Molecular Dynamics Simulation of Diffusion Behavior of CH4, CO2, and N2 in Mid-Rank Coal Vitrinite

Abstract
:
1. Introduction
2. Materials and Methods
3. Model and Calculation Method
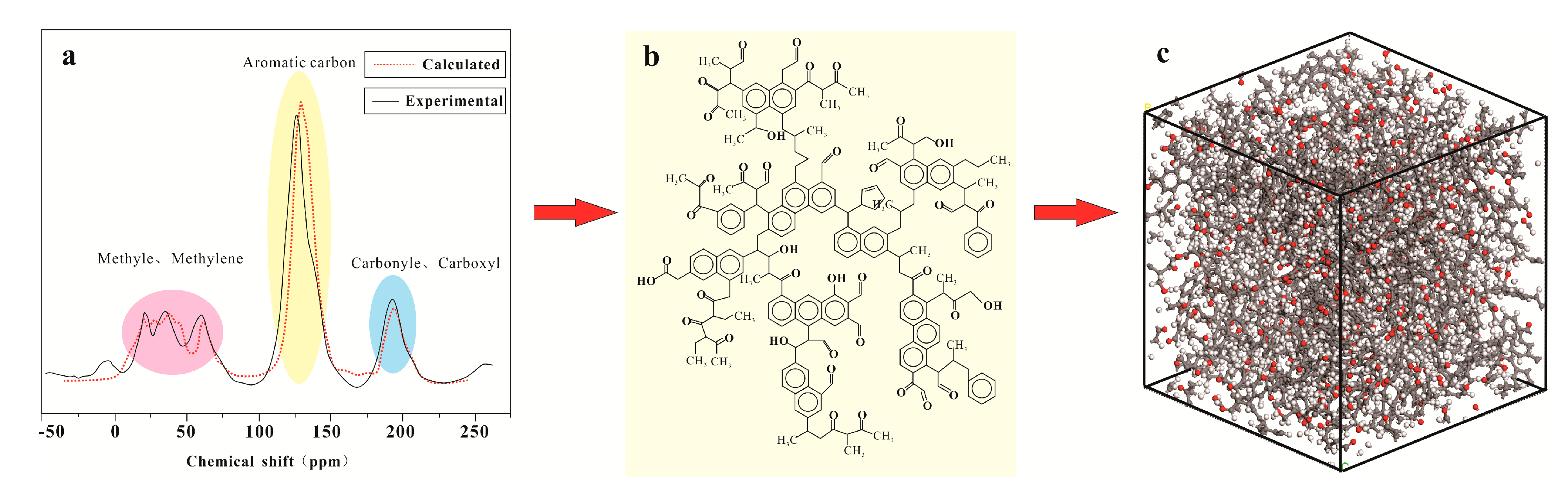
3.1. Macromolecular Structure Construction
3.2. Monte Carlo Simulation
3.3. Molecular Dynamics Simulation
3.4. Self-, Maxwell’s, and Transport Diffusions
4. Results and Discussion
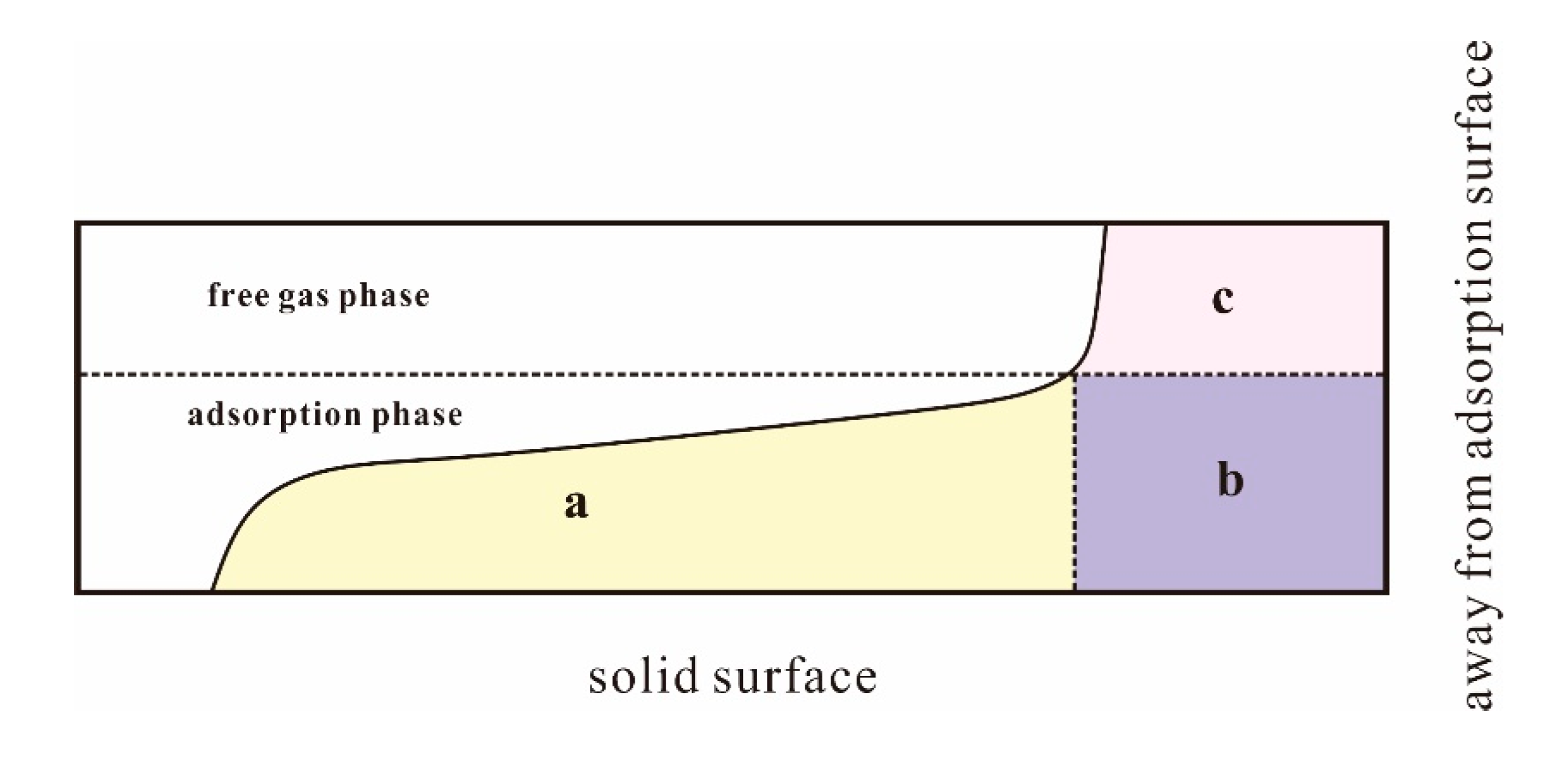
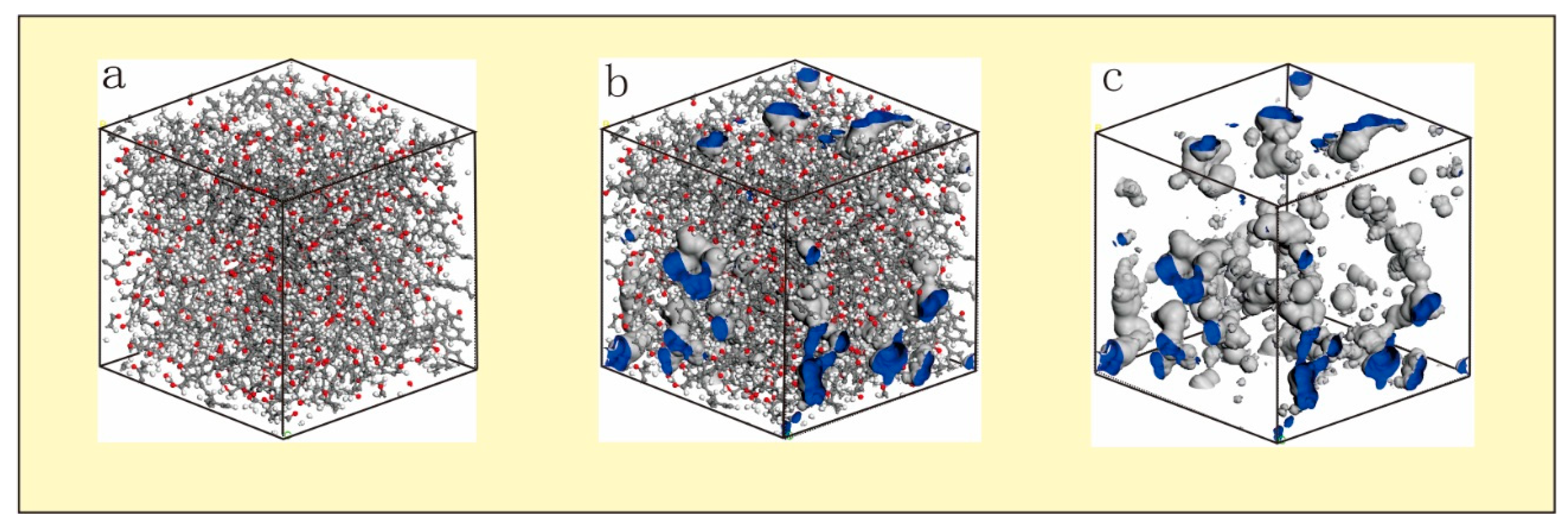
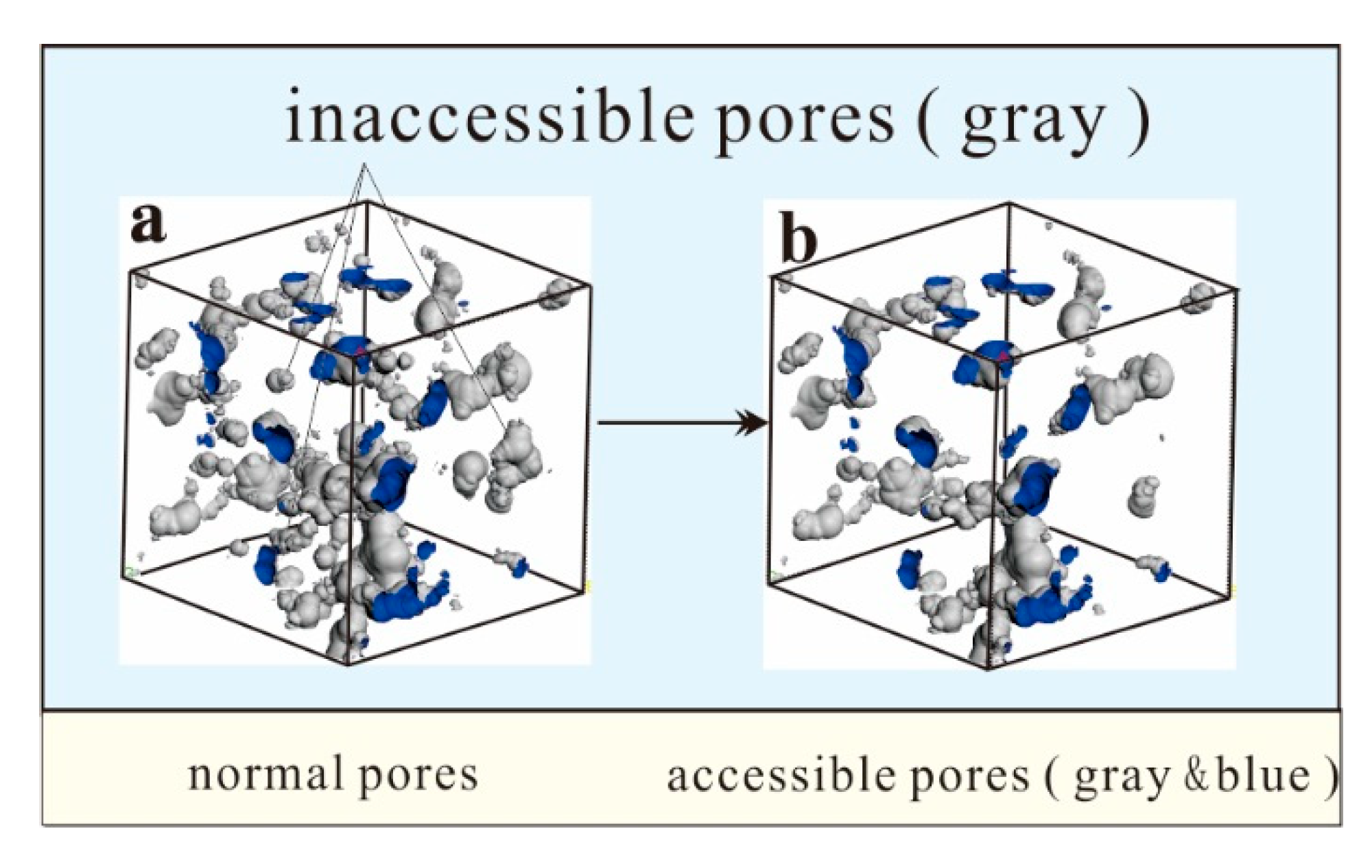
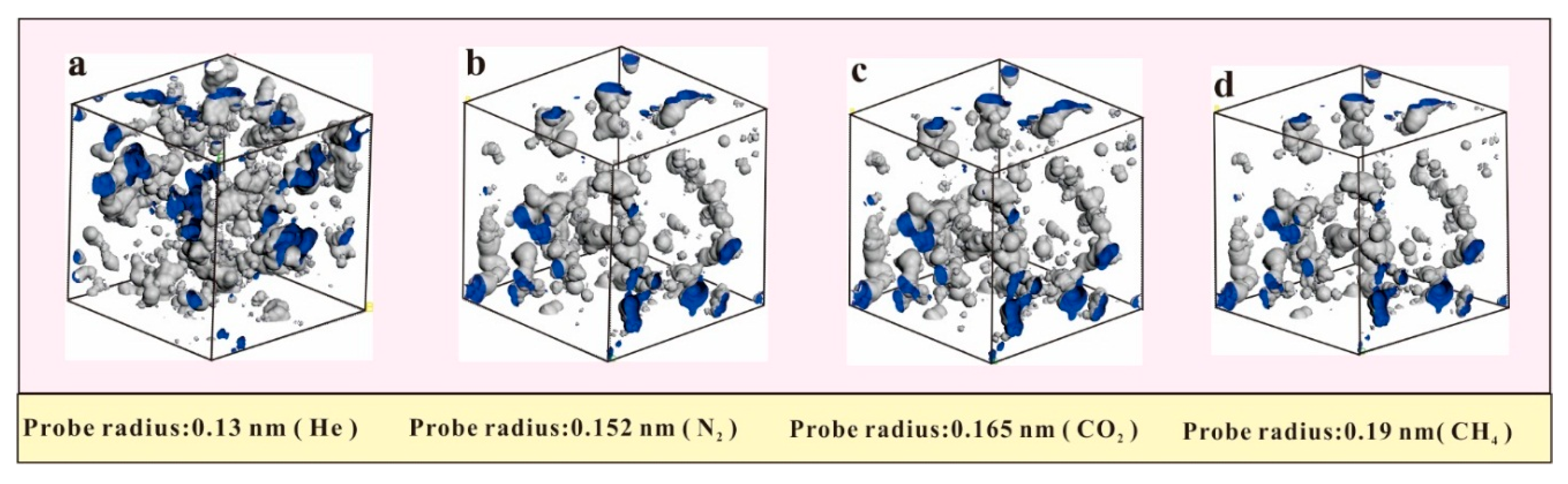
4.1. Pores in Coal Macromolecule
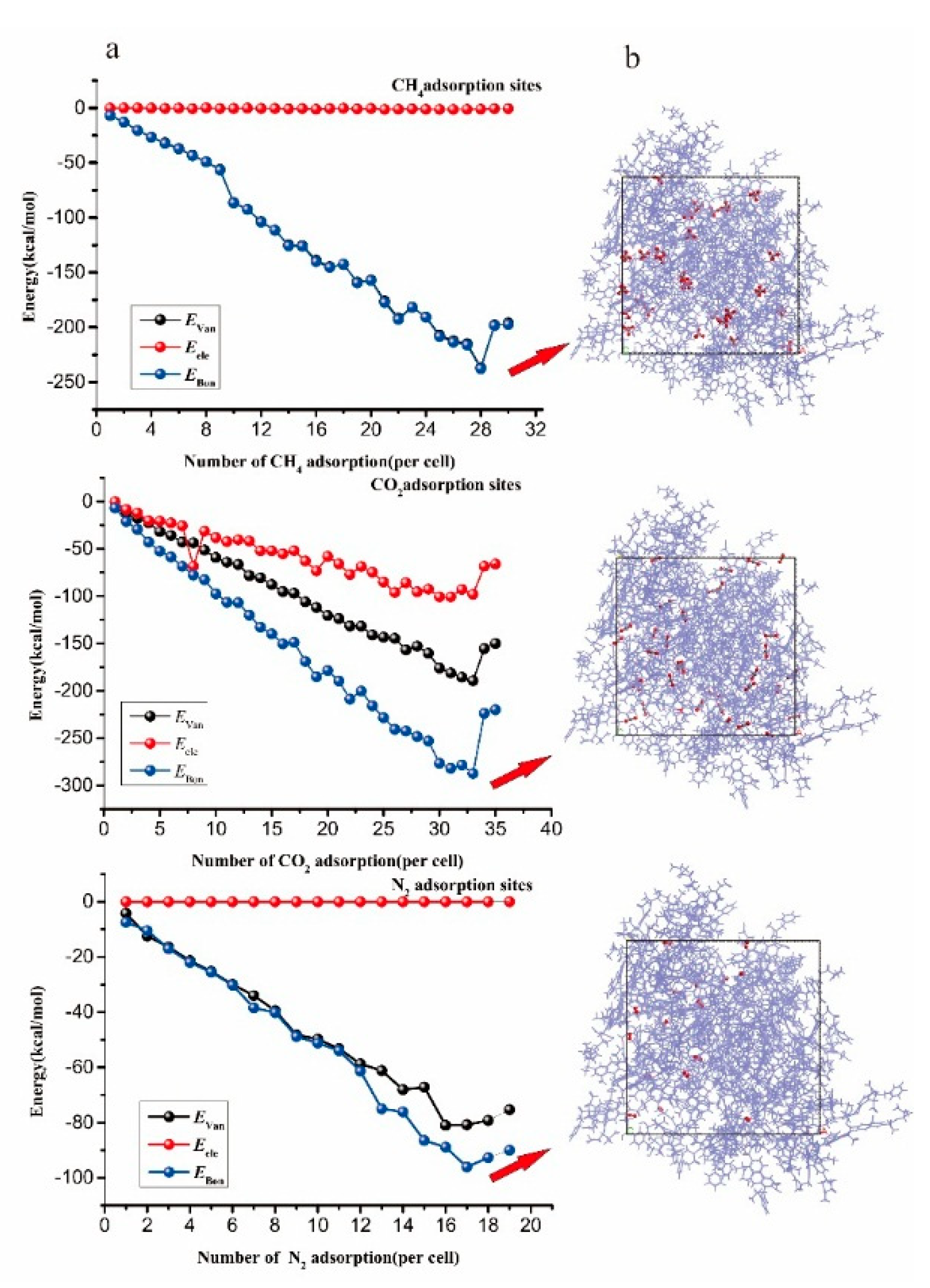
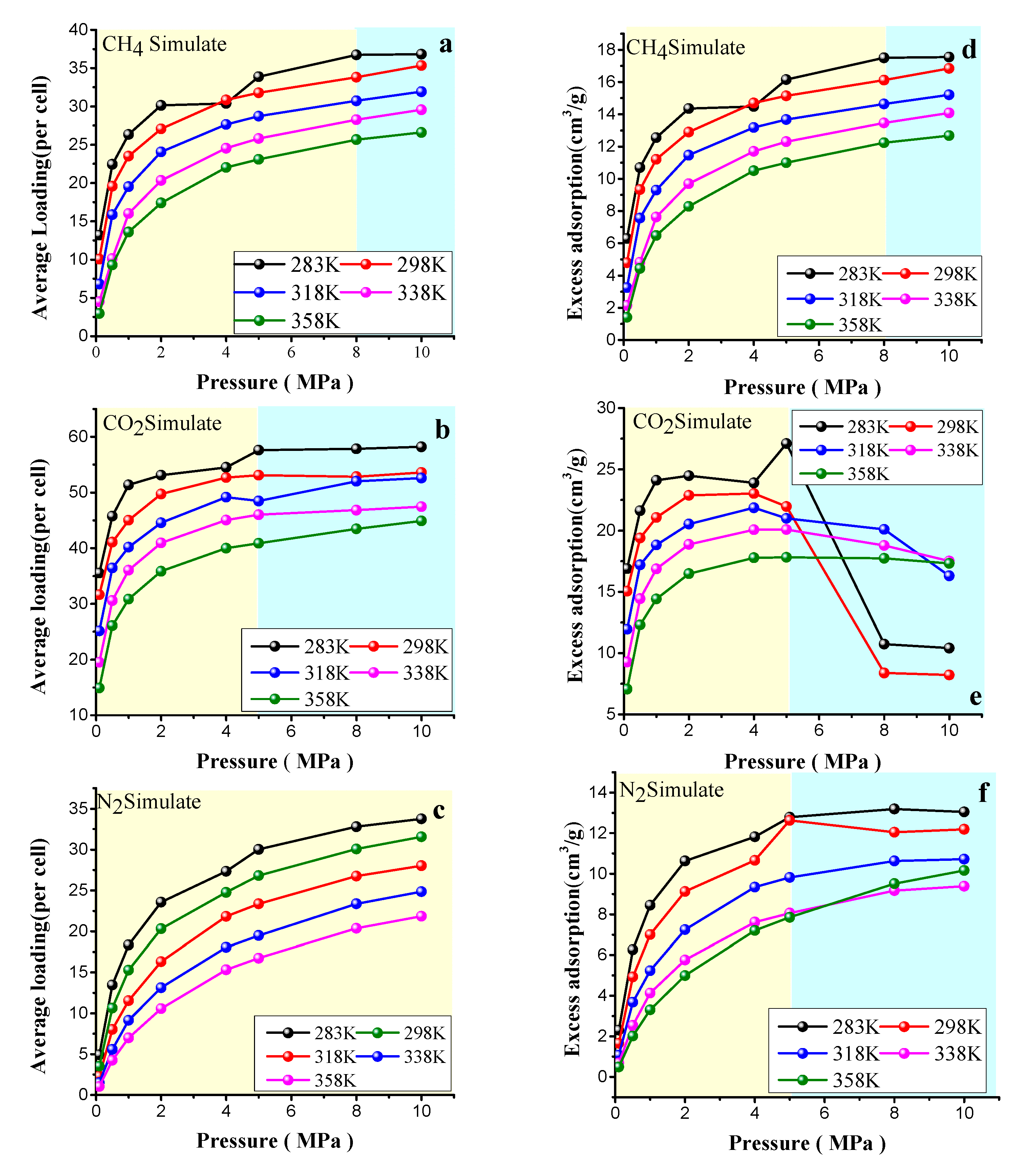
4.2. Adsorption Results
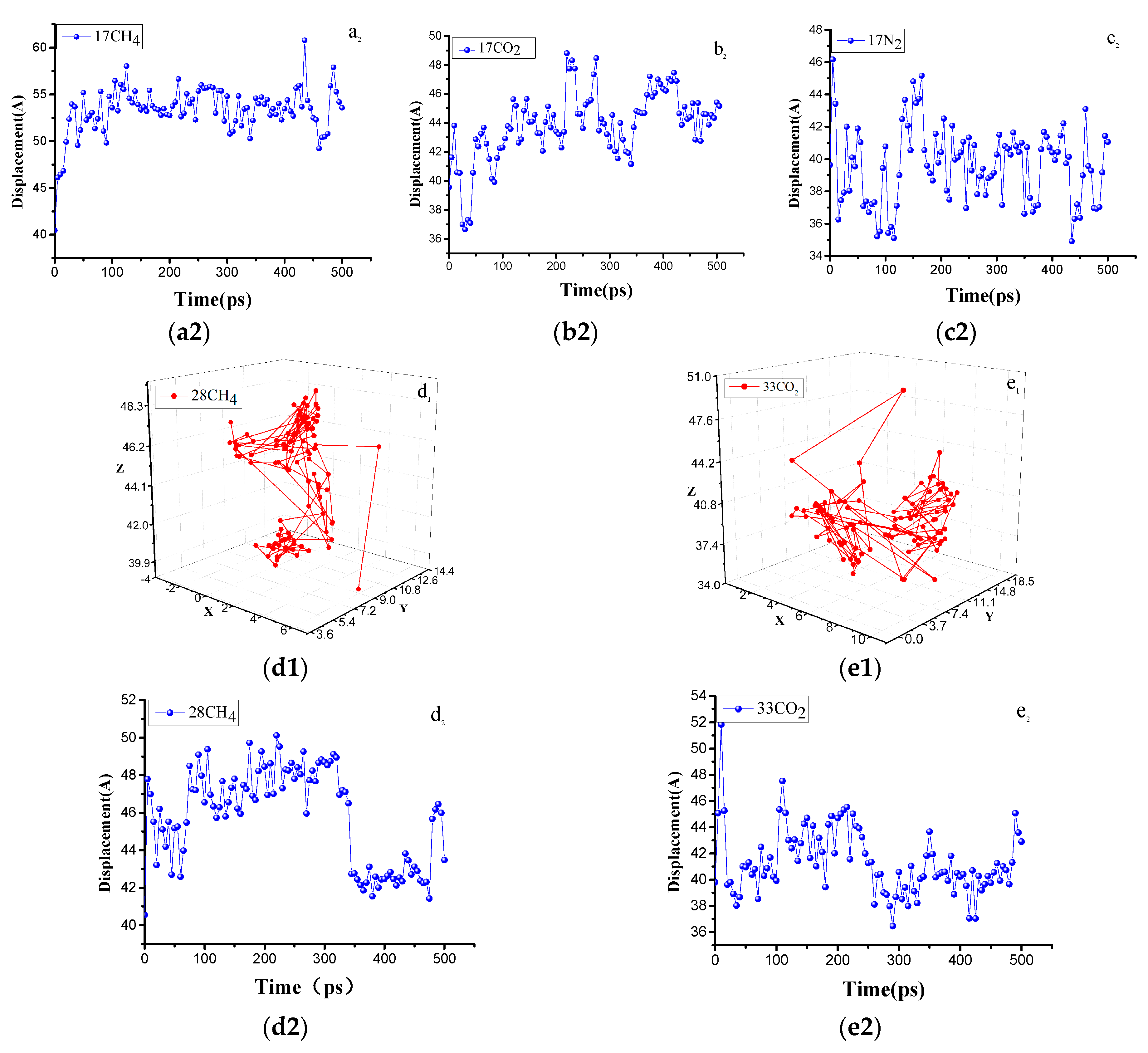
4.3. Self-, Corrected, and Transport Diffusion Coefficients
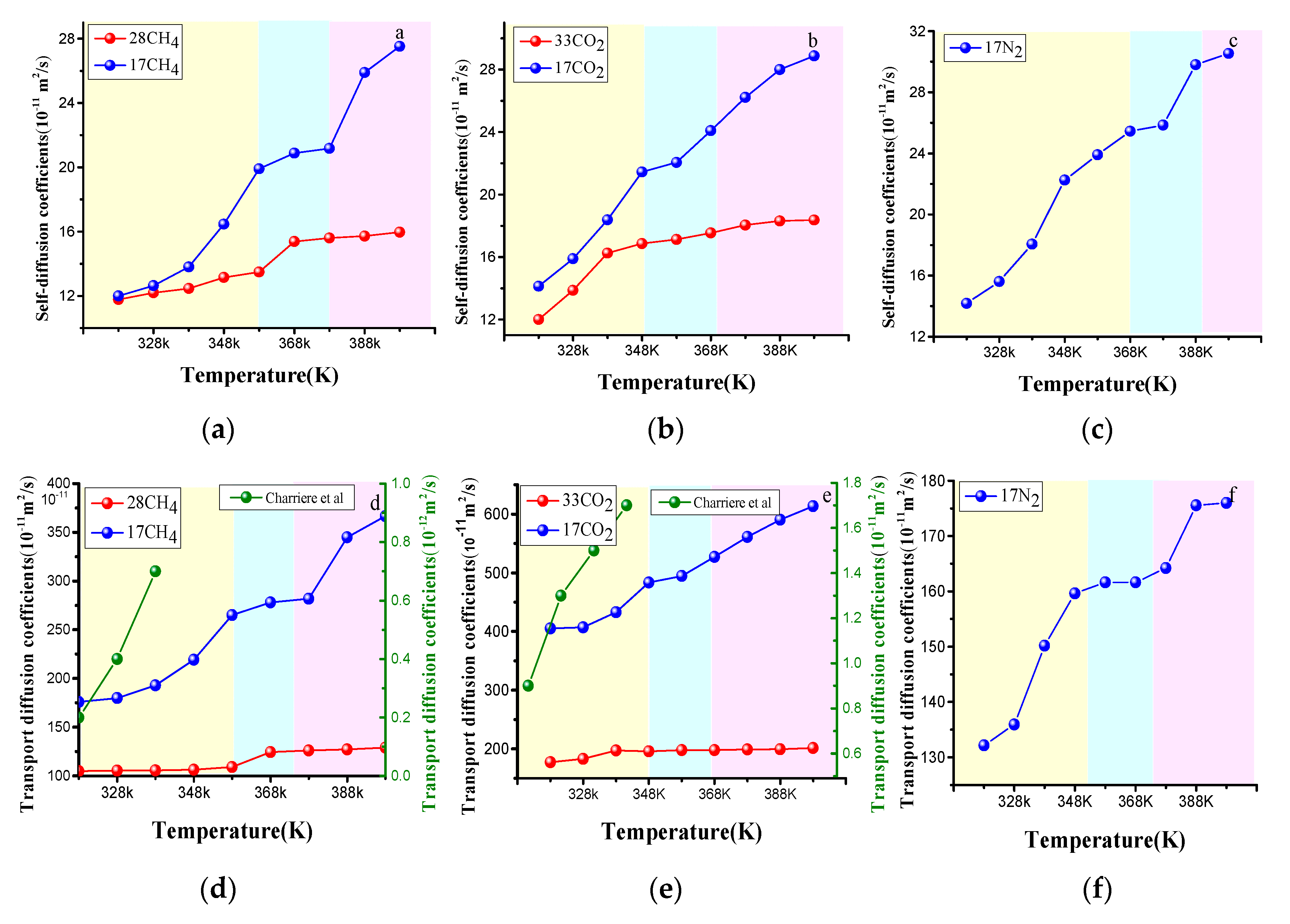
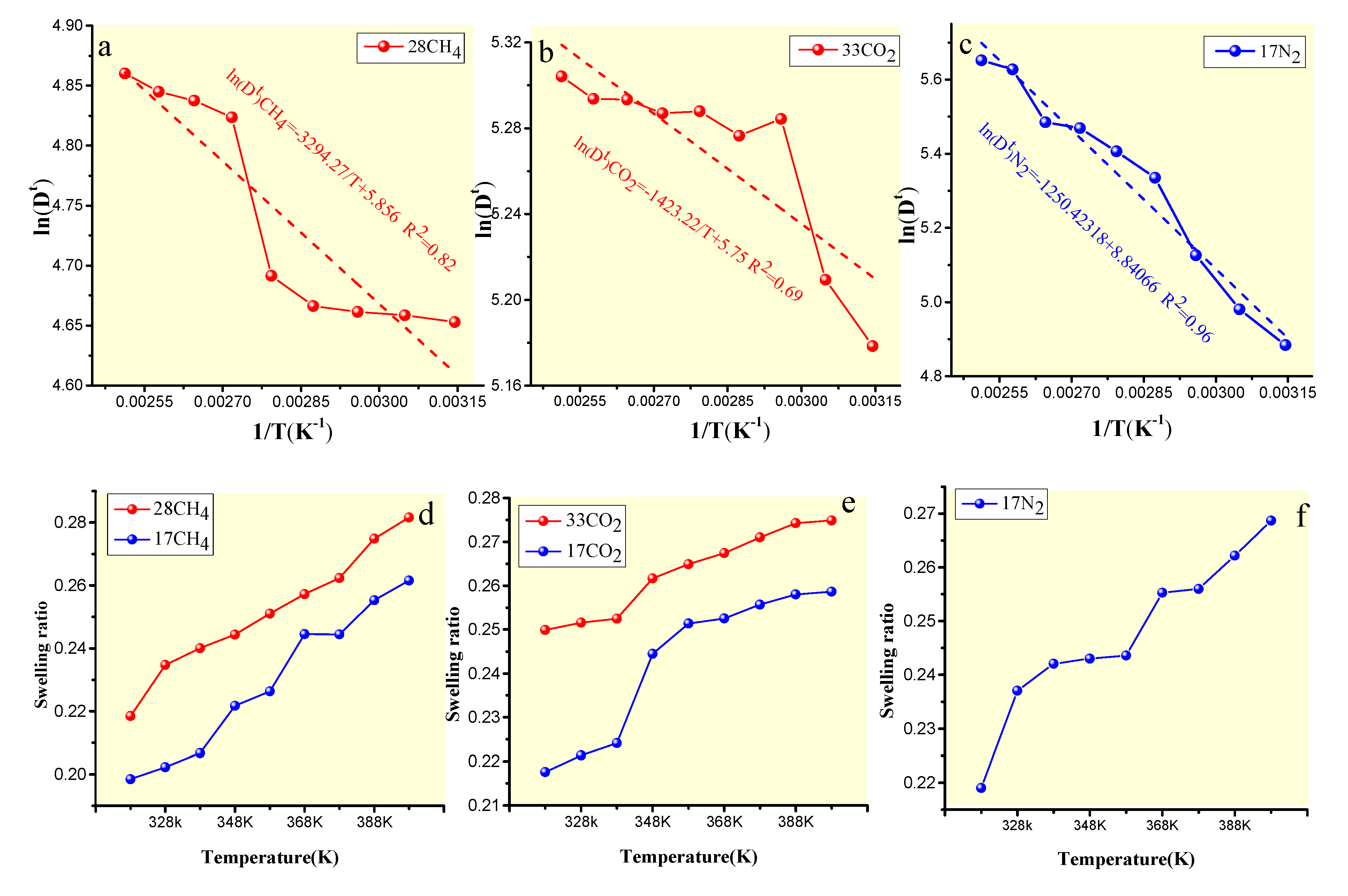
4.3.1. Effect of the Temperature on Gas Diffusion
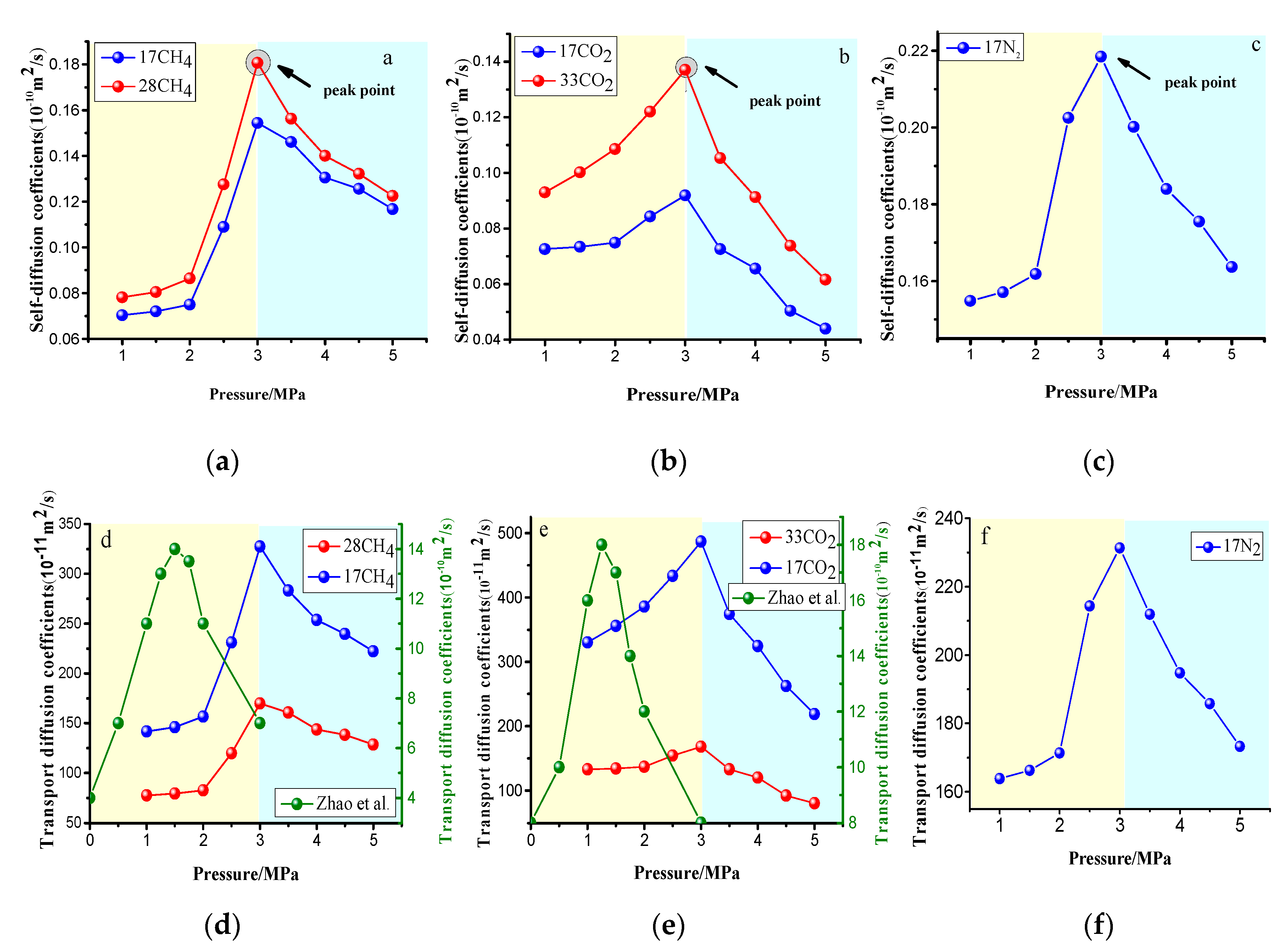
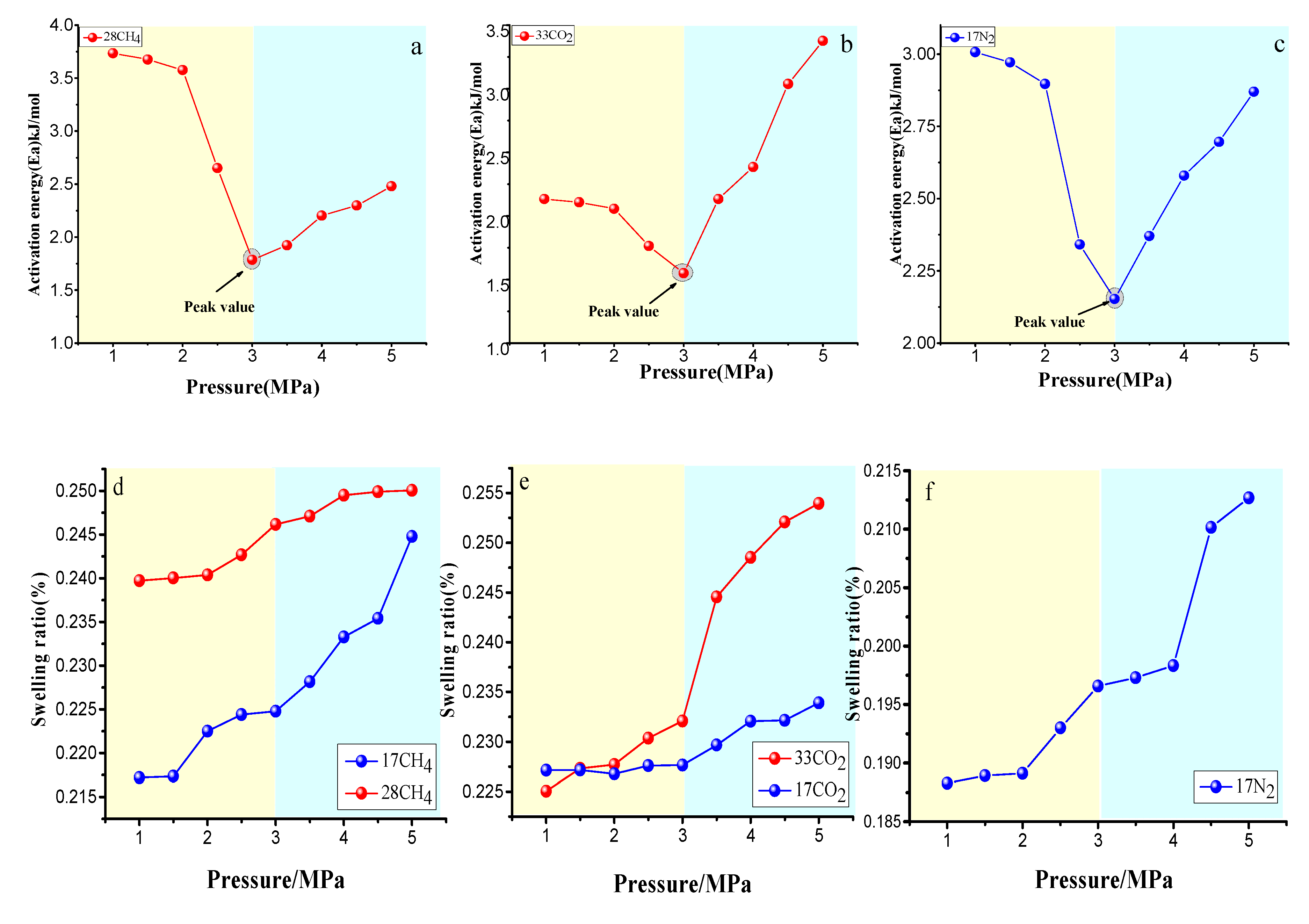
4.3.2. Effect of the Pressure on Gas Diffusion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Liu, Y.; Zhu, Y.; Li, W.; Zhang, C.; Wang, Y. Ultra micropores in macromolecular structure of subbituminous coal vitrinite. Fuel 2017, 210, 298–306. [Google Scholar] [CrossRef]
