
Polymerization in the Borstar Polypropylene Hybrid Process: Combining Technology and Catalyst for Optimized Product Performance


Abstract
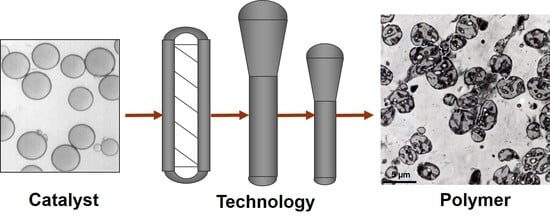
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
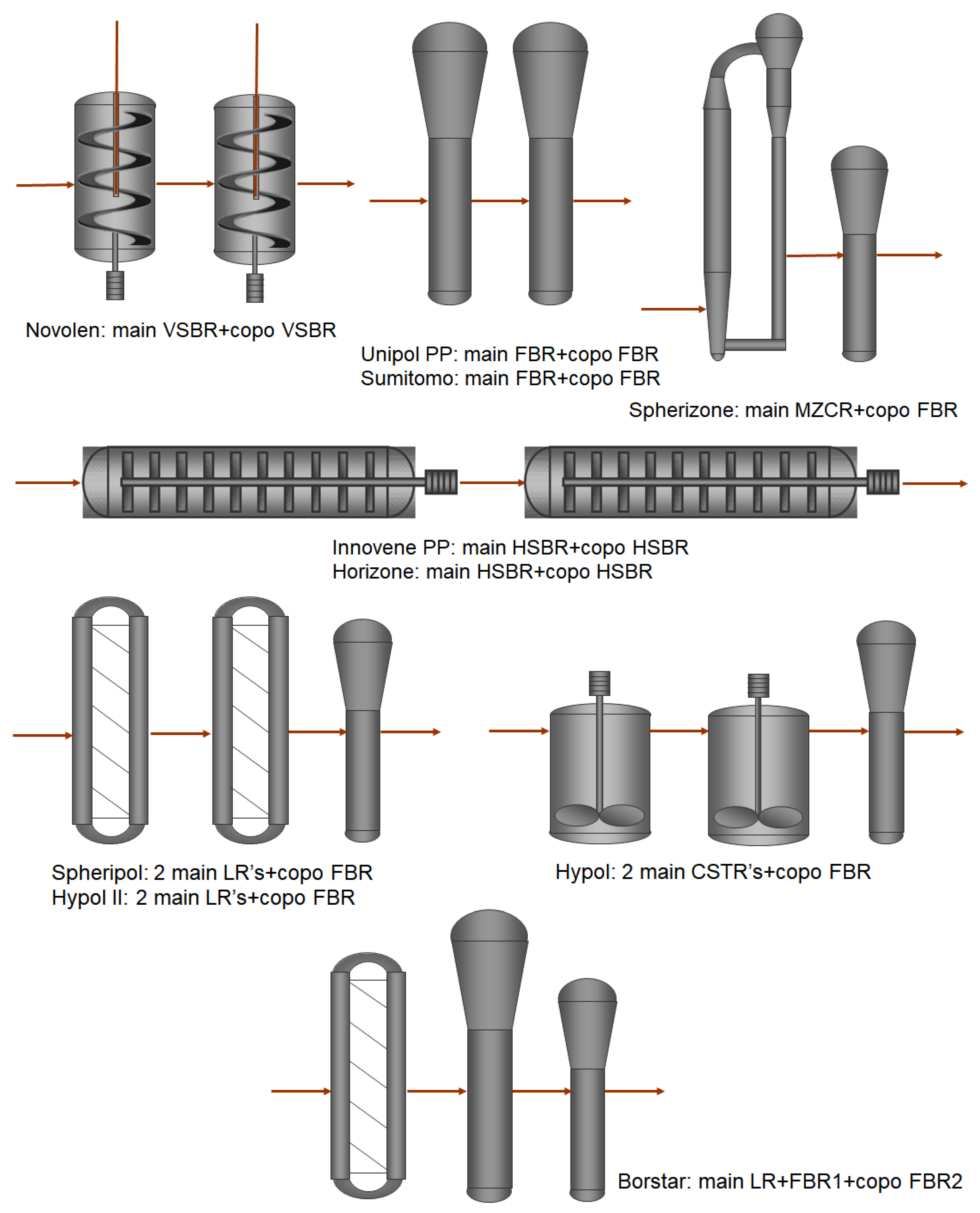
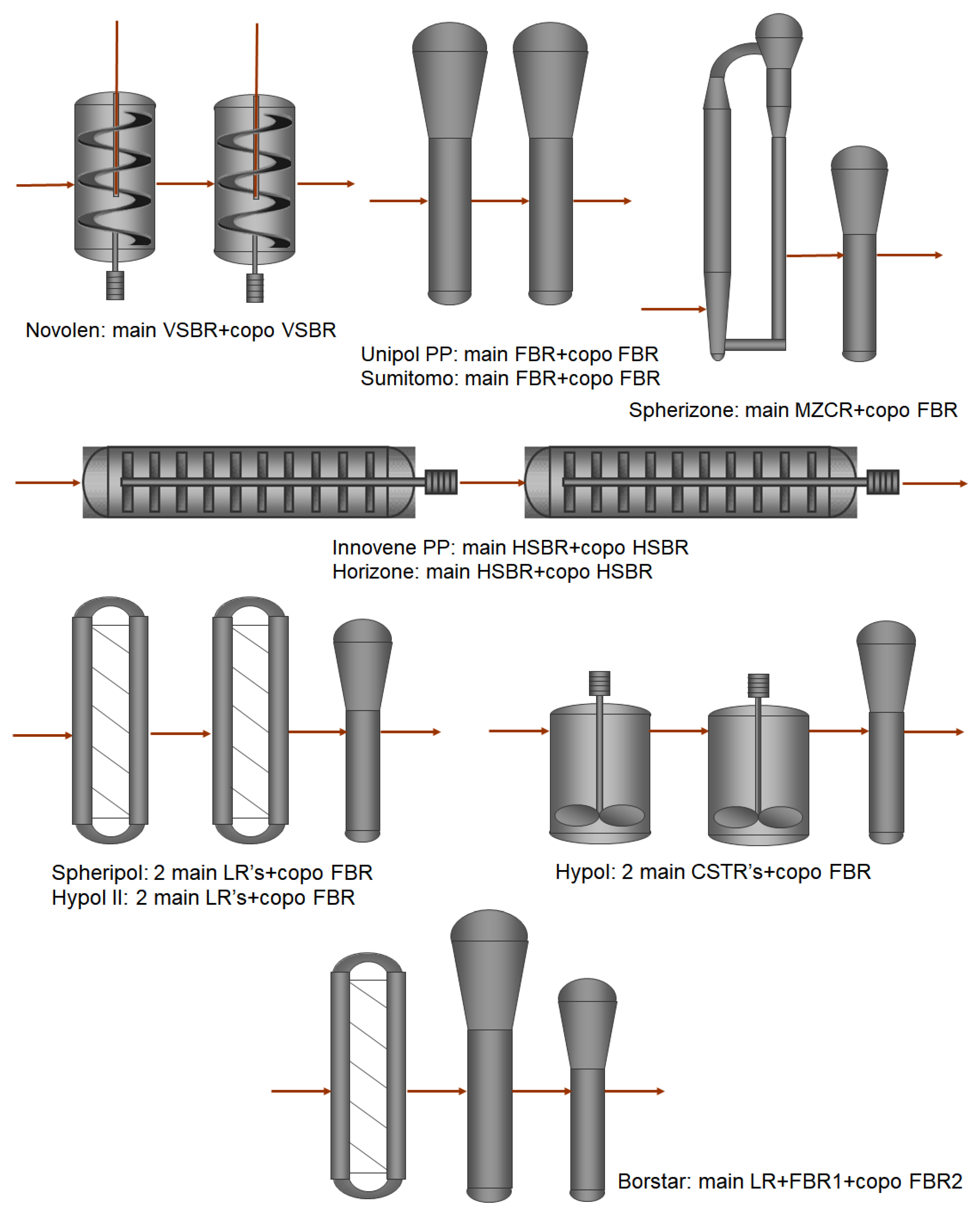
2. Borstar® PP Technology
- Feedstock preparation;
- Reactor area;
- Recovery area;
- Dry end (comprising pelletizing);
- Material handling, comprising bagging and storage area.
