
Catalyzed Ethanol Chemical Looping Gasification Mechanism on the Perfect and Reduced Fe2O3 Surfaces


Abstract
:
1. Introduction
2. Materials and Methods
3. Results and Discussion
3.1. Adsorption of Ethanol on the Perfect and Reduced Fe2O3(001)
3.2. Ethanol Deep Decomposition on Fe2O3(001)
3.3. Ethanol Deep Decomposition on Fe3O4(001)
3.4. Ethanol Deep Decomposition FeO(001)
3.5. Ethanol Deep Decomposition on FeO0.375(001)
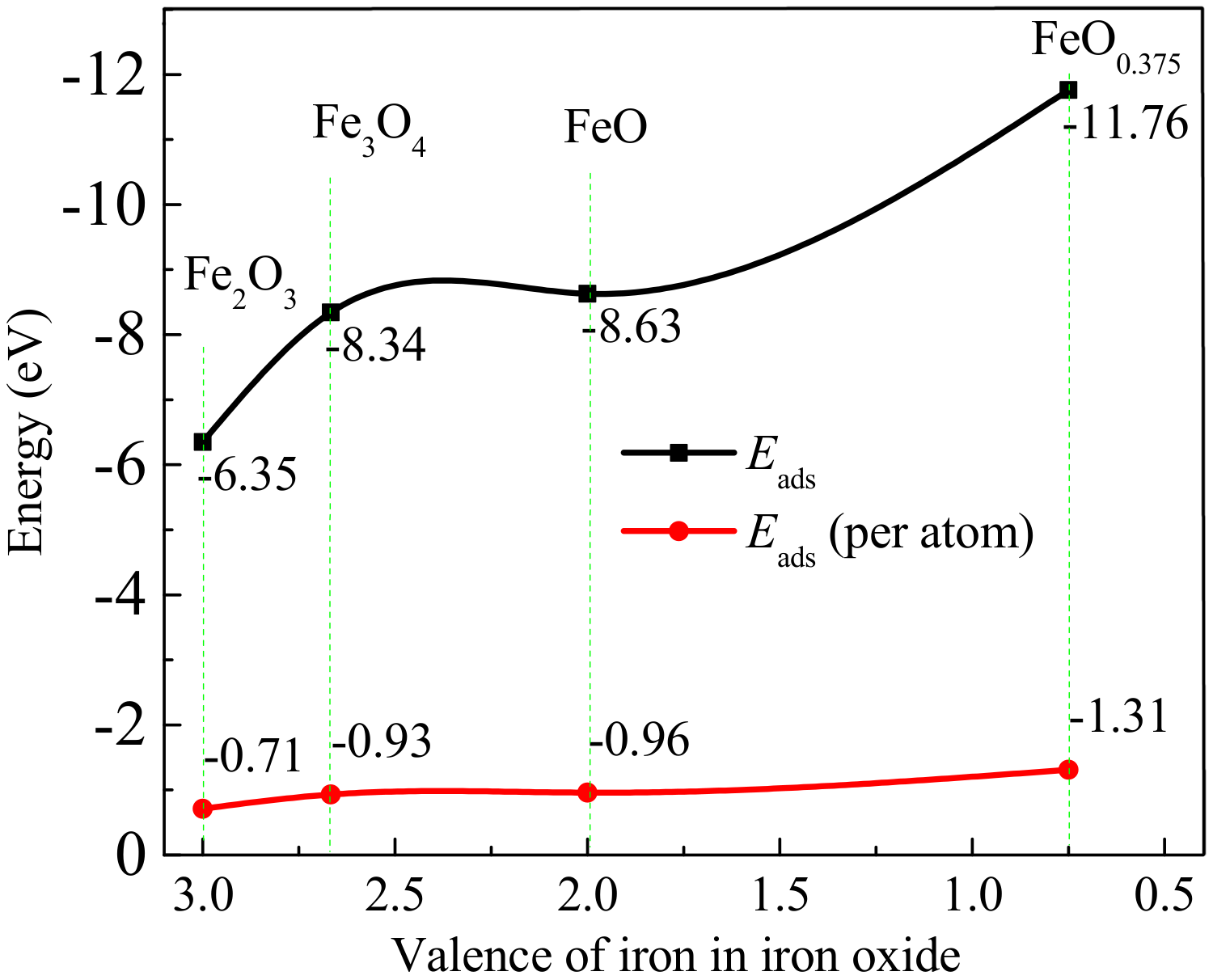
3.6. Thermodynamic Analysis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yu, Z.; Li, C.; Jing, X.; Zhang, Q.; Wang, Z.; Fang, Y.; Huang, J. Catalytic chemical looping combustion of carbon with an iron-based oxygen carrier modified by K2CO3: Catalytic mechanism and multicycle tests. Fuel Process. Technol. 2015, 135, 119–124. [Google Scholar] [CrossRef]
- Singh, L.; Wahid, Z.A. Methods for enhancing bio-hydrogen production from biological process: A review. J. Ind. Eng. Chem. 2015, 21, 70–80. [Google Scholar] [CrossRef]
- Kang, D.; Lim, H.S.; Lee, M.; Lee, J.W. Syngas production on a Ni-enhanced Fe2O3/Al2O3 oxygen carrier via chemical looping partial oxidation with dry reforming of methane. Appl. Energy 2018, 211, 174–186. [Google Scholar] [CrossRef]
- Liu, G.; Liao, Y.; Wu, Y.; Ma, X.; Chen, L. Characteristics of microalgae gasification through chemical looping in the presence of steam. Int. J. Hydrogen Energy 2017, 42, 22730–22742. [Google Scholar] [CrossRef]
- Zeng, J.; Xiao, R.; Zhang, H.; Wang, Y.; Zeng, D.; Ma, Z. Chemical looping pyrolysis-gasification of biomass for high H2/CO syngas production. Fuel Process. Technol. 2017, 168, 116–122. [Google Scholar] [CrossRef]
- Nandy, A.; Loha, C.; Gu, S.; Sarkar, P.; Karmakar, M.K.; Chatterjee, P.K. Present status and overview of chemical looping combustion technology. Renew. Sust. Energy Rev. 2016, 59, 597–619. [Google Scholar] [CrossRef]
- Yuan, Y.; Dong, X.; Ricardez-Sandoval, L. A density functional theory analysis on syngas adsorption on NiO (100) surface. Appl. Surf. Sci. 2019, 498, 143782. [Google Scholar] [CrossRef]
- Huang, Z.; He, F.; Zhao, K.; Zheng, A.Q.; Li, H.B.; Zhao, Z.L. Synthesis gas production by chemical-looping reforming of methane using lattice oxygen. Prog. Chem. 2012, 24, 1599–1609. [Google Scholar]
- Yuan, Y.; Dong, X.; Ricardez-Sandoval, L. A multi-scale simulation of syngas combustion reactions by Ni-based oxygen carriers for chemical looping combustion. Appl. Surf. Sci. 2020, 531, 147277. [Google Scholar] [CrossRef]
- Chen, L.; Kong, L.; Bao, J.; Combs, M.; Nikolic, H.S.; Fan, Z.; Liu, K. Experimental evaluations of solid-fueled pressurized chemical looping combustion—The effects of pressure, solid fuel and iron-based oxygen carriers. Appl. Energy 2017, 195, 1012–1022. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Chen, T.; Chang, D.; Chen, D.; He, H.; Frost, R.L. Catalytic cracking of tar derived from rice hull gasification over palygorskite-supported Fe and Ni. J. Mol. Catal. A Chem. 2012, 363, 304–310. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Fan, L.; Wang, G. Study on chemical looping reforming of ethanol (CLRE) for hydrogen production using NiMn2O4 spinel as oxygen carrier. J. Energy Inst. 2017, 90, 884–892. [Google Scholar] [CrossRef]