- Moore, T.A. Coalbed methane: A review. Int. J. Coal Geol. 2012, 101, 36–81. [Google Scholar] [CrossRef]
- Vishal, V.; Singh, T.N.; Ranjith, P.G. Influence of sorption time in CO2-ECBM process in Indian coals using coupled numerical simulation. Fuel 2015, 139, 51–58. [Google Scholar] [CrossRef]
- Cui, X.; Bustin, R.M.; Dipple, G. Selective transport of CO2, CH4, and N2 in coals: Insights from modeling of experimental gas adsorption data. Fule 2004, 83, 293–303. [Google Scholar] [CrossRef]
- Cuéllar-Franca, R.M.; Azapagic, A. Carbon capture, storage and utilisation technologies: A critical analysis and comparison of their life cycle environmental impacts. J. CO2 Util. 2015, 9, 82–102. [Google Scholar] [CrossRef]
- Middleton, R.S.; Keating, G.N.; Stauffer, P.H.; Jordan, A.B.; Viswanathan, H.S.; Kang, Q.J.; Carey, J.W.; Mulkey, M.L.; Sullivan, E.J.; Chu, S.P. The cross-scale science of CO2 capture and storage: From pore scale to regional scale. Energy Environ. Sci. 2012, 5, 7328–7345. [Google Scholar] [CrossRef]
- Tang, X.; Ripepi, N. High pressure supercritical carbon dioxide adsorption in coal: Adsorption model and thermodynamic characteristics. J. CO2 Util. 2017, 18, 189–197. [Google Scholar] [CrossRef] [Green Version]
