
Numerical Simulation of Hydrate Formation in the LArge-Scale Reservoir Simulator (LARS)



Abstract
:1. Introduction
2. Materials and Methods
2.1. Experimental Data
2.1.1. Hydrate Formation Experimental Schedule
2.2. Mathematical Model
2.2.1. Modeling Assumptions
- The porous medium is completely filled by pore fluid and/or CH4 hydrate, with single-phase flow considered in the entire modeling domain;
- Deformation of the porous medium (hydrate-bearing sand) is assumed to be negligible due to the applied confining pressure of 14 to 15 MPa, with the porous medium matrix being evenly compacted and homogeneous;
- Thermophysical properties of the aqueous solution do not consider the effects of the dissolved CH4, as these are negligible for the present study. The dissolved inhibitor (NaCl) influences neither the molecular structure of the formed CH4 hydrate nor the rate of hydrate formation, but fluid density, viscosity, heat conductivity and capacity as well as CH4 solubility, only;
- CH4 from the supersaturated aqueous phase is directly consumed by equilibrium hydrate formation without any intermediate phase changes and side reactions;
- Mobile components contain the aqueous phase with dissolved CH4 and NaCl. All water-soluble species and liquids are non-volatile at the applied temperature range (0–25 °C) and pressure conditions (ca. 11 MPa).
2.2.2. Numerical Model Verification
2.3. Model Implementation to Reproduce the LARS Experiments
2.3.1. Model Geometry
2.3.2. Initial and Boundary Conditions
3. Results and Discussion
3.1. Model Calibration
3.1.1. Model Calibration by Comparison of Simulated and Observed Temperature Evolution Profiles
3.1.2. Calibration of RTD Locations
3.2. Model Calibration and Validation
3.2.1. Model Calibration via Comparison of the Temporal Evolution of Simulated and Observed Temperature Profiles
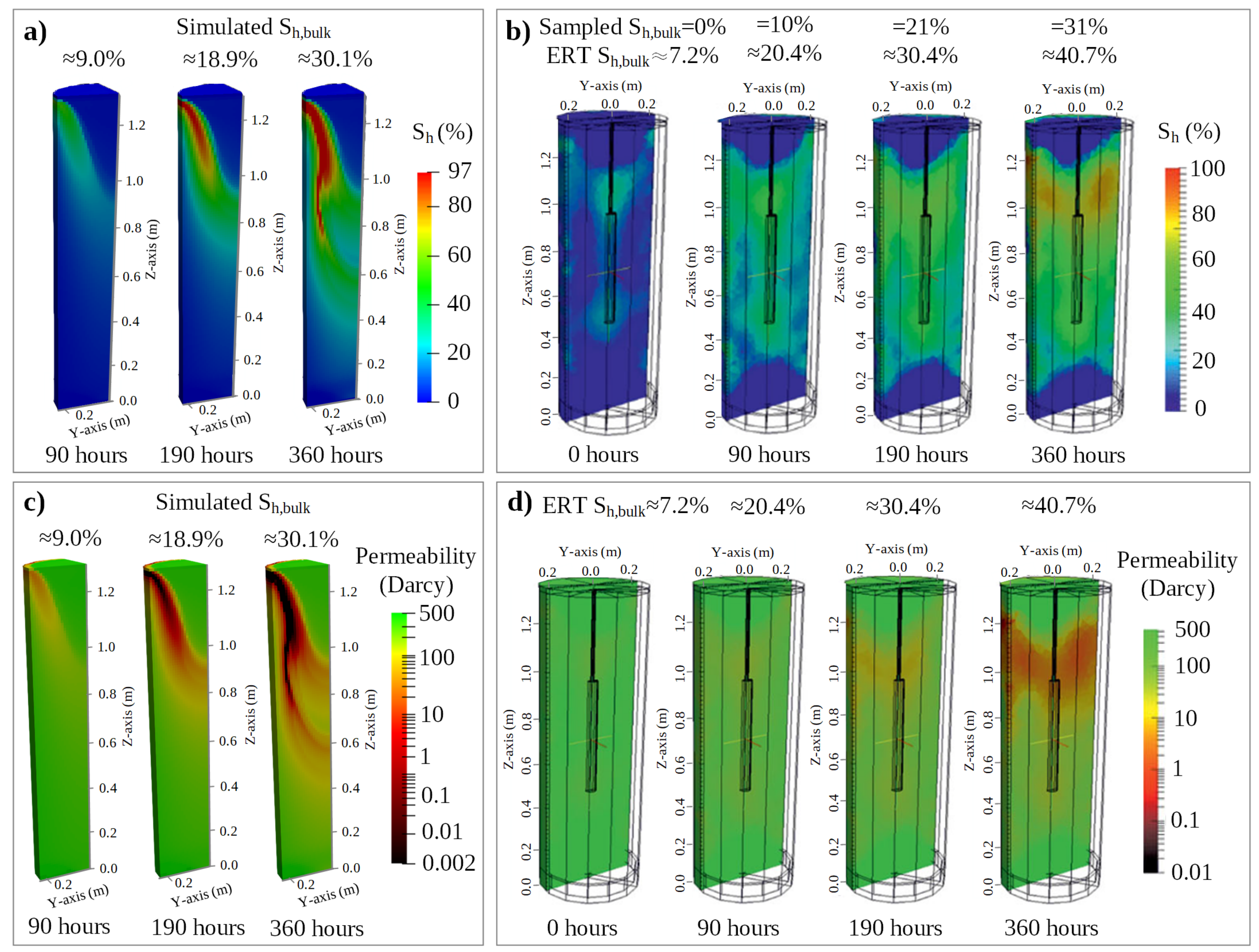
3.2.2. Model Validation by Comparison of the Temporal and Spatial Evolution of Simulated and Observed Hydrate Saturations
3.3. Uncertainties of Critical Parameters in the Experimental Study
4. Summary and Conclusions
- The general consistency of the experimental observations with the simulation results proves that the employed equilibrium CH4 hydrate formation model can represent the main processes of hydrate formation in LARS. The equilibrium reaction model is a practicable alternative to kinetic approaches at macro-scale (vessel volume > 0.2 m3) given the application of the “dissolved-gas” method. In contrast, kinetic reaction approaches tend to be irreplaceable for modeling hydrate formation by other methods, because their CH4 hydrate growth rates are orders of magnitude faster than that of the “dissolved-gas” method.
