
Hydrogenation of CO2 or CO2 Derivatives to Methanol under Molecular Catalysis: A Review



Abstract
:1. Introduction
2. Hydrogenation of CO2 or CO2 Derivatives
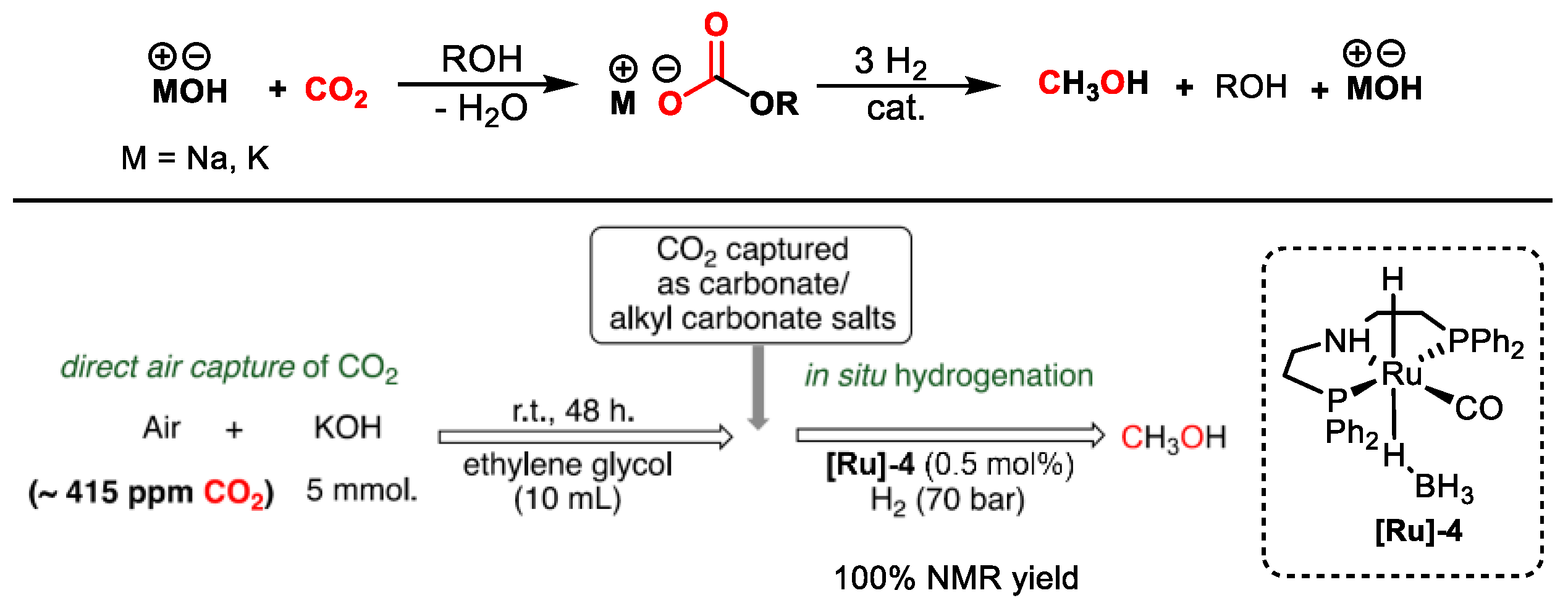