- Ranathunga, A.S.; Perera, M.S.A.; Ranjith, P.G.; Zhang, X.G.; Wu, B. Super-critical carbon dioxide flow behaviour in low rank coal: A meso-scale experimental study. J. CO2 Util. 2017, 20, 1–13. [Google Scholar] [CrossRef]
- Damen, K.; Faaij, A.; van Bergen, F.; Gale, J.; Lysen, E. Identification of early opportunities for CO2 sequestration—worldwide screening for CO2-EOR and CO2-ECBM projects. Energy 2005, 30, 1931–1952. [Google Scholar] [CrossRef]
- Jian, X.; Guan, P.; Zhang, W. Carbon dioxide sorption and diffusion in coals: Experimental investigation and modeling. Sci. China Earth Sci. 2012, 55, 633–643. [Google Scholar] [CrossRef]
- Yu, S.; Bo, J.; Meijun, Q. Molecular Dynamic Simulation of Self- and Transport Diffusion for CO2/CH4/N2 in Low-Rank Coal Vitrinite. Energy Fuels 2018, 32, 3085–3096. [Google Scholar] [CrossRef]
- Zhao, Y.; Feng, Y.; Zhang, X. Molecular simulation of CO2/CH4 self- and transport diffusion coefficients in coal. Fuel 2016, 165, 19–27. [Google Scholar] [CrossRef]
- Zhao, Y.; Liu, S.; Elsworth, D.; Jiang, Y.; Zhu, J. Pore Structure Characterization of Coal by Synchrotron Small-Angle X-ray Scattering and Transmission Electron Microscopy. Energy Fuels 2014, 28, 3704–3711. [Google Scholar] [CrossRef]
- Rouquerol, J.; Avnir, D.; Fairbridge, C.W.; Everett, D.H.; Haynes, J.M.; Pernicone, N.; Ramsay, J.D.F.; Sing, K.S.W.; Unger, K.K. Recommendations for the characterization of porous solids. Pure Appl. Chem. 1994, 66, 1739–1758. [Google Scholar] [CrossRef]