- The deviations among the experimental observations (i.e., continuously recorded temperature profiles, periodically gathered Sh,bulk, and ERT-tomography derived spatial Sh distributions) and the corresponding numerical predictions were minimized through an iterative optimization procedure. It has been indicated that the combination of the thermal properties of inflowing CH4-loaded fluid and the hydrate-bearing sand determine the spatial distribution of hydrate accumulations.
- The presented spatial Sh distribution illustrates a heterogeneous accumulation within the hydrate-bearing sand at an early experimental period when Sh,bulk < 30%, with the feature becoming less prominent until Sh,bulk > 80%.
- In the LARS hydrate formation experiment, a relatively large temperature gradient (ca. 10 °C/0.23 m) is generated between the inflow of warm brine and its surrounding coolants, leading to a heterogeneous hydrate distribution. In contrast, the sub-permafrost and sub-seafloor geothermal gradients in natural settings are substantially lower (3 °C/100 m) [75] and steady for long time periods, causing a lower and almost constant dissolved CH4 concentration gradient in the saline fluid. Therefore, relatively uniformly distributed Sh were found within the NGH intervals with ignorable lateral variations at the Mallik site. These NGH accumulation intervals could be simplified as CH4 hydrate layers formed via the continuous supply of dissolved CH4, migrating through the up-dip natural faults in the Canadian Beaufort-Mackenzie Basin region.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Nomenclature
| density | kg·m−3 | |
| effective porosity | - | |
| matrix compressibility | Pa−1 | |
| fluid compressibility | Pa−1 | |
| P and p | pressure | Pa |
| t | time | s |
| Darcy velocity vector | m·s−1 | |
| W | fluid source or sink term | kg·m−3·s−1 |
| dynamic viscosity | Pa·s | |
| k | effective permeability tensor | m2 |
| gravitational acceleration vector | m·s−2 | |
| X | mass fraction | - |
| C | concentration matrix of mobile components | kg·m−3 |
| D | hydrodynamic dispersion tensor | m2·s−1 |
| Q | NaCl and CH4 source or sink term | kg·m−3·s−1 |
| specific heat capacity | J·kg−1·K−1 | |
| T | temperature | °C |
| thermal conductivity tensor | W·m−1·K−1 | |
| H | heat source or sink term | W·m−3 |
| S | species saturation in the pore volume | - |
| intrinsic porosity | - | |
| intrinsic permeability tensor | m2 | |
| x | molality | mol·kg−1 |
| M | molecular weight | kg·mol−1 |
| h | enthalpy | J·kg−1 |
| mole fraction | - | |
| Superscripts and Subscripts | ||
| f | mobile components (pore fluid with dissolved CH4 and NaCl) | |
| r | immobile components (hydrate-bearing sediment and hydrate to be formed) | |
| h | hydrate component | |
| m | methane component (CH4) | |
| w | water component | |
| i | inhibitor component (NaCl) | |
| a | average value | |
| s | quartz sand (matrix material of the hydrate-bearing sediment) | |
| energy change during hydrate formation | ||
| equilibrium | ||
| shifted temperature | ||
Appendix A
Appendix A.1. Governing Equations for Fluid Flow as Well as Heat and Chemical Species Transport
Appendix A.2. Equations of State (EOS) for CH4 Hydrate Equilibrium Formation
References
- Kvenvolden, K.A.; Ginsburg, G.D.; Soloviev, V.A. Worldwide distribution of subaquatic gas hydrates. Geo-Mar. Lett. 1993, 13, 32–40. [Google Scholar] [CrossRef]
- Sloan, E.D., Jr.; Koh, C.A. Clathrate Hydrates of Natural Gases, 3rd ed.; CRC Press: Boca Raton, FL, USA, 2008. [Google Scholar]
- Yin, Z.; Moridis, G.; Tan, H.K.; Linga, P. Numerical analysis of experimental studies of methane hydrate formation in a sandy porous medium. Appl. Energy 2018, 220, 681–704. [Google Scholar] [CrossRef] [Green Version]
- Makogon, Y.F. Natural gas hydrates—A promising source of energy. J. Nat. Gas Sci. Eng. 2010, 2, 49–59. [Google Scholar] [CrossRef]
- Osadetz, K.G.; Dixon, J.; Dietrich, J.R.; Snowdon, L.R.; Dallimore, S.R.; Majorowicz, J.A. A Review of Mackenzie Delta-Beaufort Sea Petroleum Province Conventional and Non-Conventional (Gas Hydrate) Petroleum Reserves and Undiscovered Resources: A Contribution to the Resource Assessment of the Proposed Mackenzie Delta-Beaufort Sea Marine Protected Area. 2005. Available online: https://waves-vagues.dfo-mpo.gc.ca/Library/281345.pdf (accessed on 20 December 2021).