- Huang, Z.; Zhang, Y.; Fu, J.; Yu, L.; Chen, M.; Liu, S.; He, F.; Chen, D.; Wei, G.; Zhao, K.; et al. Chemical looping gasification of biomass char using iron ore as an oxygen carrier. Int. J. Hydrogen Energy 2016, 41, 17871–17883. [Google Scholar] [CrossRef]
- Zeng, J.; Xiao, R.; Zeng, D.; Zhao, Y.; Zhang, H.; Shen, D. High H2/CO ratio syngas production from chemical looping gasification ofsawdust in a dual fluidized bed gasifier. Energy Fuels 2015, 30, 1764–1770. [Google Scholar] [CrossRef]
- Wei, G.; He, F.; Zhao, Z.; Huang, Z.; Zheng, A.; Zhao, K.; Li, H. Performance of Fe–Ni bimetallic oxygen carriers for chemical looping gasification of biomass in a 10 kWth interconnected circulating fluidized bed reactor. Int. J. Hydrogen Energy 2015, 40, 16021–16032. [Google Scholar] [CrossRef]
- Qin, W.; Wang, Y.; Dong, C.; Zhang, J.; Chen, Q.; Yang, Y. The synergetic effect of metal oxide support on Fe2O3 for chemical looping combustion: A theoretical study. Appl. Surf. Sci. 2013, 282, 718–723. [Google Scholar] [CrossRef]
- Feng, Y.; Wang, N.; Guo, X.; Zhang, S. Reaction mechanism of Ca2Fe2O5 oxygen carrier with CO in chemical looping hydrogen production. Appl. Surf. Sci. 2020, 534, 147583. [Google Scholar] [CrossRef]
- Qin, W.; Chen, Q.; Wang, Y.; Dong, C.; Zhang, J.; Li, W.; Yang, Y. Theoretical study of oxidation–reduction reaction of Fe2O3 supported on MgO during chemical looping combustion. Appl. Surf. Sci. 2013, 266, 350–354. [Google Scholar] [CrossRef]
- Qin, W.; Chen, S.; Zhu, J.; Zhang, M.; Xiao, X. Hydrogen production by chemical looping gasification of corn stalk driven by a tert-butanol solution. Chem. Res. Chin. Univ. 2019, 35, 1012–1017. [Google Scholar] [CrossRef]
- Qin, W.; Wang, J.; Luo, L.; Liu, L.; Xiao, X.; Zheng, Z.; Sun, S.; Hu, X.; Dong, C. Chemical looping reforming of ethanol-containing organic wastewater for high ratio H2/CO syngas with iron-based oxygen carrier. Int. J. Hydrogen Energy 2018, 43, 12985–12998. [Google Scholar] [CrossRef]
- Qin, W.; Chen, S.; Ma, B.; Wang, J.; Li, J.; Liang, R.; Xu, Z.; Liu, L.; Dong, C.; Zhang, H. Methanol solution promoting cotton fiber chemical looping gasification for high H2/CO ratio syngas. Int. J. Hydrogen Energy 2019, 44, 7149–7157. [Google Scholar] [CrossRef]
- Dou, B.; Zhang, H.; Cui, G.; Wang, Z.; Jiang, B.; Wang, K.; Chen, H.; Xu, Y. Hydrogen production and reduction of Ni-based oxygen carriers during chemical looping steam reforming of ethanol in a fixed-bed reactor. Int. J. Hydrogen Energy 2017, 42, 26217–26230. [Google Scholar] [CrossRef]
- Jiang, B.; Dou, B.; Wang, K. Hydrogen production by chemical looping steam reforming of ethanol using NiO/montmorillonite oxygen carriers in a fixed-bed reactor. Chem. Eng. J. 2016, 298, 96–106. [Google Scholar] [CrossRef]
- García-Labiano, F.; García-Díez, E.; de Diego, L.F.; Serrano, A.; Abad, A.; Gayán, P.; Adánez, J.; Ruíz, J.A.C. Syngas/H2 production from bioethanol in a continuous chemical-looping reforming prototype. Fuel Process. Technol. 2015, 137, 24–30. [Google Scholar] [CrossRef] [Green Version]