- Zhao, W.; Cheng, Y.; Yuan, M.; An, F. Effect of Adsorption Contact Time on Coking Coal Particle Desorption Characteristics. Energy Fuels 2014, 28, 2287–2296. [Google Scholar] [CrossRef]
- Tambach, T.J.; Mathews, J.P.; van Bergen, F. Molecular Exchange of CH4 and CO2 in Coal: Enhanced Coalbed Methane on a Nanoscale. Energy Fuels 2009, 23, 4845–4847. [Google Scholar] [CrossRef]
- Barton, G. The Mathematics of Diffusion 2nd edn. Phys. Bull. 1975, 26, 500–501. [Google Scholar] [CrossRef]
- Ruckenstein, E.; Vaidyanathan, A.S.; Youngquist, G.R. Sorption by solids with bidisperse pore structures. Chem. Eng. Sci. 1971, 26, 1305–1318.21. [Google Scholar] [CrossRef]
- Clarkson, C.R.; Bustin, R.M. The effect of pore structure and gas pressure upon the transport properties of coal: A laboratory and modeling study. 2. Adsorption rate modeling. Fuel 1999, 78, 1345–1362. [Google Scholar] [CrossRef]
- Staib, G.; Sakurovs, R.; Gray, E.M.A. Dispersive diffusion of gases in coals. Part II: An assessment of previously proposed physical mechanisms of diffusion in coal. Fuel 2015, 143, 620–629. [Google Scholar] [CrossRef]