- Koh, C.A.; Sum, A.K.; Sloan, E.D. Gas hydrates: Unlocking the energy from icy cages. J. Appl. Phys. 2009, 106, 061101. [Google Scholar] [CrossRef]
- Milkov, A.V. Global estimates of hydrate-bound gas in marine sediments: How much is really out there? Earth-Sci. Rev. 2004, 66, 183–197. [Google Scholar] [CrossRef]
- Wallmann, K.; Schicks, J.M. Gas Hydrates as an Unconventional Hydrocarbon Resource. In Hydrocarbons, Oils and Lipids: Diversity, Origin, Chemistry and Fate. (Handbook of Hydrocarbon and Lipid Microbiology); Wilkes, H., Ed.; Springer: Cham, Switzerland, 2018; pp. 1–17. [Google Scholar] [CrossRef]
- Yin, Z.; Khurana, M.; Tan, H.K.; Linga, P. A review of gas hydrate growth kinetic models. Chem. Eng. J. 2018, 342, 9–29. [Google Scholar] [CrossRef]
- Yin, Z.; Chong, Z.R.; Tan, H.K.; Linga, P. Review of gas hydrate dissociation kinetic models for energy recovery. J. Nat. Gas Sci. Eng. 2016, 35, 1362–1387. [Google Scholar] [CrossRef]
- Hassanpouryouzband, A.; Joonaki, E.; Vasheghani Farahani, M.; Takeya, S.; Ruppel, C.; Yang, J.; English, N.J.; Schicks, J.M.; Edlmann, K.; Mehrabian, H.; et al. Gas hydrates in sustainable chemistry. Chem. Soc. Rev. 2020, 49, 5225–5309. [Google Scholar] [CrossRef]
- Handa, Y.P.; Stupin, D.Y. Thermodynamic properties and dissociation characteristics of methane and propane hydrates in 70-.ANG.-radius silica gel pores. J. Phys. Chem. 1992, 96, 8599–8603. [Google Scholar] [CrossRef]
- Winters, W.J.; Pecher, I.A.; Waite, W.F.; Mason, D.H. Physical properties and rock physics models of sediment containing natural and laboratory-formed methane gas hydrate. Am. Mineral. 2004, 89, 1221–1227. [Google Scholar] [CrossRef]
- Priest, J.A.; Rees, E.V.L.; Clayton, C.R.I. Influence of gas hydrate morphology on the seismic velocities of sands. J. Geophys. Res. Solid Earth 2009, 114, B11205. [Google Scholar] [CrossRef] [Green Version]
- Schicks, J.M.; Spangenberg, E.; Giese, R.; Steinhauer, B.; Klump, J.; Luzi, M. New approaches for the production of hydrocarbons from hydrate bearing sediments. Energies 2011, 4, 151–172. [Google Scholar] [CrossRef] [Green Version]
- Fitzgerald, G.C.; Castaldi, M.J.; Zhou, Y. Large scale reactor details and results for the formation and decomposition of methane hydrates via thermal stimulation dissociation. J. Pet. Sci. 2012, 94–95, 19–27. [Google Scholar] [CrossRef]
- Chong, Z.R.; Pujar, G.A.; Yang, M.; Linga, P. Methane hydrate formation in excess water simulating marine locations and the impact of thermal stimulation on energy recovery. Appl. Energy 2016, 177, 409–421. [Google Scholar] [CrossRef]
- Broseta, D.; Ruffine, L.; Desmedt, A. Gas Hydrates 1: Fundamentals, Characterization and Modeling; John Wiley & Sons: Hoboken, NJ, USA, 2017. [Google Scholar]
- Li, G.; Wu, D.; Li, X.; Zhang, Y.; Lv, Q.; Wang, Y. Experimental Investigation into the Production Behavior of Methane Hydrate under Methanol Injection in Quartz Sand. Energy Fuels 2017, 31, 5411–5418. [Google Scholar] [CrossRef]
- Chandrasekharan Nair, V.; Mech, D.; Gupta, P.; Sangwai, J.S. Polymer flooding in artificial hydrate bearing sediments for methane gas recovery. Energy Fuels 2018, 32, 6657–6668. [Google Scholar] [CrossRef]
- Schicks, J.M.; Strauch, B.; Heeschen, K.U.; Spangenberg, E.; Luzi-Helbing, M. From Microscale (400 μl) to Macroscale (425 L): Experimental Investigations of the CO2/N2--CH4 Exchange in Gas Hydrates Simulating the Iġnik Sikumi Field Trial. J. Geophys. Res. Solid Earth 2018, 123, 3608–3620. [Google Scholar] [CrossRef] [Green Version]
- Thoutam, P.; Rezaei Gomari, S.; Ahmad, F.; Islam, M. Comparative analysis of hydrate nucleation for methane and carbon dioxide. Molecules 2019, 24, 1055. [Google Scholar] [CrossRef] [Green Version]
- Pan, M.; Ismail, N.A.; Luzi-Helbing, M.; Koh, C.A.; Schicks, J.M. New Insights on a µm-Scale into the Transformation Process of CH4 Hydrates to CO2–Rich Mixed Hydrates. Energies 2020, 13, 5908. [Google Scholar] [CrossRef]