- Adanez, J.; Abad, A.; Garcia-Labiano, F.; Gayan, P.; de Diego, L.F. Progress in Chemical-Looping Combustion and Reforming technologies. Prog. Energ. Combust. 2012, 38, 215–282. [Google Scholar] [CrossRef] [Green Version]
- Dong, C.; Sheng, S.; Qin, W.; Lu, Q.; Zhao, Y.; Wang, X.; Zhang, J. Density functional theory study on activity of α-Fe2O3 in chemical-looping combustion system. Appl. Surf. Sci. 2011, 257, 8647–8652. [Google Scholar] [CrossRef]
- Lin, C.; Qin, W.; Dong, C. H2S adsorption and decomposition on the gradually reduced α-Fe2O3(001) surface: A DFT study. Appl. Surf. Sci. 2016, 387, 720–731. [Google Scholar] [CrossRef]
- Xiao, X.; Qin, W.; Wang, J.; Li, J.; Dong, C. Effect of surface Fe-S hybrid structure on the activity of the perfect and reduced α-Fe2O3(001) for chemical looping combustion. Appl. Surf. Sci. 2018, 440, 29–34. [Google Scholar] [CrossRef]
- Zhang, Q.; Li, L.; Jiang, B. An intelligent oxygen carrier of La2−xSrxNiO4−λ for hydrogen production by chemical looping reforming of ethanol. Int. J. Hydrogen Energy 2017, 42, 17102–17111. [Google Scholar] [CrossRef]
- Dou, B.; Wang, C.; Song, Y.; Chen, H.; Jiang, B.; Yang, M.; Xu, Y. Solid sorbents for in-situ CO2 removal during sorption-enhanced steam reforming process: A review. Renew. Sust. Energ. Rev. 2016, 53, 536–546. [Google Scholar] [CrossRef]
- Zhang, G.; Cui, G.; Dou, B.; Wang, Z.; Goula, M.A. An experimental investigation of forced convection heat transfer with novel microencapsulated phase change material slurries in a circular tube under constant heat flux. Energy Convers. Manag. 2018, 171, 699–709. [Google Scholar] [CrossRef]
- Lin, C.; Qin, W.; Dong, C. Reduction effect of α-Fe2O3 on carbon deposition and CO oxidation during chemical-looping combustion. Chem. Eng. J. 2016, 301, 257–265. [Google Scholar] [CrossRef]
- Bayham, S.C.; Kim, H.R.; Wang, D.; Tong, A.; Zeng, L.; McGiveron, O.; Kathe, M.V.; Chung, E.; Wang, W.; Wang, A.; et al. Iron-based coal direct chemical looping combustion process: 200-h continuous operation of a 25-kWth subpilot unit. Energy Fuels 2013, 27, 1347–1356. [Google Scholar] [CrossRef]
- Chein, R.; Hsu, W. Thermodynamic equilibrium analysis of H2-rich syngas production via sorption-enhanced chemical looping biomass gasification. Renew. Energy 2020, 153, 117–129. [Google Scholar] [CrossRef]
- Yan, J.; Sun, R.; Shen, L.; Bai, H.; Jiang, S.; Xiao, Y.; Song, T. Hydrogen-rich syngas production with tar elimination via biomass chemical looping gasification (BCLG) using BaFe2O4/Al2O3 as oxygen carrier. Chem. Eng. J. 2020, 387, 124107. [Google Scholar] [CrossRef]
- Zaini, I.N.; Nurdiawati, A.; Aziz, M. Cogeneration of power and H2 by steam gasification and syngas chemical looping of macroalgae. Appl. Energy 2017, 207, 134–145. [Google Scholar] [CrossRef]
- Sarafraz, M.M.; Jafarian, M.; Arjomandi, M.; Nathan, G.J. Potential use of liquid metal oxides for chemical looping gasification: A thermodynamic assessment. Appl. Energy 2017, 195, 702–712. [Google Scholar] [CrossRef] [Green Version]