- Einstein, A. On the movement of small particles suspended in a stationary liquid demanded by the molecular-kinetic theory of heat. Ann. Phys. (Leipzig) 1905, 17, 549–560. [Google Scholar] [CrossRef]
- Hu, H.; Du, L.; Xing, Y.; Li, X. Detailed study on self- and multicomponent diffusion of CO2-CH4 gas mixture in coal by molecular simulation. Fuel 2017, 187, 220–228. [Google Scholar] [CrossRef]
- Charrière, D.; Pokryszka, Z.; Behra, P. Effect of pressure and temperature on diffusion of CO2 and CH4 into coal from the Lorraine basin (France). Int. J. Coal Geol. 2010, 81, 373–380. [Google Scholar] [CrossRef]
- Saghafi, A.; Faiz, M.; Roberts, D. CO2 storage and gas diffusivity properties of coals from Sydney Basin, Australia. Int. J. Coal Geol. 2007, 34, 240–254. [Google Scholar] [CrossRef]
- Yao, Y.; Liu, D.; Xie, S. Quantitative characterization of methane adsorption on coal using a low-field NMR relaxation method. Int. J. Coal Geol. 2014, 131, 32–40. [Google Scholar] [CrossRef]
- Olague, N.E.; Smith, D.M. Diffusion of gases in American coals. Fuel 1989, 68, 1381–1387. [Google Scholar] [CrossRef]
- Wiser, W.H. Conversion of bituminous coal to liquids and gases: Chemistry and representative processes. In Magnetic Resonance; Springer: Dordrecht, The Netherlands, 1984; pp. 325–350. [Google Scholar]