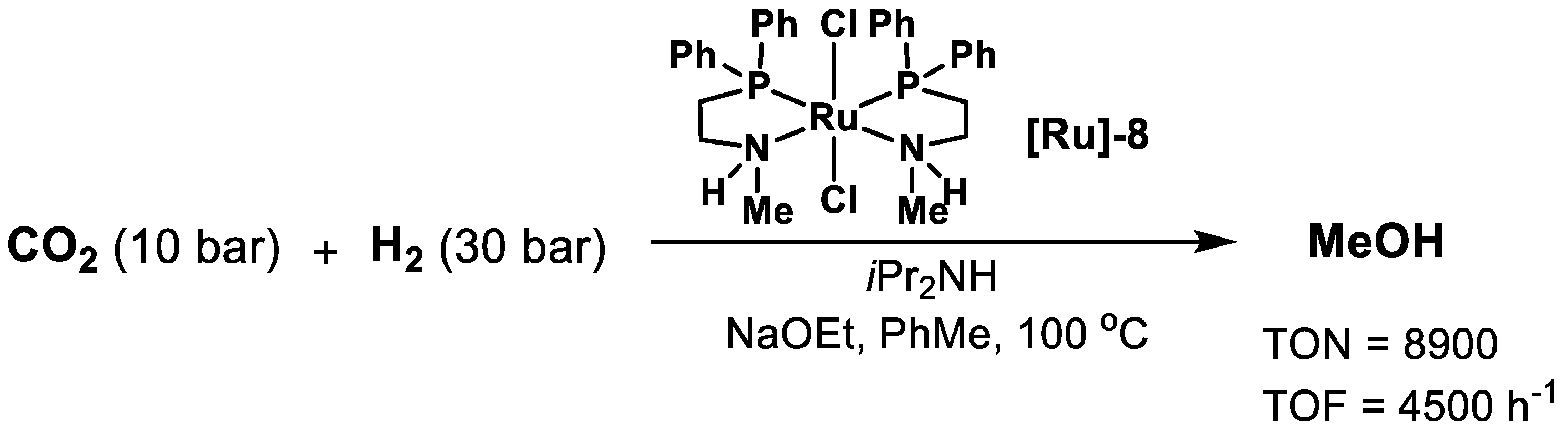
2.1. Ru-Based Catalysts
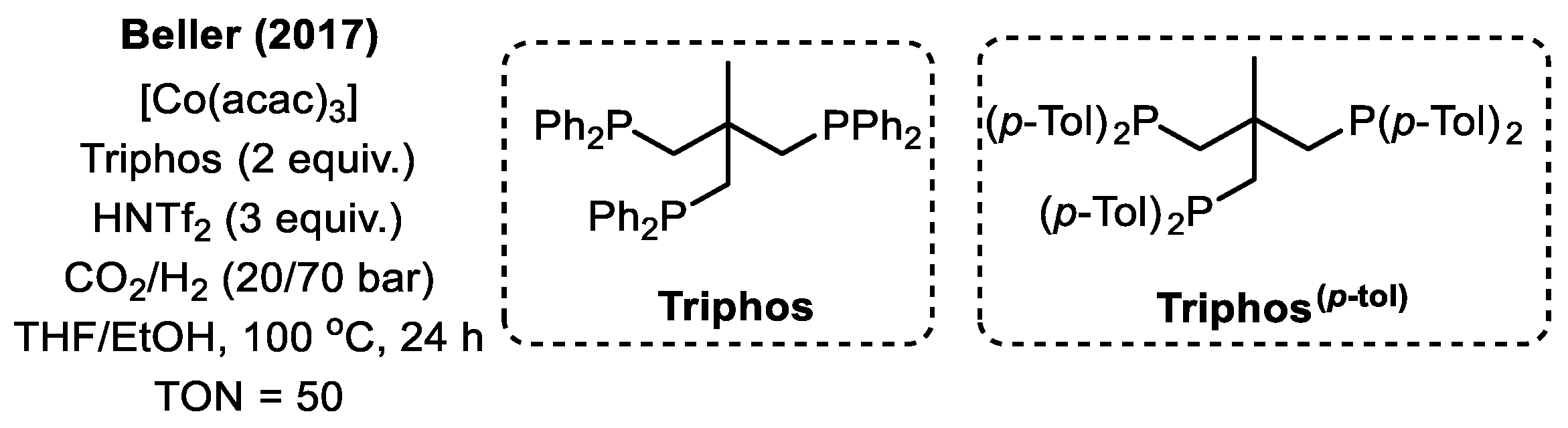