- Very high catalyst productivity due to high-temperature operation;
- Very wide product window and independent reactor control, enabling the tailoring of the MWD and CC along the MWD in both the matrix and rubber part of the product;
- High once-through monomer conversion due to propylene conversion from the loop effluent to the first GPR;
- Competitive monomer factor and energy consumption;
- Robust reactor operability and reliability.
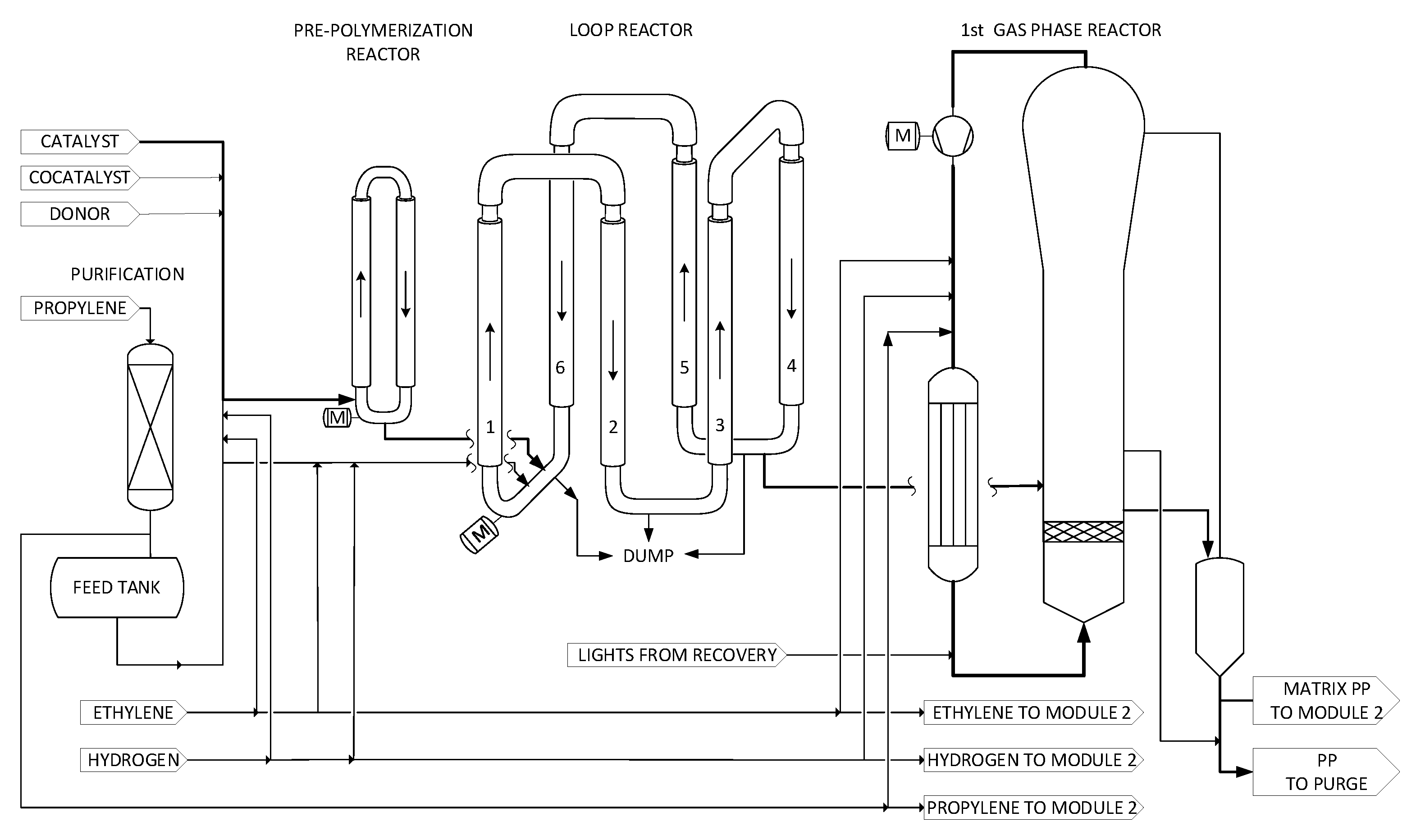
2.1. Module 1: Catalyst Preparation and Pre-Polymerization
2.2. Module 1: Loop Reactor and First GPR
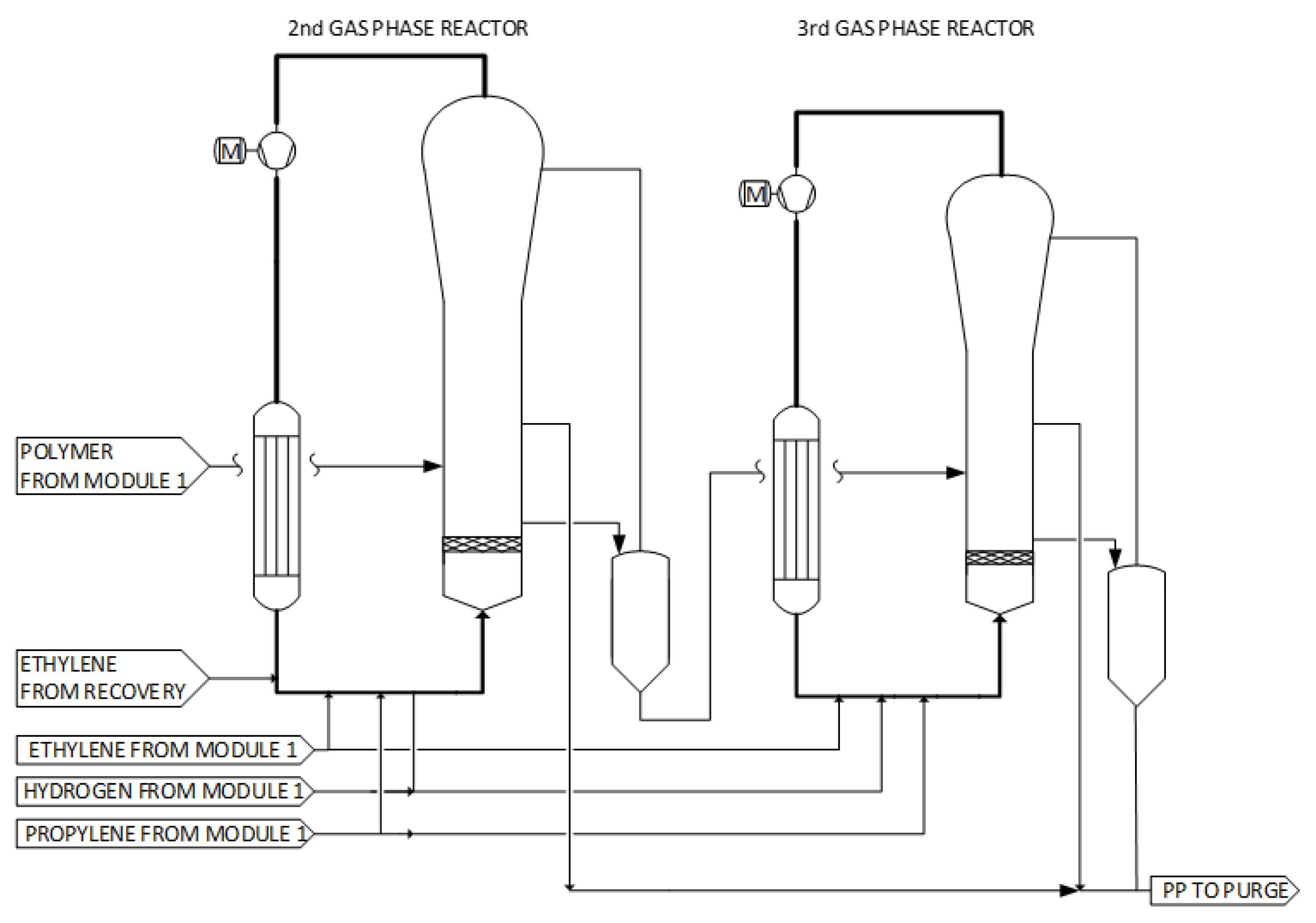
2.3. Module 2: Additional GPRs
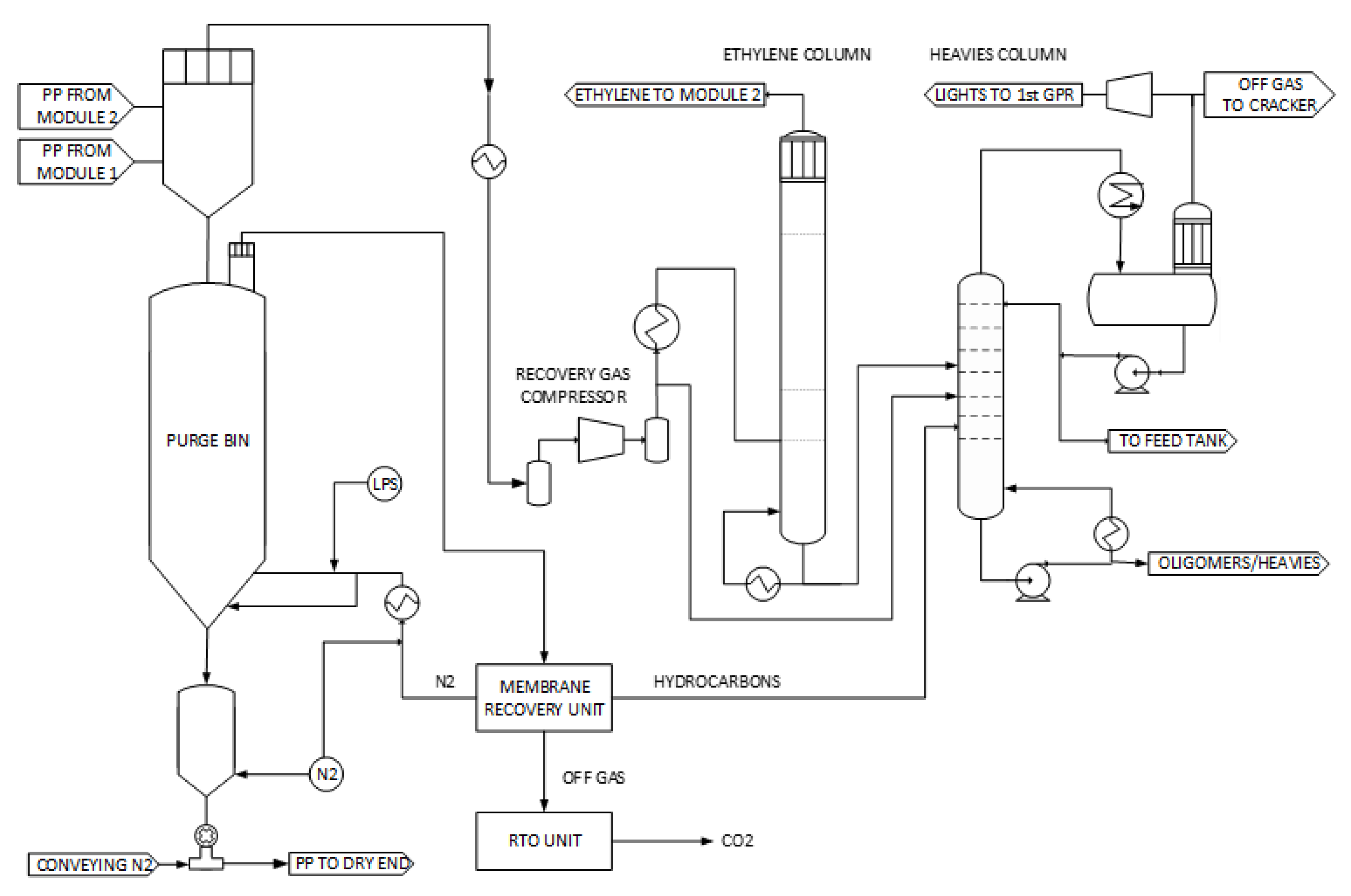
2.4. Downstream Area
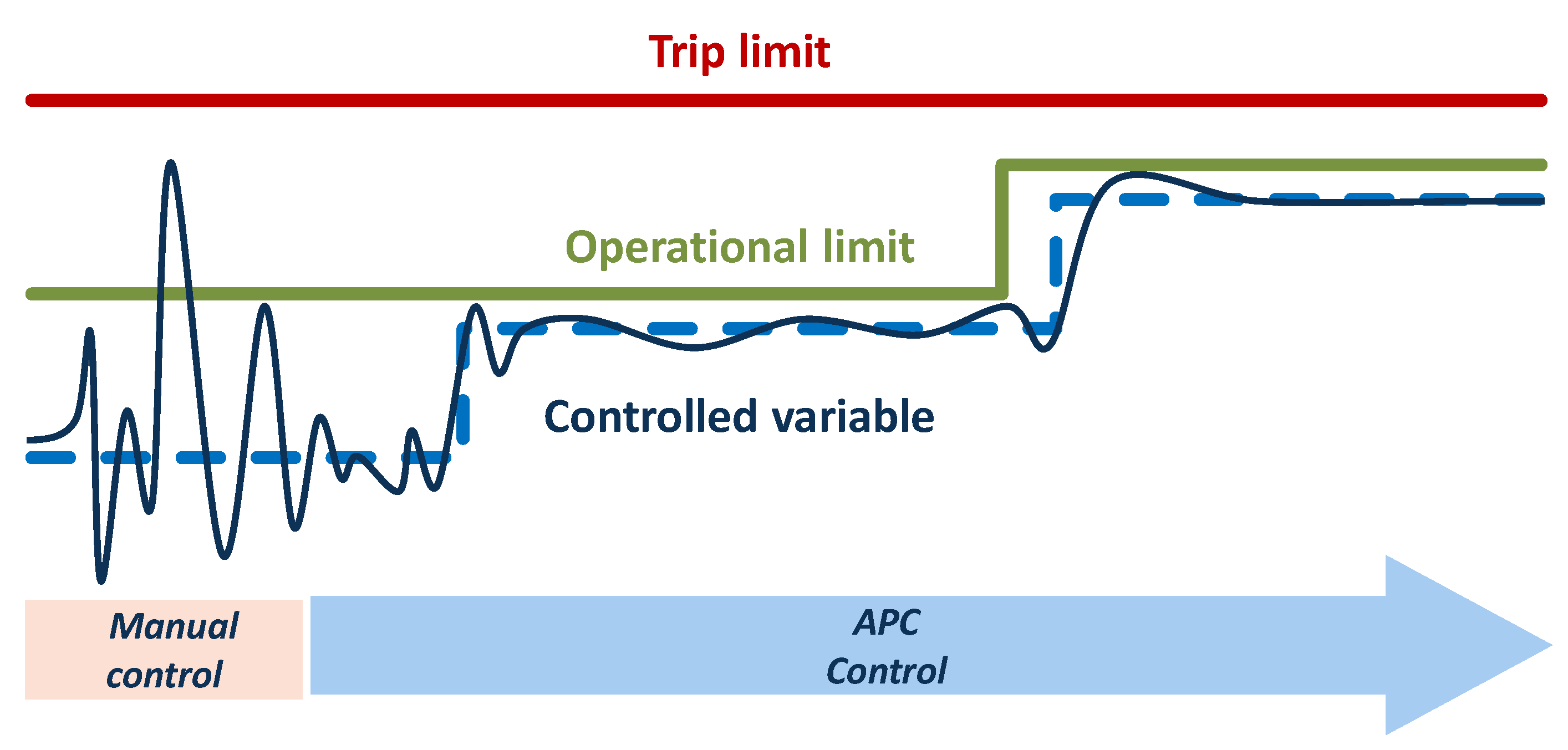
2.5. Process Control
2.6. Dry End, Quality Control and Sustainability
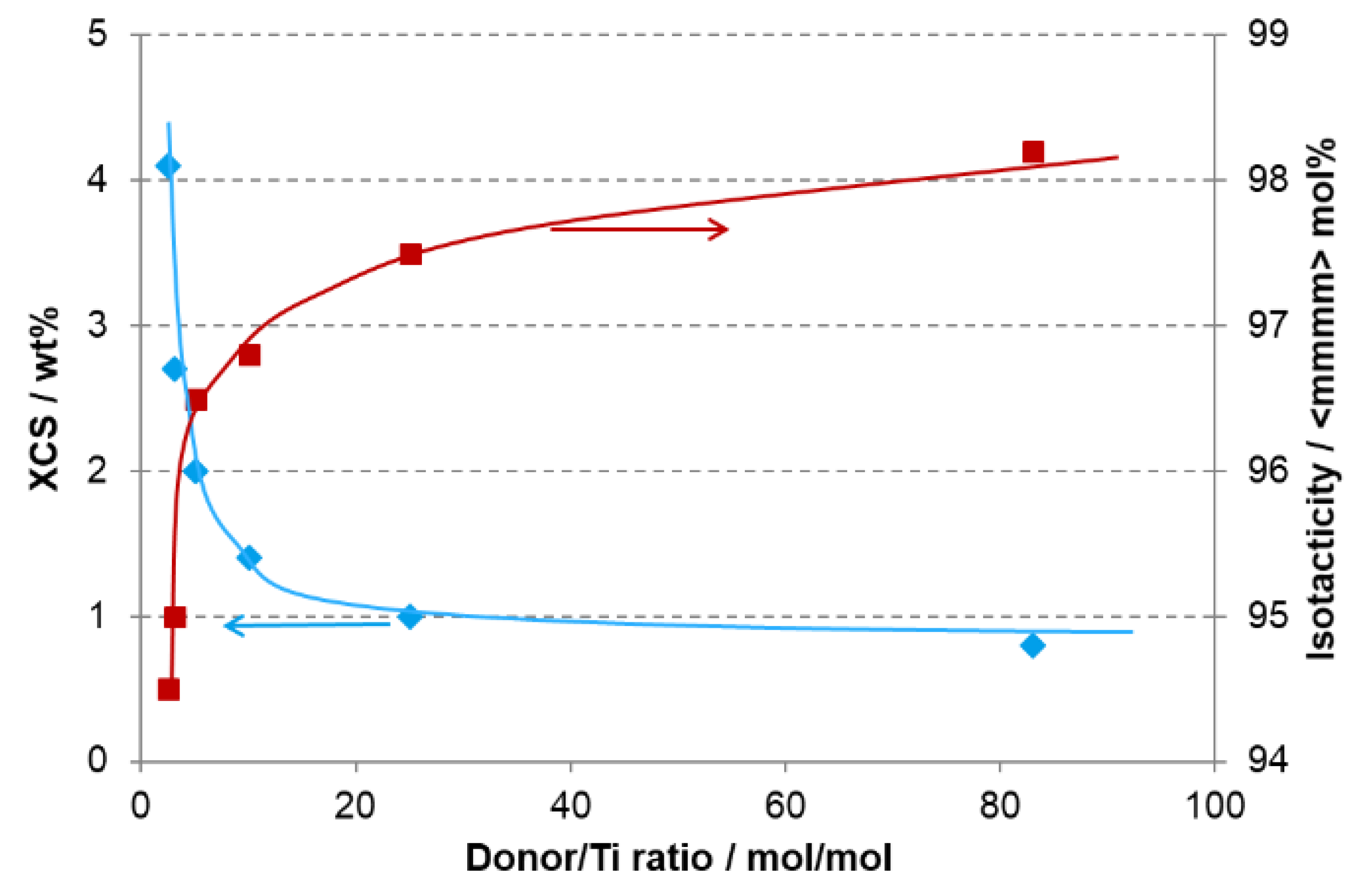
3. Borealis Catalyst Design
MgCl2 × n R3COOR1 × m TiCl4 + n TiCl3OR2
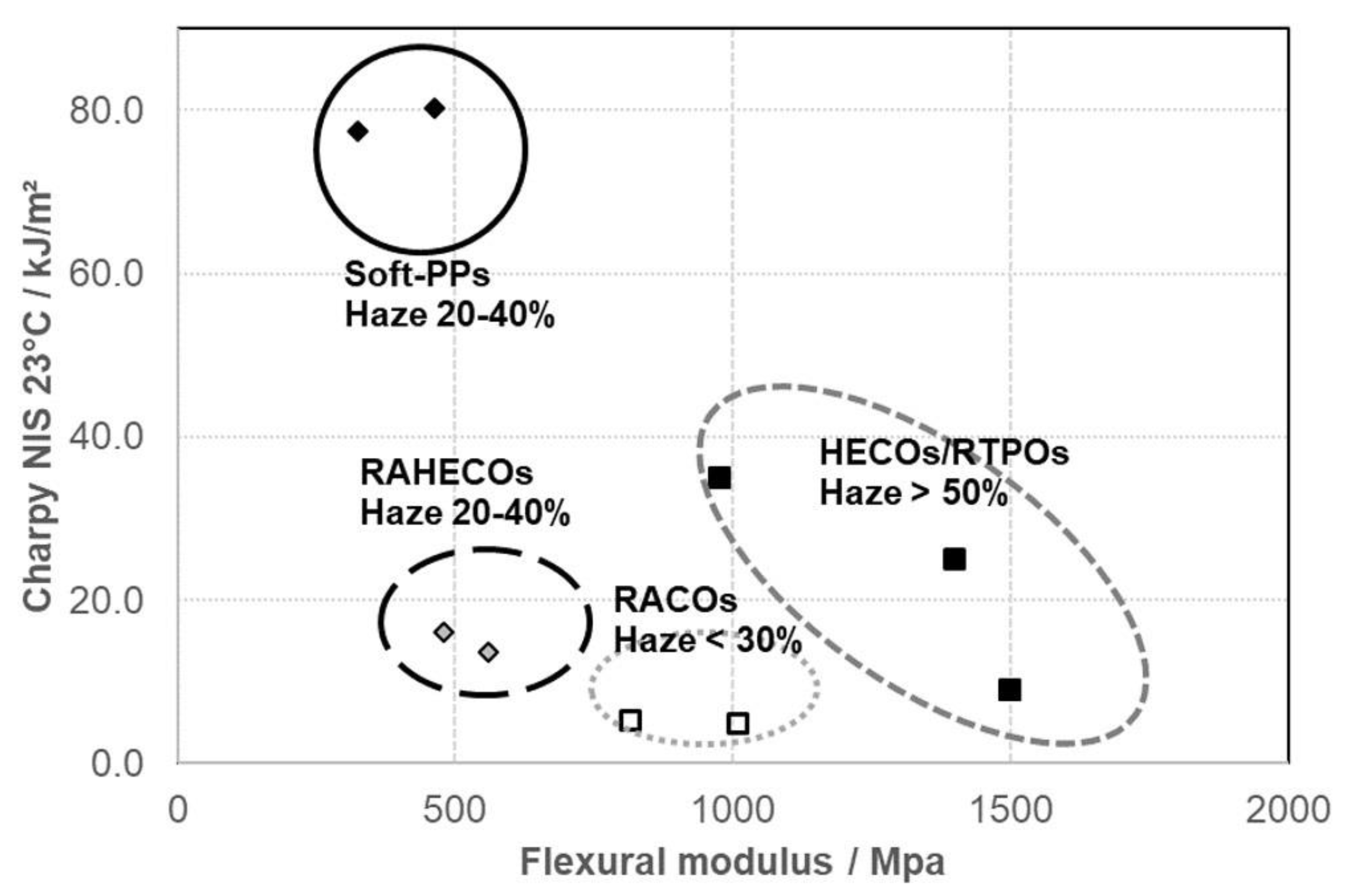
4. Borstar PP Composition and Performance Range
5. Conclusions and Outlook
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Natta, G.; Pasquon, I.; Zambelli, A.; Gatti, G. Highly stereospecific catalytic systems for the polymerization of α-olefins to isotactic polymers. J. Polym. Sci. 1961, 51, 387–398. [Google Scholar] [CrossRef]
- Natta, G.; Corradini, P.; Allegra, G. The different crystalline modifications of TiCl3, a catalyst component for the polymerization of α-olefins. I: α-, β-, γ-TiCl3. II: δ-TiCl3. J. Polym. Sci. 1961, 51, 399–410. [Google Scholar] [CrossRef]
- Jubinville, D.; Esmizadeh, E.; Saikrishnan, S.; Tzoganakis, C.; Mekonnen, T. A comprehensive review of global production and recycling methods of polyolefin (PO) based products and their post-recycling applications. Sustain. Mater. Technol. 2020, 25, e00188. [Google Scholar] [CrossRef]
- Moore, E.P. The Rebirth of Polypropylene: Supported Catalysts; Hanser: Munich, Germany, 1998; ISBN 3-446-19587-4. [Google Scholar]
- Luciani, L.; Barbe, P.C.; Kashiwa, N.; Toyoda, A. Polymerisation Catalyst. German Patent DE 2643143 A1, 6 February 1977. [Google Scholar]
- Giannini, U.; Albizzati, E.; Parodi, S. Components of Catalysts Useful for the Polymerization of α-Olefins, and Catalysts Prepared Therefrom. German Patent DE 2735672 A1, 16 February 1978. [Google Scholar]
- Albizzati, E.; Galimberti, M.; Giannini, U.; Morini, G. The Chemistry of Magnesium Chloride Supported Catalysts for Polypropylene. Die Makromol. Chem.—Macromol. Symp. 1991, 48, 223–238. [Google Scholar] [CrossRef]
- Galli, P.; Vecellio, G. Polyolefins: The Most Promising Large-Volume Materials for the 21st Century. J. Polym. Sci. A Polym. Chem. 2004, 42, 396–415. [Google Scholar] [CrossRef]
- Chadwick, J.C. Polyolefins—Catalyst and Process Innovations and their Impact on Polymer Properties. Macromol. React. Eng. 2009, 3, 428–432. [Google Scholar] [CrossRef]
- Soares, J.B.P.; McKenna, T.F.L. Polyolefin Reaction Engineering; Wiley-VCH: Weinheim, Germany, 2012; ISBN 9783527317103. [Google Scholar]
- Mei, G.; Herben, P.; Cagnani, C.; Mazzucco, A. The Spherizone Process: A New PP Manufacturing Platform. Macromol. Symp. 2006, 245, 677–680. [Google Scholar] [CrossRef]
- Martino-Andrade, A.J.; Chahoud, I. Reproductive toxicity of phthalate esters. Mol. Nutr. Food Res. 2010, 1, 148–157. [Google Scholar] [CrossRef]
- Chadwick, J.C.; Morini, G.; Balbontin, G.; Camurati, I.; Heere, J.J.R.; Mingozzi, I.; Testoni, F. Effects of Internal and External Donors on the Regio- and Stereoselectivity of Active Species in MgCl2-Supported Catalysts for Propene Polymerization. Macromol. Chem. Phys. 2001, 202, 1995–2002. [Google Scholar] [CrossRef]
- Zhang, H.-X.; Lee, Y.-J.; Park, J.-R.; Lee, D.-H.; Yoon, K.-B. Control of Molecular Weight Distribution for Polypropylene Obtained by Commercial Ziegler-Natta Catalyst: Effect of Electron Donor. Macromol. Res. 2011, 19, 622–628. [Google Scholar] [CrossRef]