- Liu, Y.; Zhu, Y.; Liu, S.; Chen, S.; Li, W.; Wang, Y. Molecular structure controls on micropore evolution in coal vitrinite during coalification. Int. J. Coal Geol. 2018, 199, 19–30. [Google Scholar] [CrossRef]
- Jianhua, X.; Fangui, Z.; Huzhen, L. Molecular Simulation of Adsorption of CH4/CO2/H2O in Coal Molecular Structure. Chin. Sci. 2014, 44, 1418–1428. [Google Scholar]
- Mathews, J.P.; Chaffee, A.L. The molecular representations of coal—A review. Fuel 2012, 96, 1–14. [Google Scholar] [CrossRef]
- Sun, H. COMPASS: An ab Initio Force-Field Optimized for Condensed-Phase Applications Overview with Details on Alkane and Benzene Compounds. J. Phys. Chem. 1998, 102, 7338–7364. [Google Scholar] [CrossRef]
- Tan, Z.; Gubbins, K.E. Adsorption in carbon micropores at supercritical temperatures. J. Phys. Chem. 1990, 94, 6061–6069. [Google Scholar] [CrossRef]
- Liu, Y.; Zhu, Y.; Liu, S.; Li, W.; Tang, X. Temperature effect on gas adsorption capacity in different sized pores of coal: Experiment and numerical modeling. J. Pet. Sci. Eng. 2018, 165, 821–830. [Google Scholar] [CrossRef]
- Zhang, C.; Chen, S.; Liu, Y.; Chen, S.; Li, X. Mechanism of methane adsorption on groove space in organic matter surface. Mol. Simul. 2019, 45, 186–198. [Google Scholar] [CrossRef]
- Gibbs, J.W. The Collected Works of JW Gibbs; Longmans, Green: New York, NY, USA, 1928. [Google Scholar]
