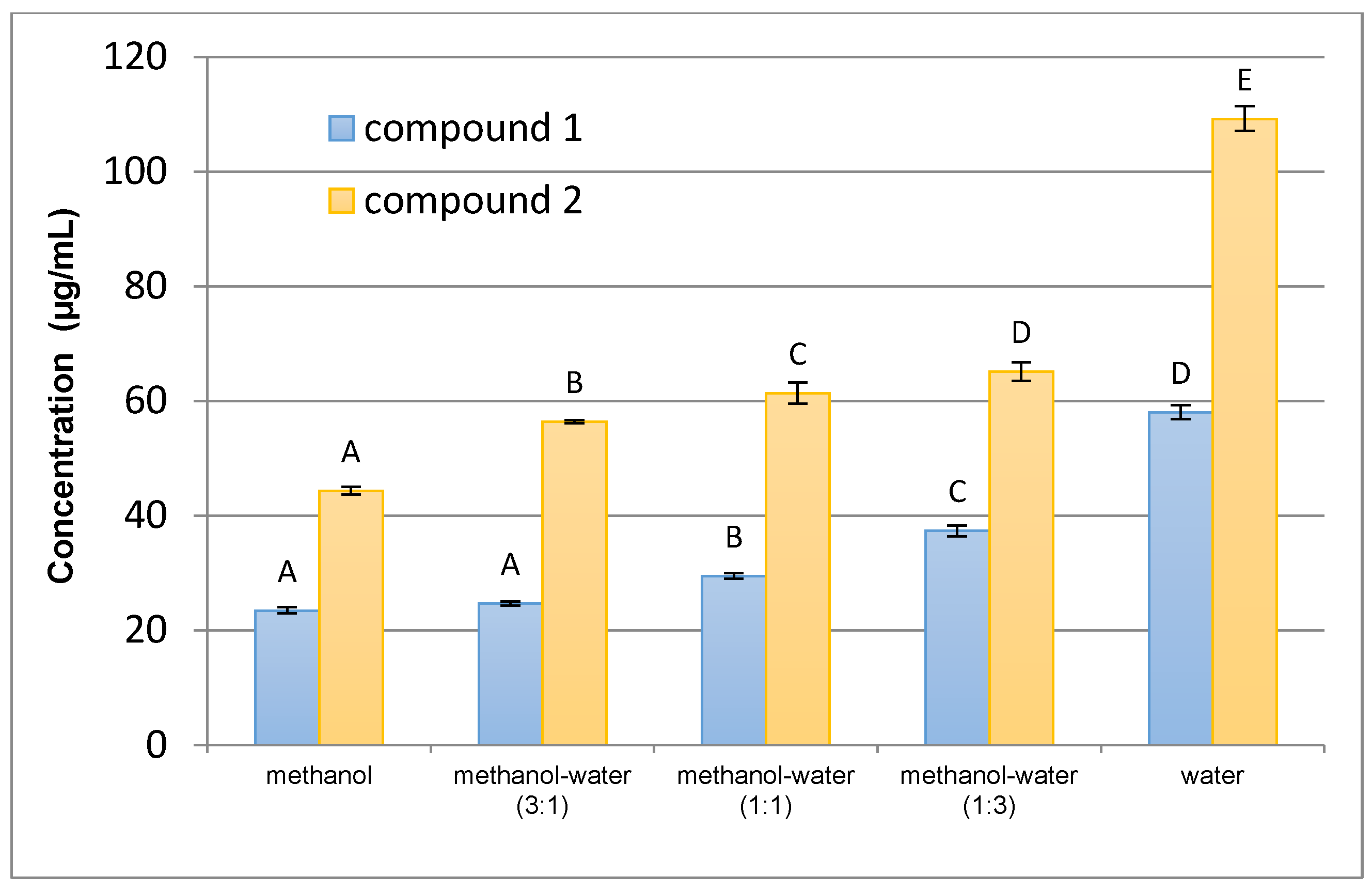
Stability of the standards was tested using standard stock solution. The solution was kept at 4 °C for 30 days and the content of compounds 1 and 2 was determined every fifth day and compared with the initial concentration.