- Severn, J.R. Recent Developments in Supported Polyolefin Catalysts: A Review. In Multimodal Polymers with Supported Catalysts. Design and Production; Albunia, A.R., Prades, F., Jeremic, D., Eds.; Springer Nature: Cham, Germany, 2019; pp. 1–53. [Google Scholar]
- Pellecchia, R.; Shutov, P.; Wang, J.; Gahleitner, M. Copolymer Structure and Performance Consequences of High-Impact Ethylene–Propylene Copolymers Based on a Ziegler–Natta Catalyst with Novel Internal Donor. Macromol. React. Eng. 2020, 14, 2000022. [Google Scholar] [CrossRef]
- Kivelä, J.; Grande, H.; Korvenoja, T. Borstar Polypropylene technology. In Handbook of Petrochemicals Production Processes; Meyers, R.A., Ed.; McGraw-Hill: New York, NY, USA, 2005. [Google Scholar]
- Elovainio, E.; Vuorikari, M.; Korhonen, E.; Leskinen, P. Process for Polymerizing Olefins in the Presence of an Olefin Polymerization Catalyst. PCT Patent WO 2006063771 A1, 22 June 2006. [Google Scholar]
- Soleimani, A.; Aigner, A.; Touloupidis, V. Single Particle Growth, Fragmentation and Morphology Modelling: A DEM Approach. Macromol. React. Eng. 2022, 16, 202200015. [Google Scholar] [CrossRef]
- Leskinen, P.; Tuominen, O. Process for the Production of Polypropylene Copolymers Using a Prepolymerized Catalyst. PCT Patent WO 2008151794 A1, 18 December 2008. [Google Scholar]
- Leskinen, P.; Hakola, S.; Alastalo, K.; Nyfors, K. Loop Reactor Providing an Advanced Production Split. PCT Patent WO 2013092342 A1, 27 June 2013. [Google Scholar]
- Harlin, A.; Alastalo, K.; Korhonen, E.; Kivelä, J. Process for Preparing Propylene Homopolymers and Copolymers. PCT Patent WO 9858975 A1, 30 December 1998. [Google Scholar]
- Heino, T.; Karvinen, S. Method and Apparatus for producing Polymers. PCT Patent WO 2005087361 A1, 22 September 2005. [Google Scholar]
- Kokko, T.; Elovainio, E.; Kivelä, J.; Nyfors, K.; Hagane, K. Process for Producing Polyolefins. PCT Patent WO 2011066892 A1, 9 June 2011. [Google Scholar]
- Nyfors, K.; Elovainio, E.; Kanellopoulos, V.; Weickert, G.; Prinsen, E.-J. Process and Apparatus for Removing Polymer Material from a Gas-Solids Olefin Polymerization Reactor. PCT Patent WO 2018233999 A1, 27 December 2018. [Google Scholar]
- Bergstra, M.F.; Eriksson, E.J.G. Impurities in polyolefin production. In Proceedings of the International Conference on the Reaction Engineering of Polyolefins, Montreal, QC, Canada, 23–27 June 2008. [Google Scholar]
- Hillestad, M.; Andersen, K.S. Model Predictive Control for Grade Transitions of a Polypropylene Reactor. In Proceedings of the ESCAPE’4, Dublin, Ireland, 28–30 March 1994. [Google Scholar]
- Haugwitz, S.; Wilsher, E.; Hofsten, K.; Andersen, K.S.; Moen, Ø.; Glemmestad, B. Commissioning of Nonlinear Model Predictive Controllers to a New-Polypropylene Plant. In Proceedings of the Reglermötet, Stockholm, Sweden, 4–5 June 2008. [Google Scholar]
- Kashiwa, N. Super Active Catalyst for Olefin Polymerization. Polym. J. 1980, 12, 603–608. [Google Scholar] [CrossRef] [Green Version]
- Chien, J.C.W.; Vizzini, J.C. Superactive and Stereospecific Catalysts. IV. Influence of Structure of Esters on MgCl2 Supported Olefin Polymerization Catalysts. J. Polym. Sci. A Polym. Chem. 1990, 28, 273–284. [Google Scholar] [CrossRef]
- Seppälä, J.V.; Härkönen, M.; Luciani, L. Effect of the structure of external alkoxysilane donors on the polymerization of propene with high activity Ziegler-Natta catalysts. Die Makromol. Chem. 1989, 190, 2535–2550. [Google Scholar] [CrossRef]
- Härkönen, M.; Seppälä, J.V.; Salminen, H. External Silane Donors in Ziegler–Natta Catalysis. A Three-Site Model Analysis of Effects on Catalyst Active Sites. Polym. J. 1995, 27, 256–261. [Google Scholar] [CrossRef] [Green Version]
- Garoff, T.; Eloranta, K. A Method for the Preparation of a Carrier for a Ziegler/Natta Polymerization Catalyst, a Carrier Prepared Using the Method, and its Use in a Polymerization Catalyst System. EP Patent EP 0364098 A2, 18 April 1990. [Google Scholar]
- Iiskola, E.; Koskinen, J. Procedure for Manufacturing Catalyst Components for Polymerizing Olefines. PCT Patent WO 8707620 A1, 17 December 1987. [Google Scholar]