- Hu, Z.; Jiang, E.; Ma, X. The effect of oxygen carrier content and temperature on chemical looping gasification of microalgae for syngas production. J. Energy. Inst. 2019, 92, 474–487. [Google Scholar] [CrossRef]
- Huang, Z.; Liu, S.; Li, D.; Zhan, Z.; He, F.; Li, H. Study on thermodynamic simulation of biomass chemical chain gasification based on Fe2O3 oxygen carrier. Act. Energ. Sol. Sin. 2017, 5, 1421–1430. [Google Scholar]
- Kment, S.; Sivula, K.; Naldoni, A.; Sarmah, S.P.; Kmentová, H.; Kulkarni, M.; Rambabu, Y.; Schmuki, P.; Zbořil, R. FeO-based nanostructures and nanohybrids for photoelectrochemical water splitting. Prog. Mater. Sci. 2020, 110, 100632. [Google Scholar] [CrossRef]
- Huang, Z.; He, F.; Zhu, H.; Chen, D.; Zhao, K.; Wei, G.; Feng, Y.; Zheng, A.; Zhao, Z.; Li, H. Thermodynamic analysis and thermogravimetric investigation on chemical looping gasification of biomass char under different atmospheres with Fe2O3 oxygen carrier. Appl. Energy 2015, 157, 546–553. [Google Scholar] [CrossRef]
- Qin, W.; Wang, Y.; Lin, C.; Hu, X.; Dong, C. Possibility of morphological control to improve the activity of oxygen carriers for chemical looping combustion. Energy Fuels 2015, 29, 1210–1218. [Google Scholar] [CrossRef]
- Zou, X.; Zhao, H.; Zheng, C. Techno-economic evaluation of chemical looping-based coal-fired power plant. Proc. CSEE 2014, 35, 34–43. [Google Scholar]
- Fan, J.; Zhu, L.; Jiang, P.; Li, L.; Liu, H. Comparative exergy analysis of chemical looping combustion thermally coupled and conventional steam methane reforming for hydrogen production. J. Clean. Prod. 2016, 131, 247–258. [Google Scholar] [CrossRef]
- Hu, Q.; Shen, Y.; Chew, J.W.; Ge, T.; Wang, C. Chemical looping gasification of biomass with Fe2O3/CaO as the oxygen carrier for hydrogen-enriched syngas production. Chem. Eng. J. 2020, 379, 122346. [Google Scholar] [CrossRef]
- Chen, H.; Zhao, C.; Li, Y.; Chen, X. CO2 capture performance of calcium-based sorbents in a pressurized carbonation/calcination loop. Energy Fuels 2010, 24, 5751–5756. [Google Scholar] [CrossRef]
- Li, Y.; Su, M.; Xie, X.; Wu, S.; Liu, C. CO2 capture performance of synthetic sorbent prepared from carbide slag and aluminum nitrate hydrate by combustion synthesis. Appl. Energy 2015, 145, 60–68. [Google Scholar] [CrossRef]
- Ma, X.; Li, Y.; Yan, X.; Zhang, W.; Zhao, J.; Wang, Z. Preparation of a morph-genetic CaO-based sorbent using paper fibre as a biotemplate for enhanced CO2 capture. Chem. Eng. J. 2019, 361, 235–244. [Google Scholar] [CrossRef]

 O and
O and  Fe).
Fe). O, Fe,
O, Fe,  H, and
H, and  C).
O, Fe, H, and C).
C).
O, Fe, H, and C).
 O, Fe, H, and C. “*” means that the group is adsorbed on the surface of Fe2O3(001)).
O, Fe, H, and C. “*” means that the group is adsorbed on the surface of Fe2O3(001)).
O, Fe, H, and C. “*” means that the group is adsorbed on the surface of Fe2O3(001)).
O, Fe, H, and C. “*” means that the group is adsorbed on the surface of Fe2O3(001)).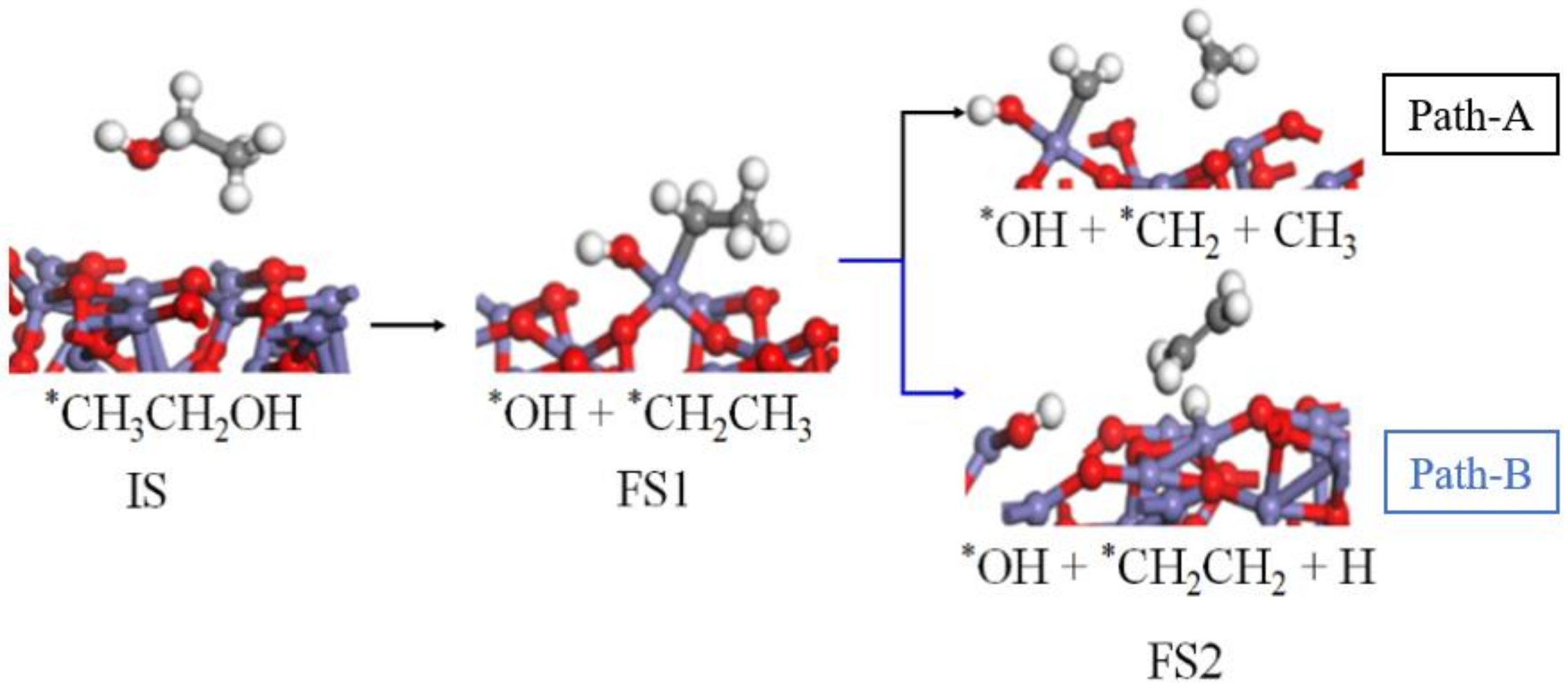
 O, Fe, H, and C).