- Schicks, J.M.; Pan, M.; Giese, R.; Poser, M.; Ismail, N.A.; Luzi-Helbing, M.; Bleisteiner, B.; Lenz, C. A new high-pressure cell for systematic in situ investigations of micro-scale processes in gas hydrates using confocal micro-Raman spectroscopy. Rev. Sci. Instrum. 2020, 91, 115103. [Google Scholar] [CrossRef]
- Heeschen, K.U.; Deusner, C.; Spangenberg, E.; Priegnitz, M.; Kossel, E.; Strauch, B.; Bigalke, N.; Luzi-Helbing, M.; Haeckel, M.; Schicks, J.M. Production Method under Surveillance: Laboratory Pilot-Scale Simulation of CH4–CO2 Exchange in a Natural Gas Hydrate Reservoir. Energy Fuels 2021, 35, 10641–10658. [Google Scholar] [CrossRef]
- Priegnitz, M.; Thaler, J.; Spangenberg, E.; Rücker, C.; Schicks, J.M. A cylindrical electrical resistivity tomography array for three-dimensional monitoring of hydrate formation and dissociation. Rev. Sci. Instrum. 2013, 84, 104502. [Google Scholar] [CrossRef] [PubMed]
- Priegnitz, M.; Thaler, J.; Spangenberg, E.; Schicks, J.M.; Schrötter, J.; Abendroth, S. Characterizing electrical properties and permeability changes of hydrate bearing sediments using ERT data. Geophys. J. Int. 2015, 202, 1599–1612. [Google Scholar] [CrossRef] [Green Version]
- Sa, J.H.; Kwak, G.H.; Han, K.; Ahn, D.; Cho, S.J.; Lee, J.D.; Lee, K.H. Inhibition of methane and natural gas hydrate formation by altering the structure of water with amino acids. Sci. Rep. 2016, 6, 31582. [Google Scholar] [CrossRef]
- Zhang, L.; Kuang, Y.; Zhang, X.; Song, Y.; Liu, Y.; Zhao, J. Analyzing the process of gas production from methane hydrate via nitrogen injection. Ind. Eng. Chem. Res. 2017, 56, 7585–7592. [Google Scholar] [CrossRef]
- Strauch, B.; Heeschen, K.U.; Schicks, J.M.; Spangenberg, E.; Zimmer, M. Application of tubular silicone (PDMS) membranes for gas monitoring in CO2–CH4 hydrate exchange experiments. Mar. Pet. Geol. 2020, 122, 104677. [Google Scholar] [CrossRef]
- Heeschen, K.U.; Janocha, J.; Spangenberg, E.; Schicks, J.M.; Giese, R. The impact of ice on the tensile strength of unconsolidated sand-A model for gas hydrate-bearing sands? Mar. Pet. Geol. 2020, 122, 104607. [Google Scholar] [CrossRef]
- Pan, M.; Schicks, J.M. Influence of Gas Supply Changes on the Formation Process of Complex Mixed Gas Hydrates. Molecules 2021, 26, 3039. [Google Scholar] [CrossRef]
- Dai, J.; Xu, H.; Snyder, F.; Dutta, N. Detection and estimation of gas hydrates using rock physics and seismic inversion: Examples from the northern deepwater Gulf of Mexico. Lead. Edge 2004, 23, 60–66. [Google Scholar] [CrossRef] [Green Version]
- Sell, K.; Saenger, E.H.; Falenty, A.; Chaouachi, M.; Haberthür, D.; Enzmann, F.; Kuhs, W.F.; Kersten, M. On the path to the digital rock physics of gas hydrate-bearing sediments—Processing of in situ synchrotron-tomography data. Solid Earth 2016, 7, 1243–1258. [Google Scholar] [CrossRef] [Green Version]
- Sell, K.; Quintal, B.; Kersten, M.; Saenger, E.H. Squirt flow due to interfacial water films in hydrate bearing sediments. Solid Earth 2018, 9, 699–711. [Google Scholar] [CrossRef] [Green Version]
- Yun, T.S.; Francisca, F.M.; Santamarina, J.C.; Ruppel, C. Compressional and shear wave velocities in uncemented sediment containing gas hydrate. Geophys. Res. Lett. 2005, 32, L10609. [Google Scholar] [CrossRef]
- Waite, W.F.; Santamarina, J.C.; Cortes, D.D.; Dugan, B.; Espinoza, D.N.; Germaine, J.; Jang, J.; Jung, J.W.; Kneafsey, T.J.; Shin, H.; et al. Physical properties of hydrate-bearing sediments. Rev. Geophys. 2009, 47. [Google Scholar] [CrossRef]
- Tupsakhare, S.S.; Fitzgerald, G.C.; Castaldi, M.J. Thermally Assisted Dissociation of Methane Hydrates and the Impact of CO2 Injection. Ind. Eng. Chem. Res. 2016, 55, 10465–10476. [Google Scholar] [CrossRef]