- Garoff, T.; Leinonen, T.; Iiskola, E. A Method for the Modification of Catalysts Intended for the Polymerization of Olefins. EP Patent EP 0491566 A2, 24 June 1992. [Google Scholar]
- Garoff, T.; Leinonen, T.; Iiskola, E. A Large-Pore Polyolefin, a Method for its Production and a Procatalyst Containing a Transesterification Product of a Lower Alcohol. EP Patent EP 0586389 A1, 16 March 1994. [Google Scholar]
- Garoff, T.; Leinonen, T.; Iiskola, E. Coarse-Grained Polyolefin, Production Thereof and a Procatalyst Containing a Transesterification Product between a Lower Alcohol and Dioctylphthalate Used Therefore. EP Patent EP 0586390 A1, 16 March 1994. [Google Scholar]
- Garoff, T.; Virkkunen, V.; Jäskeläinen, P.; Vestberg, T. A qualitative model for polymerisation of propylene with a MgCl2-supported TiCl4 Ziegler–Natta catalyst. Eur. Polym. J. 2003, 39, 1679–1685. [Google Scholar] [CrossRef]
- Gahleitner, M.; Bachner, C.; Ratajski, E.; Rohaczek, G.; Neißl, W. Effects of the Catalyst System on the Crystallization of Polypropylene. J. Appl. Polym. Sci. 1999, 73, 2507–2515. [Google Scholar] [CrossRef]
- Cancelas, A.J.; Yang, L.; Girod, R.; de Heer, J.; Kleppinger, R.; Delsman, E.; Wang, J.; Gahleitner, M.; Monteil, V.; McKenna, T.F.L. The Effect of Reactor Conditions on High-Impact Polypropylene Properties and Gas Phase Polymerization Kinetics. Macromol. React. Eng. 2018, 12, 1700063. [Google Scholar] [CrossRef]
- Rönkkö, H.-L.; Knuutila, H.; Denifl, P.; Leinonen, T.; Venäläinen, T. Structural studies on a solid self-supported Ziegler–Natta-type catalyst for propylene polymerization. J. Mol. Catal. A Chem. 2007, 278, 127–134. [Google Scholar] [CrossRef]
- Leinonen, T.; Denifl, P. Catalyst Component Comprising Magnesium, Titanium, a Halogen and an Electron Donor, Its Preparation and Use. PCT Patent WO 0238631 A1, 16 May 2002. [Google Scholar]
- Leinonen, T.; Denifl, P. Preparation of Olefin Polymerization Catalyst Component. EP Patent EP 1270610 A1, 2 January 2003. [Google Scholar]
- Abboud, M.; Denifl, P.; Reichert, K.-H. Study of the morphology and kinetics of novel Ziegler–Natta catalysts for propylene polymerization. J. Appl. Polym. Sci. 2005, 98, 2191–2200. [Google Scholar] [CrossRef]
- Rönkkö, H.-L.; Korpela, T.; Knuutila, H.; Pakkanen, T.T.; Denifl, P.; Leinonen, T.; Kemell, M.; Leskelä, M. Particle growth and fragmentation of solid self-supported Ziegler-Natta-type catalysts in propylene polymerization. J. Mol. Catal. A Chem. 2009, 309, 40–49. [Google Scholar] [CrossRef]
- Vestberg, T.; Denifl, P.; Parkinson, M.; Wilén, C.-E. Effects of External Donors and Hydrogen Concentration on Oligomer Formation and Chain End Distribution in Propylene Polymerization with Ziegler-Natta Catalysts. J. Polym. Sci. A Polym. Chem. 2010, 48, 351–358. [Google Scholar] [CrossRef]
- Vestberg, T.; Parkinson, M.; Fonseca, I.; Wilén, C.-E. Poly(propylene-co-ethylene) Produced with a Conventional and a Self-Supported Ziegler–Natta Catalyst: Effect of Ethylene and Hydrogen Concentration on Activity and Polymer Structure. J. Appl. Polym. Sci. 2012, 124, 4889–4896. [Google Scholar] [CrossRef]
- Cecchin, G.; Morini, G.; Pelliconi, A. Polypropene Product Innovation by Reactor Granule Technology. Macromol. Symp. 2001, 173, 195–210. [Google Scholar] [CrossRef]
- Abboud, M.; Denifl, P.; Reichert, K.-H. Fragmentation of Ziegler-Natta Catalyst Particles During Propylene Polymerization. Macromol. Mater. Eng. 2005, 290, 558–564. [Google Scholar] [CrossRef]
- Grein, C.; Schedenig, T. Polyolefin Compositions Having Improved Optical and Mechanical Properties. EP Patent EP 2030996 A1, 4 March 2009. [Google Scholar]
- Denifl, P.; Leinonen, T.; Haikarainen, A.; Vestberg, T. Process for the Manufacture of Heterophasic Propylene Copolymer. EP Patent EP 2065404 A1, 3 June 2009. [Google Scholar]
- Haikarainen, A.; Denifl, P.; Leinonen, T. Preparation of Precipitated ZN PP Catalysts with Internal Pore Structure Using Nanoparticles. PCT Patent WO 2010127997 A2, 11 November 2010. [Google Scholar]