- Hu, H.; Li, X.; Fang, Z.; Wei, N.; Li, Q. Small-molecule gas sorption and diffusion in coal: Molecular simulation. Energy 2010, 35, 2939–2944. [Google Scholar] [CrossRef]
- Jobic, H.; Theodorou, D.N. Quasi-elastic neutron scattering and molecular dynamics simulation as complementary techniques for studying diffusion in zeolites. Microporous Mesoporous Mater. 2007, 102, 21–50. [Google Scholar] [CrossRef]
- Krishna, R.; Wesselingh, J.A. The Maxwell-Stefan Approach to Mass Transfer; Elsevier: Amsterdam, The Netherlands, 1997; Volume 52, pp. 861–911. [Google Scholar]
- Robinson, D.P.A.D. A New Two-Constant Equation of State. Ind. Eng. Chem. Fundam. 1976, 1, 59–64. [Google Scholar]
- Shieh, J.J.; Chung, T.S. Gas permeability, diffusivity, and solubility of poly (4-vinylpyridine) film. J. Polym. Sci. Part B Polym. Phys. 1999, 37, 2851–2861. [Google Scholar] [CrossRef]
- Larsen, J.W. The effects of dissolved CO2 on coal structure and properties. Int. J. Coal Geol. 2004, 57, 63–70. [Google Scholar] [CrossRef]
- Kärger, J.; Valiullin, R. Mass transfer in mesoporous materials: The benefit of microscopic diffusion measurement. Chem. Soc. Rev. 2013, 42, 4172–4197. [Google Scholar] [CrossRef]