- Schicks, J.M.; Spangenberg, E.; Giese, R.; Luzi-Helbing, M.; Priegnitz, M.; Beeskow-Strauch, B. A Counter-Current Heat-Exchange Reactor for the Thermal Stimulation of Hydrate-Bearing Sediments. Energies 2013, 6, 3002–3016. [Google Scholar] [CrossRef]
- Heeschen, K.U.; Abendroth, S.; Priegnitz, M.; Spangenberg, E.; Thaler, J.; Schicks, J.M. Gas Production from Methane Hydrate: A Laboratory Simulation of the Multistage Depressurization Test in Mallik, Northwest Territories, Canada. Energy Fuels 2016, 30, 6210–6219. [Google Scholar] [CrossRef]
- Spangenberg, E.; Priegnitz, M.; Heeschen, K.; Schicks, J.M. Are Laboratory-Formed Hydrate-Bearing Systems Analogous to Those in Nature? J. Chem. Eng. Data 2014, 60, 258–268. [Google Scholar] [CrossRef] [Green Version]
- Waite, W.; Winters, W.; Mason, D. Methane hydrate formation in partially water-saturated Ottawa sand. Am. Mineral. 2004, 89, 1202–1207. [Google Scholar] [CrossRef]
- Waite, W.; Bratton, P.M.; Mason, D.H. Laboratory formation of non-cementing, methane hydrate-bearing sands. In Proceedings of the 7th International Conference on Gas Hydrates (ICGH 2011), Edinburgh, UK, 17–21 July 2011. [Google Scholar]
- Choi, J.H.; Dai, S.; Cha, J.H.; Seol, Y. Laboratory formation of noncementing hydrates in sandy sediments. Geochem. Geophys. Geosystems 2014, 15, 1648–1656. [Google Scholar] [CrossRef]
- Yin, Z.; Chong, Z.R.; Hoon, K.T.; Linga, P. Effect of Multi-Stage Cooling on the Kinetic Behavior of Methane Hydrate Formation in Sandy Medium. Energy Procedia 2019, 158, 5374–5381. [Google Scholar] [CrossRef]
- Yin, Z.; Moridis, G.; Chong, Z.R.; Linga, P. Effectiveness of multi-stage cooling processes in improving the CH4–hydrate saturation uniformity in sandy laboratory samples. Appl. Energy 2019, 250, 729–747. [Google Scholar] [CrossRef] [Green Version]
- Kono, H.O.; Narasimhan, S.; Song, F.; Smith, D.H. Synthesis of methane gas hydrate in porous sediments and its dissociation by depressurizing. Powder Technol. 2002, 122, 239–246. [Google Scholar] [CrossRef]
- Fitzgerald, G.C.; Castaldi, M.J.; Schicks, J.M. Methane Hydrate Formation and Thermal Based Dissociation Behavior in Silica Glass Bead Porous Media. Ind. Eng. Chem. Res. 2014, 53, 6840–6854. [Google Scholar] [CrossRef]
- Gambelli, A.M.; Castellani, B.; Nicolini, A.; Rossi, F. Experimental study on natural gas hydrate exploitation: Optimization of methane recovery, carbon dioxide storage and deposit structure preservation. J. Pet. Sci. Eng. 2019, 177, 594–601. [Google Scholar] [CrossRef]
- Feng, J.C.; Li, B.; Li, X.S.; Wang, Y. Effects of depressurizing rate on methane hydrate dissociation within large-scale experimental simulator. Appl. Energy 2021, 304, 117750. [Google Scholar] [CrossRef]
- Cui, J.; Li, K.; Cheng, L.; Li, Q.; Sun, Z.; Xiao, P.; Li, X.; Chen, G.; Sun, C. Experimental investigation on the spatial differences of hydrate dissociation by depressurization in water-saturated methane hydrate reservoirs. Fuel 2021, 292, 120277. [Google Scholar] [CrossRef]
- Spangenberg, E.; Kulenkampff, J.; Naumann, R.; Erzinger, J. Pore space hydrate formation in a glass bead sample from methane dissolved in water. Geophys. Res. Lett. 2005, 32, L24301. [Google Scholar] [CrossRef]
- Waite, W.F.; Spangenberg, E. Gas hydrate formation rates from dissolved-phase methane in porous laboratory specimens. Geophys. Res. Lett. 2013, 40, 4310–4315. [Google Scholar] [CrossRef]
- Berge, L.I.; Jacobsen, K.A.; Solstad, A. Measured acoustic wave velocities of R11 (CCl3F) hydrate samples with and without sand as a function of hydrate concentration. J. Geophys. Res. Solid Earth 1999, 104, 15415–15424. [Google Scholar] [CrossRef]
- Stern, L.A.; Kirby, S.H.; Durham, W.B. Peculiarities of Methane Clathrate Hydrate Formation and Solid-State Deformation, Including Possible Superheating of Water Ice. Science 1996, 273, 1843–1848. [Google Scholar] [CrossRef]