- Vestberg, T.; Denifl, P.; Wilén, C.-E. Increased Rubber Content in High-Impact Polypropylene via a Sirius Ziegler-Natta Catalyst Containing Nanoparticles. J. Polym. Sci. A Polym. Chem. 2013, 51, 2040–2048. [Google Scholar] [CrossRef]
- Iiskola, E.; Mills, K.; Garoff, T.; Leinonen, T. A Procatalyst Composition Containing Substituted Maleic or Fumaric Acid Esters as an Electron Donor for Olefin Polymerization. PCT Patent WO 9307182 A1, 15 April 1993. [Google Scholar]
- David, R.M.; Gans, G. Summary of mammalian toxicology and health effects of phthalate esters. In The Handbook of Environmental Chemistry Vol. 3, Part Q; Staples, C., Ed.; Springer Nature: Cham, Switzerland, 2003; pp. 299–316. [Google Scholar]
- Chadwick, J.C.; van der Burgt, F.P.T.J.; Rastogi, S.; Busico, V.; Cipullo, V.; Talarico, G.; Heere, J.J.R. Influence of Ziegler-Natta Catalyst Regioselectivity on Polypropylene Molecular Weight Distribution and Rheological and Crystallization Behavior. Macromolecules 2004, 37, 9722–9727. [Google Scholar] [CrossRef]
- Yin, X.; Qin, Y.; Dong, J.-Y. Investigation of Chain Microstructure of Polypropylene Polymerized by Ziegler–Natta Catalysts with Diester and Diether Compound as Internal Donor via Hydrogen Chain Transfer. Ind. Eng. Chem. Res. 2020, 59, 1836–1844. [Google Scholar] [CrossRef]
- Haikarainen, A.; Denifl, P.; Leinonen, P. Catalyst Component. EP Patent EP 2415790 A1, 8 February 2012. [Google Scholar]
- Wang, J.; Lilja, J.; Horill, T.; Gahleitner, M.; Denifl, P.; Leinonen, T. Low Emission Propylene Homopolymer with High Melt Flow. EP Patent EP 3071606 A1, 28 September 2016. [Google Scholar]
- Wang, J.; Lilja, J.; Gahleitner, M. Propylene Copolymer for Thin-Wall Packaging. EP Patent EP 2999721 A1, 30 March 2016. [Google Scholar]
- Menyhárd, A.; Gahleitner, M.; Varga, J.; Bernreitner, K.; Jääskeläinen, P.; Øysæd, H.; Pukánszky, B. The influence of nucleus density on optical properties in nucleated isotactic polypropylene. Eur. Polym. J. 2009, 45, 3138–3148. [Google Scholar] [CrossRef]
- Alczar, D.; Ruan, J.; Thierry, A.; Lotz, B. Structural Matching between the Polymeric Nucleating Agent Isotactic Poly(vinylcyclohexane) and Isotactic Polypropylene. Macromolecules 2006, 39, 2832–2840. [Google Scholar] [CrossRef]
- Abe, A.; Hama, T. Structure and Properties of Poly(Vinyl cyclohexane). Polym. Lett. 1969, 7, 427–435. [Google Scholar] [CrossRef]
- Ishihara, N.; Kuramoto, M. Vinylcyclohexane-Based Polymers and Process for Production Thereof. EP Patent EP 0322731 A2, 5 July 1989. [Google Scholar]
- Wege, V.; Dujardin, R.; Chen, Y.; Rechner, J.; Bruder, F.-K. Amorphous Vinyl Cyclohexane Polymers. US Patent US 6365694 B1, 2 April 2002. [Google Scholar]
- Shiga, A.; Kakugo, M.; Kojima, J.; Wakatsuki, K. Crystalline Propylene Polymer Composition. EP Patent EP 0151883 A1, 21 August 1985. [Google Scholar]
- Kakugo, M. Recent progress in polyolefin materials and processing. Macromol. Symp. 2001, 174, 295–300. [Google Scholar] [CrossRef]
- Karbasi, A.; Leskinen, P.; Jääskeläinen, P.; Malm, B.; Pitkänen, P.; Härkönen, M.; Haugen, J. Process for Preparing Polypropylene. PCT Patent WO 9924478 A1, 20 May 1999. [Google Scholar]
- Karbasi, A.; Leskinen, P.; Jääskeläinen, P.; Malm, B.; Pitkänen, P.; Härkönen, M.; Haugen, J. Novel Propylene Polymers and Products Thereof. PCT Patent WO 9924779 A1, 20 May 1999. [Google Scholar]
- Lee, D.-H.; Yoon, K.-B. Effect of polycyclopentene on Crystallization of Isotactic Polypropylene. J. Appl. Polym. Sci. 1994, 54, 1507–1511. [Google Scholar] [CrossRef]
- De Rosa, C.; Auriemma, F.; Tarallo, O.; Di Girolamo, R.; Troisi, E.M.; Esposito, S.; Liguori, D.; Piemontesi, F.; Vitale, G.; Morini, G. Tailoring the properties of polypropylene in the polymerization reactor using polymeric nucleating agents as prepolymers on the Ziegler-Natta catalyst granule. Polym. Chem. 2017, 8, 655–660. [Google Scholar] [CrossRef]