O, Fe, H, and C).
O, Fe, H, and C).
O, Fe, H, and C).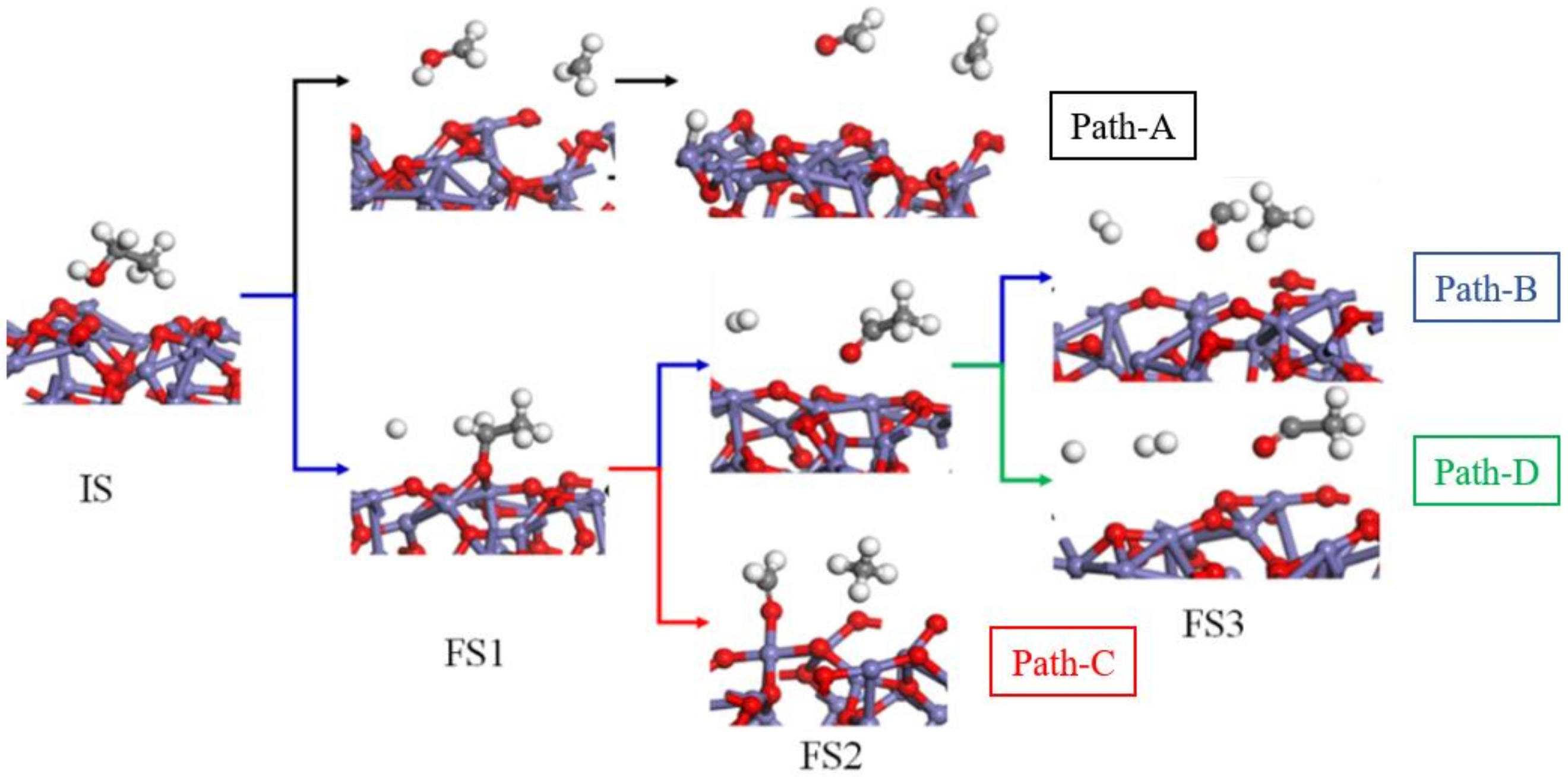
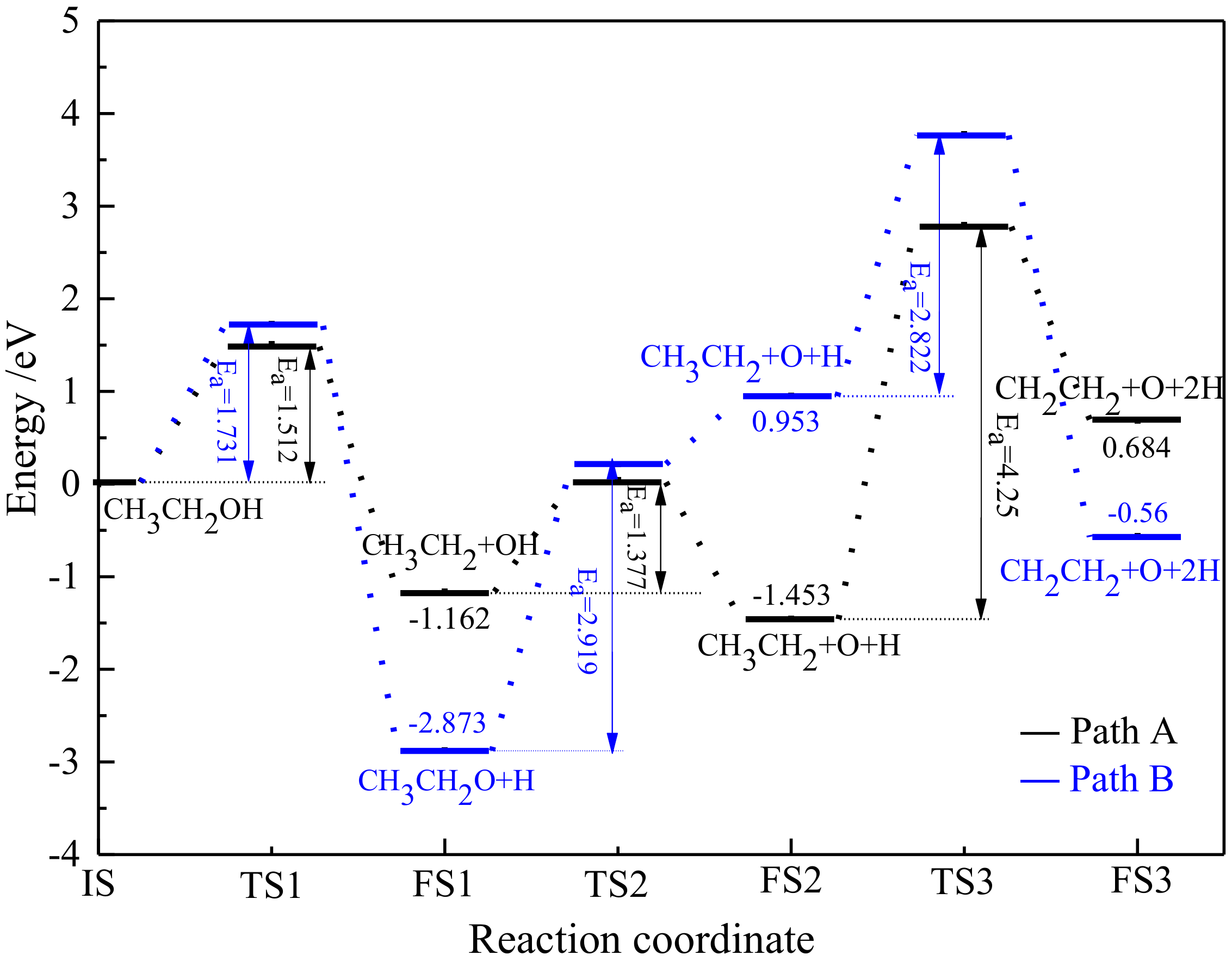 