The precision tests were performed in triplicate using standard solution at two concentration levels (10% and 100% of the stock concentration) and the real sample of the rhizome of G. rivale. The replicate solutions were injected three times into the HPLC system and the RSD values were calculated for both retention time and peak area, and considered as a measure of precision. The repeatability (intra-day variability) was determined by analysing each sample within 24 h, while the reproducibility (inter-day variability) was measured on three non-consecutive days.
The accuracy of the method was tested in the extract of
G. rivale rhizome by means of the standard addition/recovery procedure at three different levels (0.25, 1.25, or 2.5 mg) of each standard, corresponding to the investigated analytical range. The samples were prepared in triplicate according to the procedure described above (
Section 2.4.2) with the standards added to the plant material prior to the extraction. The replicate samples were injected in triplicate, and the accuracy was evaluated by calculating the mean recovery of the analytes from the spiked extracts versus the non-spiked samples.
2.4.4. Cytotoxic Activity Assay
Cytotoxicity of the compounds was assessed by the mitochondrial reduction assay [
21] on two human leukaemia cell lines (promyelocytic HL-60 and lymphoblastic NALM-6) and on human melanoma cell line WM 115. Cells were purchased from the European Collection of Cell Cultures (ECACC, Salisbury, UK). Leukaemia cells were cultured in RPMI 1640 medium, while WM 115 cells in DMEM (Dulbecco’s Modified Eagle Medium), both supplemented with 10% heat-inactivated foetal bovine serum (Invitrogen, Paisley, UK) and antibiotics (100 mg/mL streptomycin and 100 U/mL penicillin). Cells were grown in 37°C, in a humidified atmosphere of 5% CO
2 in air. Exponentially growing cells were seeded at 8 × 103 per well on 96-well plates (Nunc, Roskilde, Denmark). After 24 h, the tested compounds (freshly prepared in DMSO and diluted with complete culture medium to obtain the concentration range from 0.1 to 1000 μM) were added and the plates were incubated for 48 h. Afterwards, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT, 5 mg/mL in PBS) was added and incubation was continued for 2 h. The metabolically active cells reduced MTT to blue formazan crystals. MTT-formazan crystals were dissolved in 20% SDS and 50% DMF at pH 4.7 and absorbance was read at 570 nm on an ELISA-plate reader (ELX 800, Bio-Tek, Winooski, VT, USA) and compared with that of the control (untreated cells). The IC
50 values (the concentrations of the test compounds required to reduce the cells survival by 50% in comparison to the untreated cells) were calculated from concentration-response curves and used as a measure of cellular sensitivity to a given treatment. Unless stated otherwise, all reagents used in the cytotoxicity tests were from Sigma-Aldrich (Germany/USA).
2.4.5. Statistical and Data Analysis
The results were expressed as means ± standard deviation (SD) of replicate determinations. The statistical analyses (calculation of SD, one-way analysis of variance, HSD Tukey tests, linearity studies) were performed using the Statistica12 PL software for Windows (StatSoft, Krakow, Poland), with p values less than 0.05 being regarded as significant.
4. Discussion
Due to their specific physicochemical and biological characteristics, sulphated phenolic conjugates are undoubtedly an interesting class of compounds. Unfortunately, they are generally not very common in nature. Sulphated flavonoids, which are by far the most prevalent group, have been found in over 250 species [
23]. Some sulphate derivatives of phenolic acids, anthraquinones, and coumarins have also been identified [
24]. Only eight species from five different families were reported to contain ellagic acid sulphates. This paper is the first report of that type of compounds in the genus
Geum, and second in the Rosaceae family. Compound
1 has previously only been isolated from
Tamarix tetragyna and
Euphorbia sororia, whereas compound
2 has previously been isolated from the latter species and
Potentilla candicans [
9,
10,
13].
The results of quantitative analysis confirmed that the rhizome of G. rivale is a good source of the investigated compounds. On the other hand, only trace amounts of the analytes were found in the aerial parts of the plant, which demonstrates that accumulation of the sulphates is organ-specific. As the production of particular metabolites is often taxonomically dependent, the rhizome and aerial parts of closely related species—G. urbanum—were also investigated. The compounds were detectable only in the rhizome of G. urbanum, but the content below the quantification limit excludes the plant material as an alternative source of the analytes.