- Mileva, D.; Gahleitner, M.; Shutov, P.; Vestberg, T.; Wang, J.; Androsch, R. Effect of Supercooling on Crystal Structure of Nucleated Isotactic Polypropylene. Thermochim. Acta 2019, 677, 194–197. [Google Scholar] [CrossRef]
- Kirchberger, M.; Jääskeläinen, P.; Gahleitner, M. Non-Oriented Polypropylene Film. PCT Patent WO 03031174 A2, 17 April 2003. [Google Scholar]
- Mileva, D.; Wang, J.; Gahleitner, M.; Doshev, P.; Androsch, R. Crystallization behaviour of heterophasic propylene-ethylene copolymer at rapid cooling conditions. Polymer 2016, 102, 214–220. [Google Scholar] [CrossRef]
- Aasetre, S.; Kamfjord, T. Polypropylene Compositions. PCT Patent WO 2007093376 A1, 23 August 2007. [Google Scholar]
- Wang, J.; Leskinen, P.; Vestberg, T.; Gahleitner, M. Nucleated Polypropylene Composition. EP Patent EP 2960279 A1, 30 December 2015. [Google Scholar]
- Gahleitner, M.; Tranninger, C.; Doshev, P. Polypropylene Copolymers. In Polypropylene Handbook: Morphology, Blends and Composites; Karger-Kocsis, J., Bárány, T., Eds.; Springer International: Berlin, Germany, 2019; pp. 295–355. ISBN 978-3-030-12902-6. [Google Scholar]
- Paulik, C.; Tranninger, C.; Wang, J.; Shutov, P.; Mileva, D.; Gahleitner, M. Catalyst type effects on structure/property relations of polypropylene random copolymers. Macromol. Chem. Phys. 2021, 222, 2100302. [Google Scholar] [CrossRef]
- Pantani, R.; Balzano, L.; Peters, G.W.M. Flow-Induced Morphology of iPP Solidified in a Shear Device. Macromol. Mater. Eng. 2011, 296, 60–67. [Google Scholar] [CrossRef]
- Kumaraswamy, G.; Verma, R.K.; Kornfield, J.A.; Yeh, F.; Hsiao, B.S. Shear-Enhanced Crystallization in Isotactic Polypropylene. In-Situ Synchrotron SAXS and WAXD. Macromolecules 2004, 37, 9005–9017. [Google Scholar] [CrossRef]
- Housmans, J.-W.; Steenbakkers, R.J.A.; Roozemond, P.C.; Peters, G.W.M.; Meijer, H.E.H. Saturation of Pointlike Nuclei and the Transition to Oriented Structures in Flow-Induced Crystallization of Isotactic Polypropylene. Macromolecules 2009, 42, 5728–5740. [Google Scholar] [CrossRef]
- Mileva, D.; Tranchida, D.; Gahleitner, M. Designing polymer crystallinity: An industrial perspective. Polym. Cryst. 2018, 1, e10009. [Google Scholar] [CrossRef]
- Jääskeläinen, P.; Hafner, N.; Pitkänen, P.; Gahleitner, M.; Tuominen, O.; Töltsch, W. Propylene Random Copolymer. EP Patent EP 1270628 A1, 2 January 2003. [Google Scholar]
- Hafner, N.; Töltsch, W.; Gahleitner, M.; Pitkänen, P. Propylene Polymer Composition with Improved Balance of Mechanical and Optical Properties. EP Patent EP 1428853 A1, 16 June 2004. [Google Scholar]
- Gahleitner, M.; Grein, C.; Blell, R.; Wolfschwenger, J.; Koch, T.; Ingolic, E. Sterilization of propylene/ethylene random copolymers: Annealing effects on crystalline structure and transparency as influenced by polymer structure and nucleation. Express Polym. Lett. 2011, 5, 788–798. [Google Scholar] [CrossRef]
- Doshev, P.; Jääskeläinen, P.; Kheirandish, S.; Leskinen, P.; Johnsen, G.K.; Ding, H. High Flow Polypropylene Composition. EP Patent EP 2449026 A1, 9 May 2012. [Google Scholar]
- Doshev, P.; Leskinen, P.; Gahleitner, M.; Potter, E.; Kheirandish, S. Random Propylene Copolymer with High Stiffness and Low Haze. EP Patent EP 2527594 A1, 28 November 2012. [Google Scholar]
- Boragno, L.; Resconi, L.; Lilja, J.; Gahleitner, M. Propylene Copolymer with High Impact Properties. EP Patent EP 2978782 A1, 3 February 2016. [Google Scholar]
- Bergstra, M.; Malm, B.; Leskinen, P.; Kock, C.; Sundholm, T. Random Propylene Copolymers for Pipes. EP Patent EP 2539398 A1, 2 January 2013. [Google Scholar]
- Grein, C.; Gahleitner, M.; Knogler, B.; Nestelberger, S. Melt viscosity effects in ethylene–propylene copolymers. Rheol. Acta 2007, 46, 1083–1089. [Google Scholar] [CrossRef]
- Grein, C.; Schedenig, T. Sterilisable and Tough Impact Polypropylene Composition. EP Patent EP 2176340 A1, 21 April 2010. [Google Scholar]
- Aarnio-Winterhof, M.; Doshev, P.; Seppälä, J.; Gahleitner, M. Structure-property relations of heterophasic ethylene-propylene copolymers based on a single-site catalyst. Express Polym. Lett. 2017, 11, 152–161. [Google Scholar] [CrossRef]