O, Fe, H, and C).
O, Fe, H, and C).
O, Fe, H, and C).
O, Fe, H, and C).
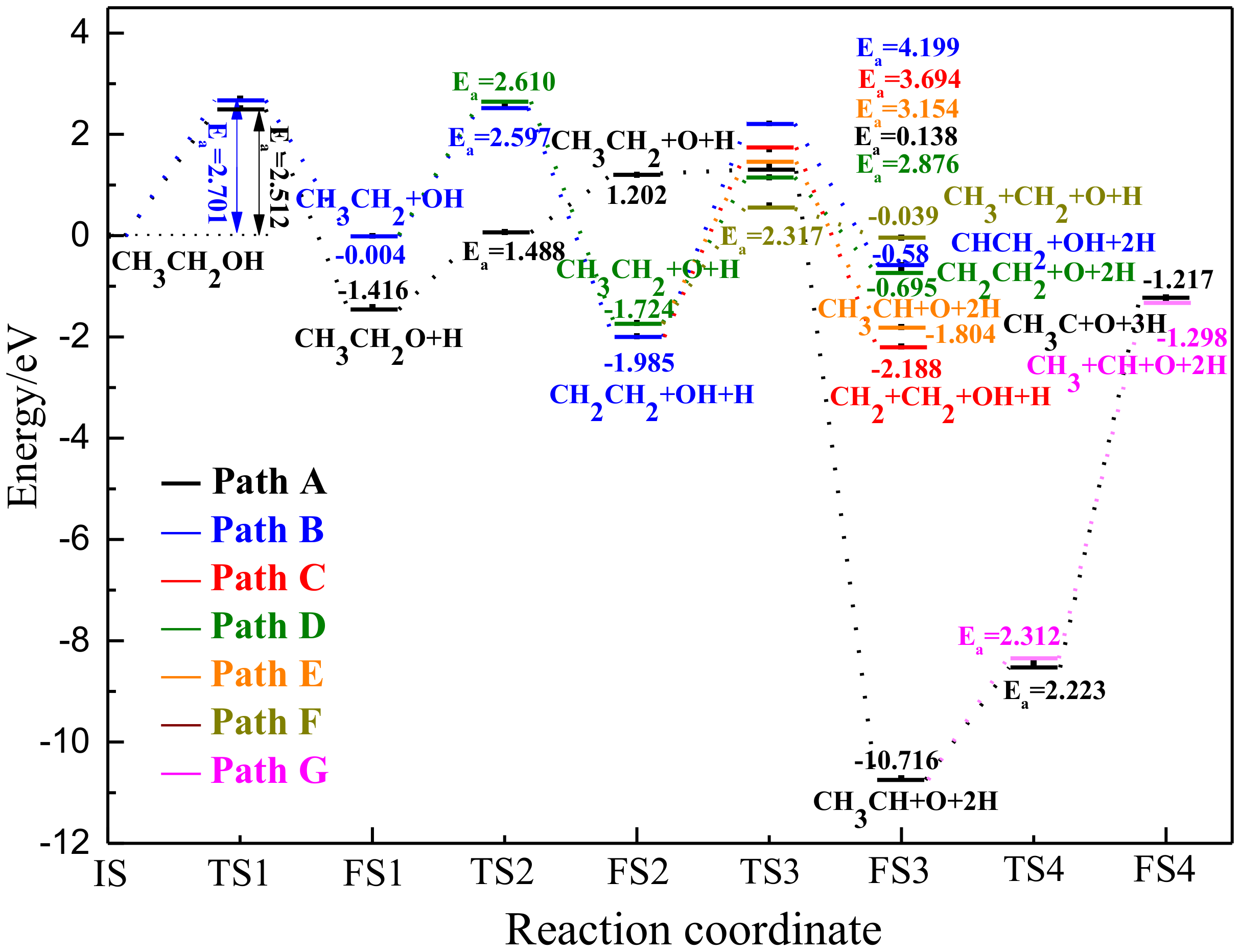 O, Fe, H, and C).