The relatively high yields from the isolation procedure (6.00 mg/g; 71.5% of the factual content) indicate that the developed simple separation procedure might be successfully applied for cost-effective production and purification of these natural chemicals. Previously, the highest yield was obtained from
Potentilla candicans—1.6 g of compound
2 was isolated from 5 kg of dry plant material [
9] which corresponds to the content of only 0.3 mg/g dw. However, the methods used in the study were multi-step general isolation procedures, not targeted at any particular compound. Thus, the recovery of those methods may underestimate the actual content of the isolates.
The role of phenolic sulphates in plant metabolism remains yet to be fully explained although some specific compounds were found to have significance in plant growth, development, and adaptation to stress [
25,
26]. It was postulated before for sulphated flavonoids that their occurrence in plants is rather an ecological adaptation than a taxonomical trait [
27]. The differences in the sulphates content between two closely related species examined in our study seems to confirm this assumption. However, the most common hypothesis about their role in adaptation to water-stress [
27,
28], does not appear to be valid here—in contrast to many of the plants containing sulphate derivatives of ellagic acid, e.g.,
Tamarix sp.,
Reaumuria vermiculata, and
Frankenia laevis that thrive on dry or salty soils [
12,
15,
29],
G. rivale prefers slow draining soils where water is in abundance [
1].
As far as pharmacological effect on human organism is considered, several biological activities were found for sulphated phenolics including anticoagulant, antiviral, antitumor, antibacterial, and anti-inflammatory capacity. On the other hand, the knowledge on the biological properties of sulphate ellagic acid derivatives is very limited. Apart from considerable inhibitory activity towards aldose reductase noticed for compound
2 isolated from
P. candicans [
9], compounds
1 and
2 were also found to exhibit moderate antimicrobial potential against
Bacillus subtilis and
Staphylococcus aureus with MIC values in the range of 22.54–50.83 μg/mL [
13]. They were, however, not effective against
Escherichia coli [
13]. So far, there has been no research about their cytotoxic activity.
Ellagic acid is a well-known chemopreventive agent protecting cells against DNA damage and mutations caused by various carcinogenic factors such as radiation, aflatoxins, benzo[a]pyrene, and
N-nitrosobenzylmethylamine [
7]. There are also plenty of data confirming its direct apoptotic and anticancer activity towards a number of human tumours including breast, colon, prostate, and oral carcinoma, although in most cases its cytotoxicity, with the IC
50 values estimated in the range of 10–350 μM, was moderate as compared with cytostatics used in clinical practice [
16,
17]. This study’s results (
Table 4) obtained for the free acid from leukaemia and melanoma cell lines are consistent with the above findings.
The data concerning substituted derivatives of ellagic acid in general are scarce and to some extent contradictory. Nesser et al. [
30] found that methylation of hydroxyl groups in the molecule leads to reduction of the cytotoxicity towards mice macrophages and postulated it could be connected with substitution of hydroxyl groups at C-3 and C-3′ positions, important for inhibitory activity towards topoisomerases [
31]. The more polar glycosidic methylated derivatives were even less effective [
31]. Similar findings were presented by Manayi et al. [
32]—glycosylated and methylated derivatives had very weak cytotoxicity with IC
50 values in the range of 374.5–711.7 μg/mL (1.09–1.41 mM). On the other hand, 3,3′-dimethylellagic acid 4′-
O-
β-
d-xylopyranoside from
Euphorbia hylonoma has been claimed to be effective against HepG2 human hepatocellular carcinoma cell line [
33]. As this research shows, methylation connected with sulphation has negative impact on ellagic acid cytotoxicity against leukaemia and melanoma cell lines, the present work generally confirmed the majority of previous studies. However, a more detailed study would be needed for a series of ellagic acid derivatives to differentiate activity effects between methoxy and sulphoxy groups at various positions. Nevertheless, the observed decrease in cytotoxicity might be indeed partly connected with blockage of hydroxyl groups responsible for intermolecular interactions and hydrogen bond formation. The difficulties in trans-membrane transport may also be the cause. As was found in the case of flavonoid derivatives, the conjugation with strong polar groups such as sugar or sulphate moieties decreases the lipid solubility, the partition coefficient and thus the probability of effective passage across cell membranes [
34].







{kind=link}
{kind=link}
{kind=link}
{kind=link}