- Staib, G.; Sakurovs, R.; Gray, E.M.A. A pressure and concentration dependence of CO2 diffusion in two Australian bituminous coals. Int. J. Coal Geol. 2013, 116–117, 106–116. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Name | VR (%) | Proximate Analysis (wt.%) | Total Sulfur (wt.%) | Ultimate Analysis (wt.%) | ||||
|---|---|---|---|---|---|---|---|---|
| Mad | Ad | Cdaf | Hdaf | Odaf | Ndaf | |||
| TL-1 | 1.3 | 0.82 | 6.01 | 0.55 | 87.82 | 5.02 | 5.52 | 1.42 |
| Diffusion Coefficients (10−11 m2/s) | Configuration | DS | Dc | Dt |
|---|---|---|---|---|
| CO2 | 33 | 7.26 | 19.15 | 132.81 |
| 17 | 9.23 | 47.27 | 327.79 | |
| CH4 | 28 | 7.03 | 21.87 | 76.55 |
| 17 | 7.82 | 40.06 | 140.19 | |
| N2 | 17 | 15.48 | 79.29 | 198.22 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, J.; Li, S.; Wang, Y. Molecular Dynamics Simulation of Diffusion Behavior of CH4, CO2, and N2 in Mid-Rank Coal Vitrinite. Energies 2019, 12, 3744. https://doi.org/10.3390/en12193744
Liu J, Li S, Wang Y. Molecular Dynamics Simulation of Diffusion Behavior of CH4, CO2, and N2 in Mid-Rank Coal Vitrinite. Energies. 2019; 12(19):3744. https://doi.org/10.3390/en12193744
Chicago/Turabian StyleLiu, Jing, Shike Li, and Yang Wang. 2019. "Molecular Dynamics Simulation of Diffusion Behavior of CH4, CO2, and N2 in Mid-Rank Coal Vitrinite" Energies 12, no. 19: 3744. https://doi.org/10.3390/en12193744