- Gahleitner, M.; Doshev, P.; Tranninger, C. Heterophasic copolymers of polypropylene—Development, design principles and future challenges. J. Appl. Polym. Sci. 2013, 130, 3028–3037. [Google Scholar] [CrossRef]
- Wang, J.; Gahleitner, M.; Potter, E.; Aho, J.; Bernreitner, K.; Monissen, L.; Cigon, M. Process for Preparing a Polypropylene Composition. PCT Patent WO 2019002346 A1, 3 January 2019. [Google Scholar]
- Amiar, N.; Bouzid, D.; McKenna, T.F.L. Influence of the rubber content and particle morphology on the mechanical properties of the (hiPP). J. Appl. Polym. Sci. 2016, 133, 44197. [Google Scholar] [CrossRef]
- Grein, C.; Bernreitner, K.; Hauer, A.; Gahleitner, M.; Neißl, W. Impact modified isotactic polypropylene with controlled rubber intrinsic viscosities: Some new aspects about morphology and fracture. J. Appl. Polym. Sci. 2002, 87, 1702–1712. [Google Scholar] [CrossRef]
- Doshev, P.; Lohse, G.; Henning, S.; Krumova, M.; Heuvelsland, A.; Michler, G.H.; Radusch, H.-J. Phase interactions and structure evolution of heterophasic ethylene–propylene copolymers as a function of system composition. J. Appl. Polym. Sci. 2006, 101, 2825–2837. [Google Scholar] [CrossRef]
- Kim, G.-M.; Michler, G.H.; Gahleitner, M.; Mülhaupt, R. Influence of Morphology on the Toughening Mechanisms of Polypropylene Modified with Core-Shell Particles derived from Thermoplastic Elastomers. Polym. Adv. Technol. 1989, 9, 709–715. [Google Scholar] [CrossRef]
- Rungswang, W.; Saendee, P.; Thitisuk, B.; Pathaweeisariyakul, T.; Cheevasrirungruang, W. Role of Crystalline Ethylene-Propylene Copolymer on Mechanical Properties of Impact Polypropylene Copolymer. J. Appl. Polym. Sci. 2013, 128, 3131–3140. [Google Scholar] [CrossRef]
- Doshev, P.; Kona, B.R.; Bernreitner, K.; Grein, C. High Flowable Heterophasic Polypropylene. EP Patent EP 2174980 A1, 14 April 2010. [Google Scholar]
- Tranninger, M. Polyolefin Composition with Reduced Occurrence of Flow Marks. EP Patent EP 2599829 A1, 5 June 2013. [Google Scholar]
- Gahleitner, M.; Grestenberger, G.; Grein, C.; Kock, C. High Flow Polyolefin Composition with Low Shrinkage and CLTE. EP Patent EP 2731988 A1, 21 May 2014. [Google Scholar]
- Tranninger, C. Transparent Thin Wall Packaging Material with Improved Stiffness and Flowability. EP Patent EP 2936343 A2, 28 October 2015. [Google Scholar]
- Bergstra, M.; Leskinen, P.; Malm, B.; Tranninger, C. Heterophasic Polypropylene with Excellent Creep Performance. EP Patent EP 2550324 A1, 30 January 2013. [Google Scholar]
- Tranninger, C.; Doshev, P.; Potter, E.; Gsellmann, G.; Leskinen, P. Heterophasic Propylene Copolymer with Excellent Impact/Stiffness Balance. EP Patent EP 2601260 A1, 12 June 2013. [Google Scholar]
- Tranninger, C.; Lummerstorfer, T.; Horill, T.; Gahleitner, M.; Wang, J.; Bernreitner, K.; Berger, F.; Leskinen, P. Multimodal Polypropylene Composition with High Stiffness and High Flowability and Process for Its Production. EP Patent EP 3945112 A1, 2 February 2022. [Google Scholar]
- Paulik, C.; Gahleitner, M.; Neißl, W. Flexible, tough and resilient PP-Copolymers. Kunstst/PlastEurope 1996, 86, 16–17. [Google Scholar]
- Wang, J.; Shutov, P.; Lilja, J.; Gahleitner, M. Soft and Transparent Impact Copolymers. EP Patent EP 3102634 A1, 14 December 2016. [Google Scholar]
- Starke, J.U.; Michler, G.H.; Grellmann, W.; Seidler, S.; Gahleitner, M.; Fiebig, J.; Nezbedova, E. Fracture Toughness of Polypropylene Copolymers: Influence of Interparticle Distance and Temperature. Polymer 1998, 39, 75–82. [Google Scholar] [CrossRef]
- Kotter, I.; Grellmann, W.; Koch, T.; Seidler, S. Morphology–Toughness Correlation of Polypropylene/Ethylene–Propylene Rubber Blends. J. Appl. Polym. Sci. 2006, 100, 3364–3371. [Google Scholar] [CrossRef]
- Cecchin, G.; Marchetti, E.; Baruzzi, G. On the Mechanism of Polypropene Growth over MgCl2/TiCl4 Catalyst Systems. Macromol. Chem. Phys. 2001, 202, 1987–1994. [Google Scholar] [CrossRef]