- Spangenberg, E.; Heeschen, K.U.; Giese, R.; Schicks, J.M. “Ester”—A new ring-shear-apparatus for hydrate-bearing sediments. Rev. Sci. Instrum. 2020, 91, 064503. [Google Scholar] [CrossRef] [PubMed]
- Moridis, G.J. User’s Manual for the Hydrate v1.5 Option of TOUGH+ v1.5: A Code for the Simulation of System Behavior in Hydrate-Bearing Geologic Media; Technical Report; Lawrence Berkeley National Lab. (LBNL): Berkeley, CA, USA, 2014. [Google Scholar]
- White, M.; Kneafsey, T.; Seol, Y.; Waite, W.F.; Uchida, S.; Lin, J.; Myshakin, E.; Gai, X.; Gupta, S.; Reagan, M.; et al. An international code comparison study on coupled thermal, hydrologic and geomechanical processes of natural gas hydrate-bearing sediments. Mar. Pet. Geol. 2020, 120, 104566. [Google Scholar] [CrossRef]
- Moridis, G.J.; Kowalsky, M.B.; Pruess, K. HydrateResSim Users Manual: A Numerical Simulator for Modeling the Behavior of Hydrates in Geologic Media; Technical Report; Lawrence Berkeley National Lab.(LBNL): Berkeley, CA USA, 2005. [Google Scholar]
- Gamwo, I.K.; Liu, Y. Mathematical Modeling and Numerical Simulation of Methane Production in a Hydrate Reservoir. Ind. Eng. Chem. Res. 2010, 49, 5231–5245. [Google Scholar] [CrossRef]
- Zheng, R.; Li, S.; Li, X. Sensitivity analysis of hydrate dissociation front conditioned to depressurization and wellbore heating. Mar. Pet. Geol. 2018, 91, 631–638. [Google Scholar] [CrossRef]
- Wu, C.Y.; Hsieh, B.Z. Comparisons of different simulated hydrate designs for Class-1 gas hydrate deposits. J. Nat. Gas. Sci. Eng. 2020, 77, 103225. [Google Scholar] [CrossRef]
- Kempka, T. Verification of a Python-based TRANsport Simulation Environment for density-driven fluid flow and coupled transport of heat and chemical species. Adv. Geosci. 2020, 54, 67–77. [Google Scholar] [CrossRef]
- Uddin, M.; Wright, F.; Dallimore, S.; Coombe, D. Gas hydrate dissociations in Mallik hydrate bearing zones A, B, and C by depressurization: Effect of salinity and hydration number in hydrate dissociation. J. Nat. Gas. Sci. Eng. 2014, 21, 40–63. [Google Scholar] [CrossRef]
- Kowalsky, M.B.; Moridis, G.J. Comparison of kinetic and equilibrium reaction models in simulating gas hydrate behavior in porous media. Energy Convers. Manag. 2007, 48, 1850–1863. [Google Scholar] [CrossRef] [Green Version]
- Moridis, G. Numerical Studies of Gas Production From Methane Hydrates. SPE J. 2003, 8, 359–370. [Google Scholar] [CrossRef]
- Kashchiev, D.; Firoozabadi, A. Driving force for crystallization of gas hydrates. J. Cryst. Growth 2002, 241, 220–230. [Google Scholar] [CrossRef]
- Malagar, B.R.; Lijith, K.; Singh, D. Formation & dissociation of methane gas hydrates in sediments: A critical review. J. Nat. Gas Sci. Eng. 2019, 65, 168–184. [Google Scholar] [CrossRef]
- Smits, K.M.; Sakaki, T.; Limsuwat, A.; Illangasekare, T.H. Thermal conductivity of sands under varying moisture and porosity in drainage–wetting cycles. Vadose Zone J. 2010, 9, 172–180. [Google Scholar] [CrossRef]
- Huang, D.; Fan, S.; Liang, D.; Feng, Z. Gas Hydrate Formation and Its Thermal Conductivity Measurement. Chin. J. Geophys. 2005, 48, 1201–1207. [Google Scholar] [CrossRef]
- Gupta, A.; Lachance, J.; Sloan, E., Jr.; Koh, C.A. Measurements of methane hydrate heat of dissociation using high pressure differential scanning calorimetry. Chem. Eng. Sci. 2008, 63, 5848–5853. [Google Scholar] [CrossRef]
- Hu, X.; Guan, J.; Wang, Y.; Keating, A.; Yang, S. Comparison of boundary and size effect models based on new developments. Eng. Fract. Mech. 2017, 175, 146–167. [Google Scholar] [CrossRef]
- Dong, H.; Sun, J.; Lin, Z.; Fang, H.; Li, Y.; Cui, L.; Yan, W. 3D pore-type digital rock modeling of natural gas hydrate for permafrost and numerical simulation of electrical properties. J. Geophys. Eng. 2018, 15, 275–285. [Google Scholar] [CrossRef] [Green Version]
- Wetzel, M.; Kempka, T.; Kühn, M. Diagenetic Trends of Synthetic Reservoir Sandstone Properties Assessed by Digital Rock Physics. Minerals 2021, 11, 151. [Google Scholar] [CrossRef]