O, Fe, H, and C).
O, Fe, H, and C).
O, Fe, H, and C).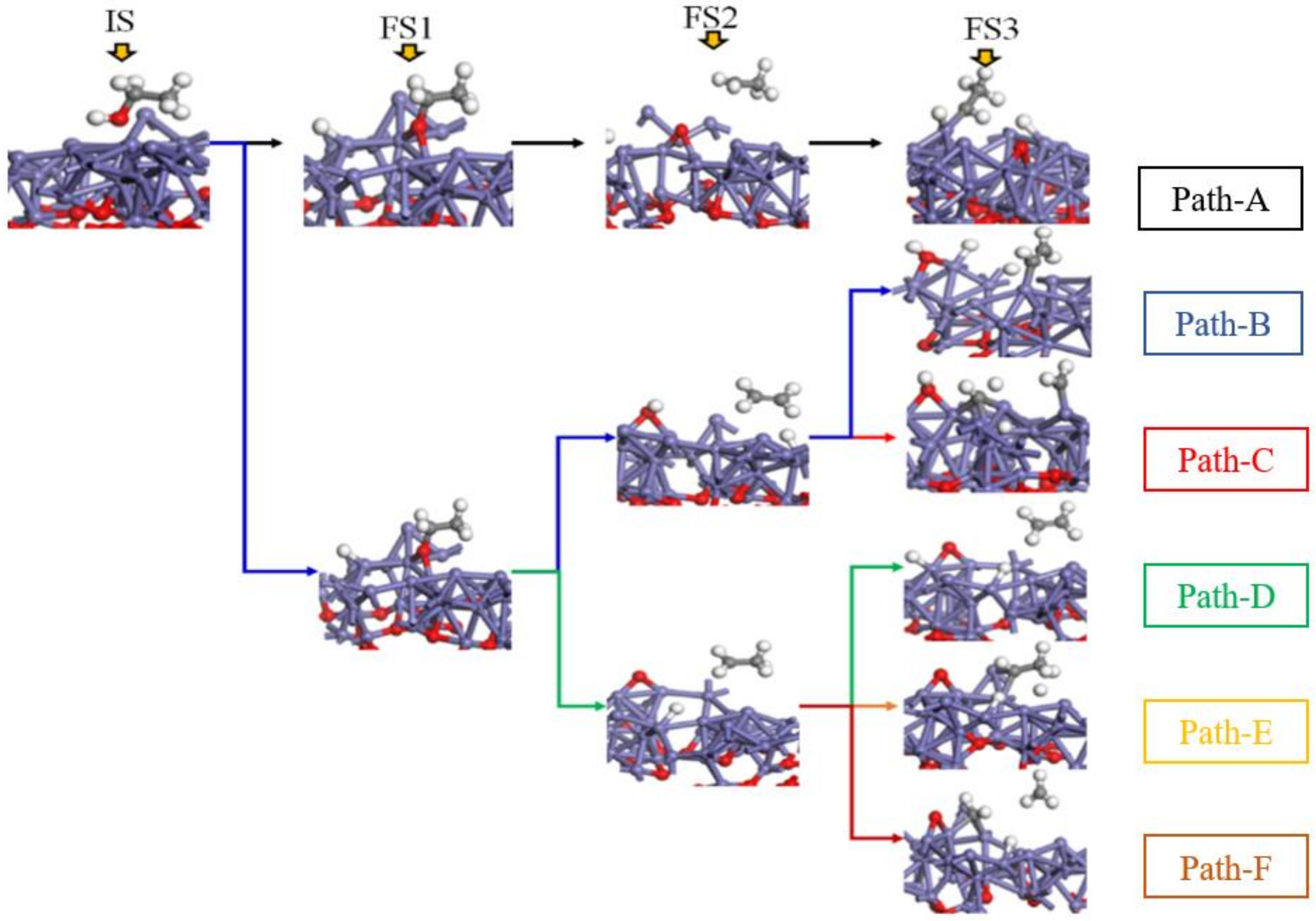
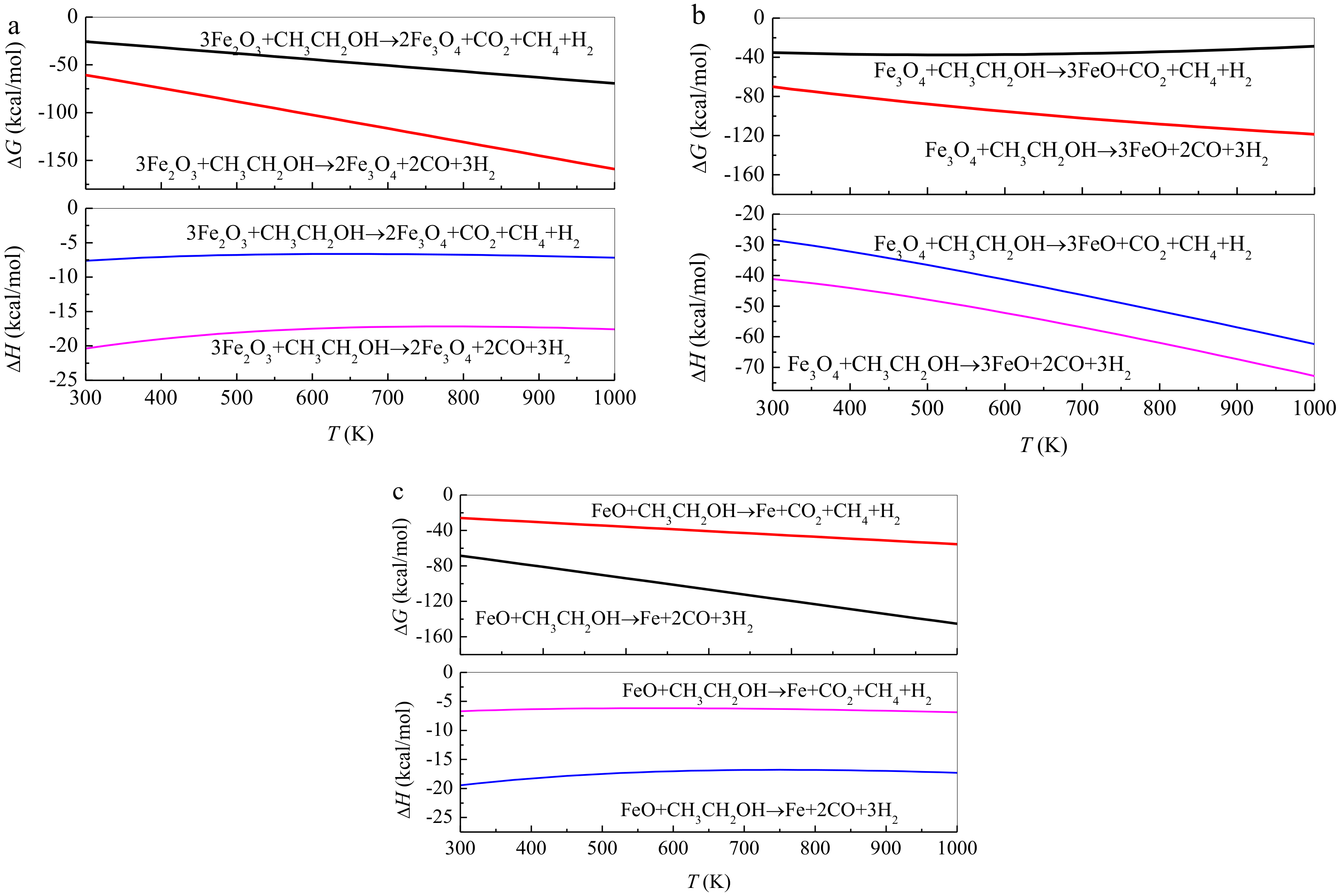

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Different Reduction Surfaces of Iron Oxide | C1-H(Å) | C2-H(Å) | C-C(Å) | C-O(Å) | O-H(Å) |
|---|---|---|---|---|---|
| Bond Length | |||||
| Fe2O3(001) | 1.101 | 1.107 | 1.520 | 1.442 | 0.974 |
| Fe3O4(001) | 1.099 | 1.106 | 1.516 | 1.454 | 0.975 |
| FeO(001) | 1.097 | 1.105 | 1.505 | 1.477 | 0.975 |
| FeO0.375(001) | 1.101 | 1.104 | 1.514 | 1.481 | 0.983 |
| Atom | Ethanol Molecule | Ethanol Molecule on Fe2O3(001) | Ethanol Molecule on Fe3O4(001) | Ethanol Molecule on FeO(001) | Ethanol Molecule on FeO0.375(001) |
|---|---|---|---|---|---|
| Charge | |||||
| C1 | −0.14 | −0.73 | −0.71 | −0.7 | −0.65 |
| H1 | 0.045 | 0.19 | 0.19 | 0.17 | 0.1 |
| H2 | 0.06 | 0.23 | 0.2 | 0.18 | 0.17 |
| H3 | 0.061 | 0.25 | 0.26 | 0.28 | 0.25 |
| C2 | 0.147 | −0.28 | −0.29 | −0.31 | −0.28 |