- Grof, Z.; Kosek, J.; Marek, M. Modeling of Morphogenesis of Growing Polyolefin Particles. AIChE J. 2005, 51, 2048–2067. [Google Scholar] [CrossRef]
- Zubov, A.; Pechackova, L.; Seda, L.; Bobak, M.; Kosek, J. Transport and reaction in reconstructed porous polypropylene particles: Model validation. Chem. Eng. Sci. 2010, 65, 2361–2372. [Google Scholar] [CrossRef]
- Sandholzer, M.; Bernreitner, K.; Klimke, K.; Gahleitner, M. Propylene Copolymer for Injection Molded Articles or Films. EP Patent EP 2794689 A1, 29 October 2014. [Google Scholar]
- Braun, J.; Edler, M.; Emig, J.; Gahleitner, M.; Kahlen, S.; Sandholzer, M.; Scriba, M.; Tranninger, M. Polypropylene (PP): News and Trends from the Reactor to the Recycled Material. Kunstst. Int. 2017, 107, 260596. [Google Scholar]
- Fink, G.; Steinmetz, B.; Zechlin, J.; Przybyla, C.; Tesche, B. Propene Polymerization with Silica-Supported Metallocene/MAO Catalysts. Chem. Rev. 2000, 100, 1377–1390. [Google Scholar] [CrossRef]
- Gahleitner, M.; Kastner, M.; Siebert, H.; Stadlbauer, M. Low Emission Polymer Composition. EP Patent EP 2262858 A1, 8 April 2009. [Google Scholar]
- Paavilainen, J.; Doshev, P.; Reichelt, K. Propylene/1-Hexene Copolymer Composition with Low Sealing Temperature. EP Patent EP 2561016 A1, 27 February 2013. [Google Scholar]
- Nenseth, S.; Doshev, P. Heterophasic Polypropylene with High Flowability and Excellent Low Temperature Impact Properties. EP Patent EP 2075284 A1, 1 July 2009. [Google Scholar]
- Chum, P.S.; Swogger, K.W. Olefin polymer technologies—History and recent progress at The Dow Chemical Company. Prog. Polym. Sci. 2008, 33, 797–819. [Google Scholar] [CrossRef]
- Siracusa, V.; Blanco, I. Bio-Polyethylene (Bio-PE), Bio-Polypropylene (Bio-PP) and Bio-Poly(ethylene terephthalate) (Bio-PET): Recent Developments in Bio-Based Polymers Analogous to Petroleum-Derived Ones for Packaging and Engineering Applications. Polymers 2020, 12, 1641. [Google Scholar] [CrossRef] [PubMed]
- Vollmer, I.; Jenks, M.J.F.; Roelands, M.C.P.; White, R.J.; van Harmelen, T.; de Wild, P.; van der Laan, G.P.; Meirer, F.; Keurentjes, J.T.F.; Weckhuysen, B.M. Beyond Mechanical Recycling: Giving New Life to Plastic Waste. Angew. Chem.—Int. Ed. 2020, 59, 15402–15423. [Google Scholar] [CrossRef] [PubMed]
- Coates, G.W.; Getzler, Y.D.Y.L. Chemical recycling to monomer for an ideal, circular polymer economy. Nat. Rev. Mater. 2020, 5, 501–516. [Google Scholar] [CrossRef]
- Lazzari, S.; Lischewski, A.; Orlov, Y.; Deglmann, P.; Daiss, A.; Schreiner, E.; Vale, H. Chapter Six—Toward a digital polymer reaction engineering. Adv. Chem. Eng. 2020, 56, 187–227. [Google Scholar]















Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bergstra, M.F.; Denifl, P.; Gahleitner, M.; Jeremic, D.; Kanellopoulos, V.; Mileva, D.; Shutov, P.; Touloupidis, V.; Tranninger, C. Polymerization in the Borstar Polypropylene Hybrid Process: Combining Technology and Catalyst for Optimized Product Performance. Polymers 2022, 14, 4763. https://doi.org/10.3390/polym14214763
Bergstra MF, Denifl P, Gahleitner M, Jeremic D, Kanellopoulos V, Mileva D, Shutov P, Touloupidis V, Tranninger C. Polymerization in the Borstar Polypropylene Hybrid Process: Combining Technology and Catalyst for Optimized Product Performance. Polymers. 2022; 14(21):4763. https://doi.org/10.3390/polym14214763
Chicago/Turabian StyleBergstra, Michiel F., Peter Denifl, Markus Gahleitner, Dusan Jeremic, Vasileios Kanellopoulos, Daniela Mileva, Pavel Shutov, Vasileios Touloupidis, and Cornelia Tranninger. 2022. "Polymerization in the Borstar Polypropylene Hybrid Process: Combining Technology and Catalyst for Optimized Product Performance" Polymers 14, no. 21: 4763. https://doi.org/10.3390/polym14214763
APA StyleBergstra, M. F., Denifl, P., Gahleitner, M., Jeremic, D., Kanellopoulos, V., Mileva, D., Shutov, P., Touloupidis, V., & Tranninger, C. (2022). Polymerization in the Borstar Polypropylene Hybrid Process: Combining Technology and Catalyst for Optimized Product Performance. Polymers, 14(21), 4763. https://doi.org/10.3390/polym14214763