- Majorowicz, J.A.; Jones, F.W.; Judge, A.S. Deep subpermafrost thermal regime in the Mackenzie Delta basin, northern Canada—Analysis from petroleum bottom-hole temperature data. Geophysics 1990, 55, 362–371. [Google Scholar] [CrossRef]
- Spangenberg, E. Modeling of the influence of gas hydrate content on the electrical properties of porous sediments. J. Geophys. Res. Solid Earth 2001, 106, 6535–6548. [Google Scholar] [CrossRef]
- Kleinberg, R.L.; Flaum, C.; Griffin, D.D.; Brewer, P.G.; Malby, G.E.; Peltzer, E.T.; Yesinowski, J.P. Deep sea NMR: Methane hydrate growth habit in porous media and its relationship to hydraulic permeability, deposit accumulation, and submarine slope stability. J. Geophys. Res. Solid Earth 2003, 108. [Google Scholar] [CrossRef]
- Delli, M.L.; Grozic, J.L. Prediction Performance of Permeability Models in Gas-Hydrate-Bearing Sands. SPE J. 2013, 18, 274–284. [Google Scholar] [CrossRef]
- Nagashima, A. Viscosity of water substance—New international formulation and its background. J. Phys. Chem. Ref. Data 1977, 6, 1133–1166. [Google Scholar] [CrossRef] [Green Version]
- Phillips, S.L. A Technical Databook for Geothermal Energy Utilization; Technical Report; Lawrence Berkeley National Lab. (LBNL): Berkeley, CA, USA, 1981. [Google Scholar]
- Wagner, W.; Pruß, A. The IAPWS Formulation 1995 for the Thermodynamic Properties of Ordinary Water Substance for General and Scientific Use. J. Phys. Chem. Ref. Data 2002, 31, 387–535. [Google Scholar] [CrossRef] [Green Version]
- Saul, A.; Wagner, W. A Fundamental Equation for Water Covering the Range from the Melting Line to 1273 K at Pressures up to 25 000 MPa. J. Phys. Chem. Ref. Data 1989, 18, 1537–1564. [Google Scholar] [CrossRef]
- Kell, G.S. Density, thermal expansivity, and compressibility of liquid water from 0.deg. to 150.deg. Correlations and tables for atmospheric pressure and saturation reviewed and expressed on 1968 temperature scale. J. Phys. Chem. Ref. Data 1975, 20, 97–105. [Google Scholar] [CrossRef]
- O’Sullivan, M.; Bodvarsson, G.; Pruess, K.; Blakeley, M. Fluid and Heat Flow In Gas-Rich Geothermal Reservoirs. Soc. Pet. Eng. J. 1985, 25, 215–226. [Google Scholar] [CrossRef] [Green Version]
- Michaelides, E.E. Thermodynamic properties of geothermal fluids. Trans.-Geotherm. Resour. Counc. 1981, 5, 316–364. [Google Scholar]
- Gudmundsson, J.S.; Thráinsson, H. Power potential of two-phase geothermal wells. Geothermics 1989, 18, 357–366. [Google Scholar] [CrossRef]
- Kamath, V.A. Study of Heat Transfer Characteristics during Dissociation of Gas Hydrates in Porous Media. Ph.D. Thesis, University of Pittsburgh, Pittsburgh, PA, USA, 1984. [Google Scholar]
- Battistelli, A.; Calore, C.; Pruess, K. The simulator TOUGH2/EWASG for modelling geothermal reservoirs with brines and non-condensible gas. Geothermics 1997, 26, 437–464. [Google Scholar] [CrossRef]
- Cramer, S.D. Solubility of methane in brines from 0 to 300.degree.C. Ind. Eng. Chem. Process Des. Dev. 1984, 23, 533–538. [Google Scholar] [CrossRef]
- Makogon, Y.F. Hydrates of Hydrocarbons; PennWell Publishing Co.: Tulsa, OK, USA, 1997. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Experimental Systems | LSHV [16,38] | LARS [15,26,27,30,39,40,41] | GHASTLI [13,42] | USGS-DOE [43,44] | NUS [3,17,45,46] |
|---|---|---|---|---|---|
| Sample volume (L) | 70 | 210 | 0.5 | 0.24 | 0.98 |
| Specimen materials | Quartz sand | Quartz sand | Ottawa sand | Quartz sand | Silica sand |
| Gas hydrate-bearing sediment types | Gas-rich permafrost sediments | Hydrate-rich permafrost sediments | Gas-rich sediments | Hydrate-rich marine sediments | Water-dominated sediments |
| Hydrate-forming gas | CH4 | CH4 | CH4 | CH4 | CH4 |
| Hydrate formation methods | “excess-gas” | “dissolved-gas” | “excess-gas” | “excess-gas” /“dissolved-gas” | “excess-water” |