| H4 | 0.034 | 0.24 | 0.26 | 0.27 | 0.22 |
| H5 | 0.036 | 0.27 | 0.27 | 0.28 | 0.27 |
| O of Hydroxyl | −0.488 | −0.76 | −0.71 | −0.66 | −0.61 |
| H of Hydroxyl | 0.246 | 0.51 | 0.48 | 0.48 | 0.44 |
| Net charge | 0.001 | −0.08 | −0.05 | −0.01 | −0.09 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luo, L.; Zheng, X.; Wang, J.; Qin, W.; Xiao, X.; Zheng, Z. Catalyzed Ethanol Chemical Looping Gasification Mechanism on the Perfect and Reduced Fe2O3 Surfaces. Energies 2021, 14, 1663. https://doi.org/10.3390/en14061663
Luo L, Zheng X, Wang J, Qin W, Xiao X, Zheng Z. Catalyzed Ethanol Chemical Looping Gasification Mechanism on the Perfect and Reduced Fe2O3 Surfaces. Energies. 2021; 14(6):1663. https://doi.org/10.3390/en14061663
Chicago/Turabian StyleLuo, Laixing, Xing Zheng, Jianye Wang, Wu Qin, Xianbin Xiao, and Zongming Zheng. 2021. "Catalyzed Ethanol Chemical Looping Gasification Mechanism on the Perfect and Reduced Fe2O3 Surfaces" Energies 14, no. 6: 1663. https://doi.org/10.3390/en14061663