| Hydrate habits | Load-bearing /Cementing | Pore-filling | Cementing | Cementing /Pore-filling | Load-bearing |
| Maximum bulk hydrate saturation (% of pore space) | ∼33 | ∼90 | ∼70 | - | ∼40 |
| T0 | T1 | T2 | T3 | T4 | T5 | |
|---|---|---|---|---|---|---|
| Location (m) (radius, height) | (0.15, 1.28) | (0.15, 1.20) | (0.02, 1.05) | (0.16, 1.05) | (0.14, 0.85) | (0.15, 0.59) |
| Correction of measured T (°C) | 3.2 | 3.0 | 3.0 | 3.1 | 3.2 | 3.3 |
| T6 | T7 | T8 | T9 | T11 | T12 | |
| Location (m) (radius, height) | (0.14, 0.59) | (0.16, 0.44) | (0.06, 0.44) | (0.03, 0.44) | (0.22, 0.44) | (0.0, 0.35) |
| Correction of measured T (°C) | 3.3 | 3.5 | 3.3 | 3.6 | 3.7 | 3.7 |
| Parameters | Value | Unit | Reference |
|---|---|---|---|
| Intrinsic permeability of porous medium | 500 | Darcy | [40] |
| Intrinsic porosity of porous medium | 0.35 | - | [40] |
| Salinity of pore fluid | 5 | kg m−3 | [40] |
| Initial pore pressure | 11 | MPa | [27] |
| Density of quartz sand | 2650 | kg m−3 | [3] |
| Thermal conductivity of wet sand | 2.36 | W m−1 K−1 | [69] |
| Thermal conductivity of CH4 hydrate | 0.68 | W m−1 K−1 | [70] |
| Specific heat of quartz sand | 830 | J kg−1 K−1 | [37] |
| Specific heat of CH4 hydrate | 2100 | J kg−1 K−1 | [37] |
| Diffusion coefficient | m2 s−1 | Assumed | |
| Density of inhibitor (NaCl) | 2160 | kg m−3 | [59] |
| Compressibility of porous medium | Pa−1 | Assumed |
| Variable | Range | Precision | Unit |
|---|---|---|---|
| Fluid pressure | [11, 11.1] | MPa | |
| External coolant temperature | [3.5, 4.0] | °C | |
| Inflowing fluid temperature | [13.6, 16] | °C | |
| Dissolved CH4 concentration | [60, 90] | % of CH4 solubility limit at given p-T conditions | |
| Initial inflowing fluid rate | [50, 100] | Liters per day |
| T0 | T1 | T2 | T3 | T4 | T5 | |
|---|---|---|---|---|---|---|
| Revised location (m) (radius, height) | (0.18, 1.27) | (0.14, 1.2) | (0.02, 1.05) | (0.15, 1.07) | (0.13, 0.84) | (0.14, 0.6) |
| Displacement of relocation (m) | 0.03 | 0.01 | 0.008 | 0.02 | 0.016 | 0.013 |
| T6 | T7 | T8 | T9 | T11 | T12 | |
| Revised location (m) (radius, height) | (0.13, 0.6) | (0.14, 0.44) | (0.07, 0.43) | (0.03, 0.44) | (0.18, 0.43) | (0.08, 0.34) |
| Displacement of relocation (m) | 0.028 | 0.018 | 0.028 | 0.036 | 0.032 | 0.085 |
| Substage | Interval (hours) | Inflowing Fluid Temperatures (°C) | Inflowing Fluid Rates (Liters per Day) | Dissolved CH4 Concentrations (kg m−3) |
|---|---|---|---|---|
| I-1-1 | 0–0.8–33.4 | 13.6 | 97.0 | 0–2.69 |
| I-1-2 | 33.4–47.5 | - | - | - |
| I-2-1 | 47.5–48.5–60.0 | 12.5 | 64.7 | 1.20–2.41 |
| I-2-2 | 60.0–95.3 | - | - | - |
| II-1 | 95.3–97.0–144.5–153.2 | 13.8 | 56.7–55.9–56.7 | 1.20–2.55–2.41 |
| II-2 | 97.0–193.5 | - | - | - |
| III-1 | 193.5–195.0–215.0–249.0–265.0–282.0–310.5–314.0–333.8–335.0 | 14.5–14.3–14.3–14.5–14.0–14.5 –16.0–15.5–15.5 | 76.8–76.8–68.3–52.7–49.8–46.1–58.5–57.6–59.5 | 1.20–2.03–2.01–2.03–2.01–1.96–1.89–1.93–1.20 |
| III-2 | 335.0–360 | - | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Z.; Spangenberg, E.; Schicks, J.M.; Kempka, T. Numerical Simulation of Hydrate Formation in the LArge-Scale Reservoir Simulator (LARS). Energies 2022, 15, 1974. https://doi.org/10.3390/en15061974
Li Z, Spangenberg E, Schicks JM, Kempka T. Numerical Simulation of Hydrate Formation in the LArge-Scale Reservoir Simulator (LARS). Energies. 2022; 15(6):1974. https://doi.org/10.3390/en15061974
Chicago/Turabian StyleLi, Zhen, Erik Spangenberg, Judith M. Schicks, and Thomas Kempka. 2022. "Numerical Simulation of Hydrate Formation in the LArge-Scale Reservoir Simulator (LARS)" Energies 15, no. 6: 1974. https://doi.org/10.3390/en15061974
APA StyleLi, Z., Spangenberg, E., Schicks, J. M., & Kempka, T. (2022). Numerical Simulation of Hydrate Formation in the LArge-Scale Reservoir Simulator (LARS). Energies, 15(6), 1974. https://doi.org/10.3390/en15061974