2.2. Co-Based Catalysts
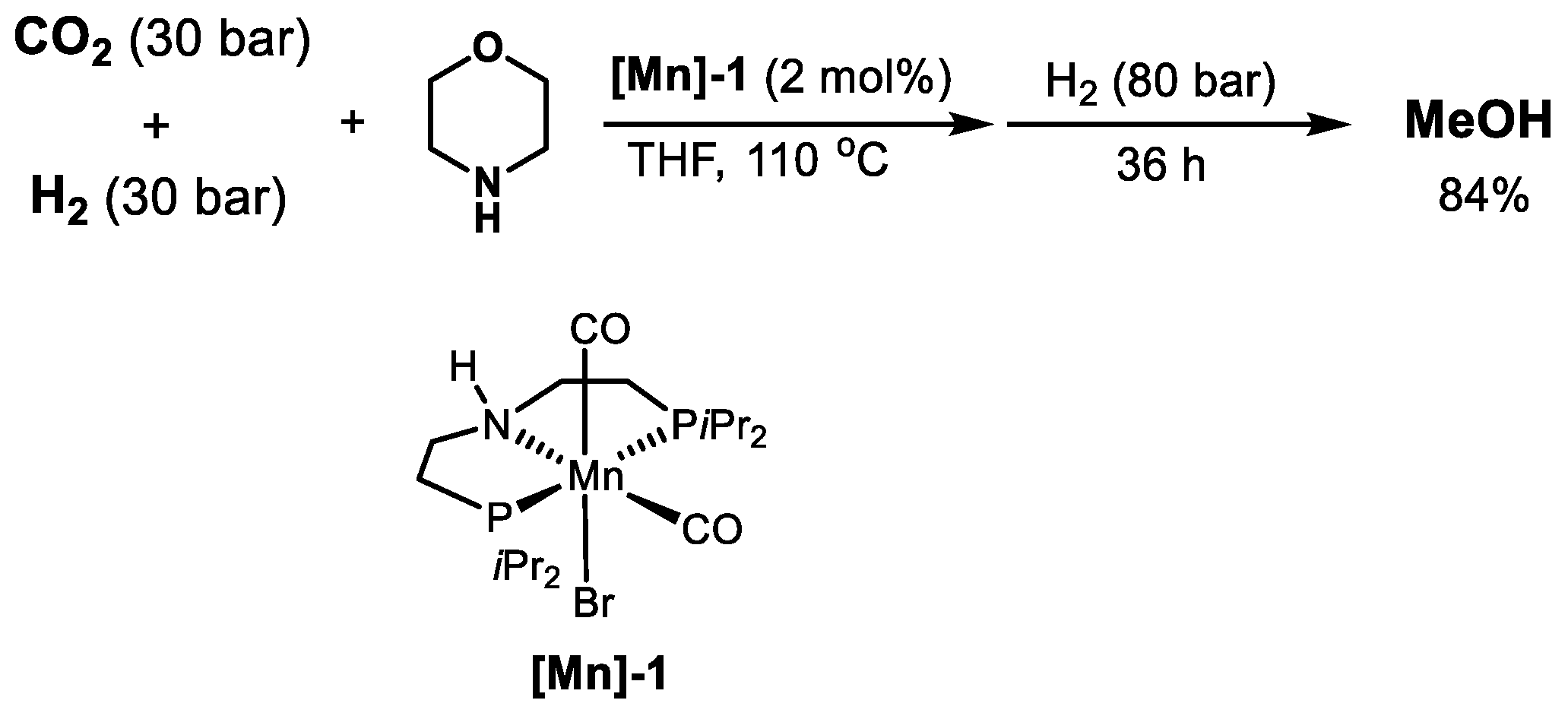
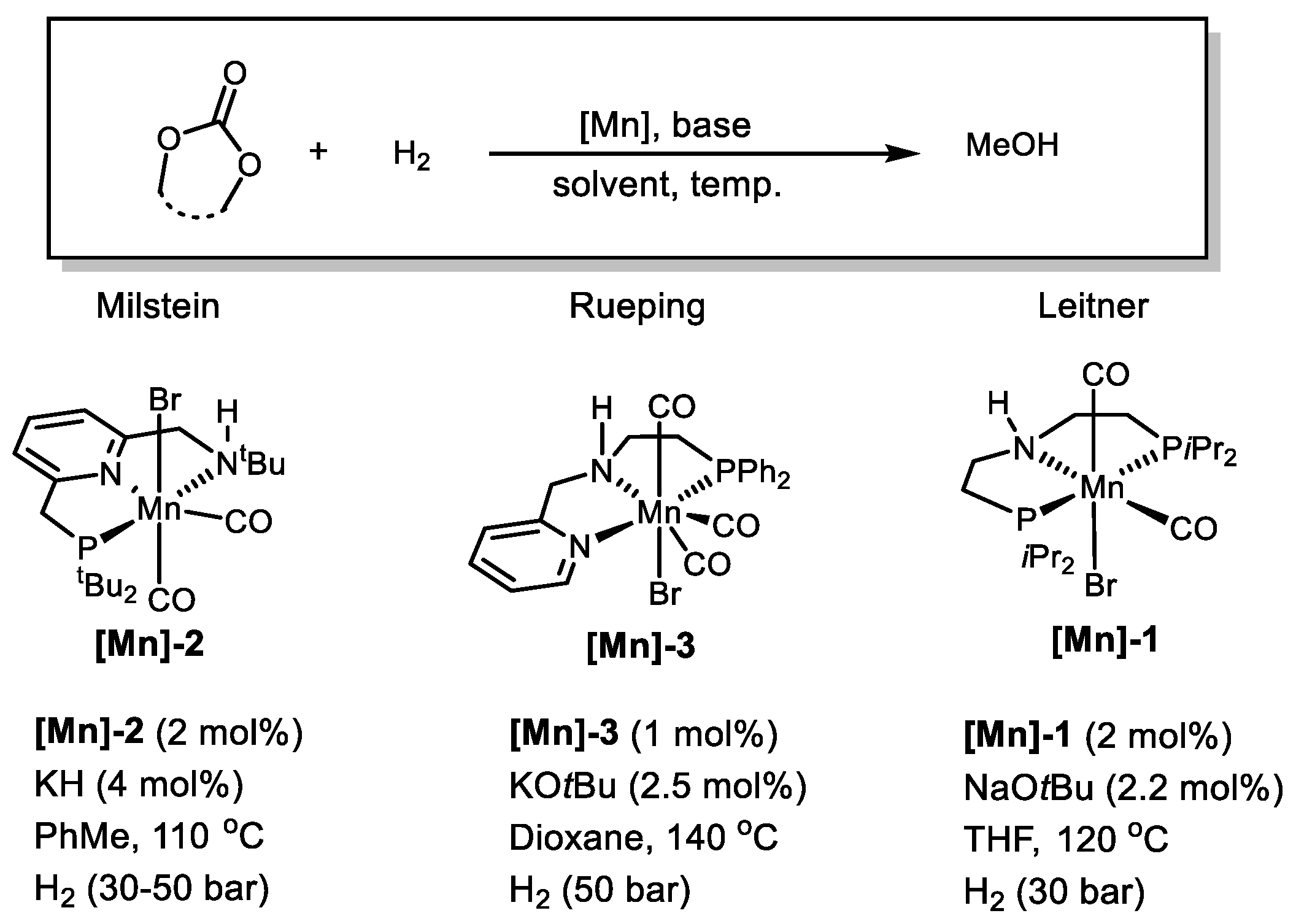
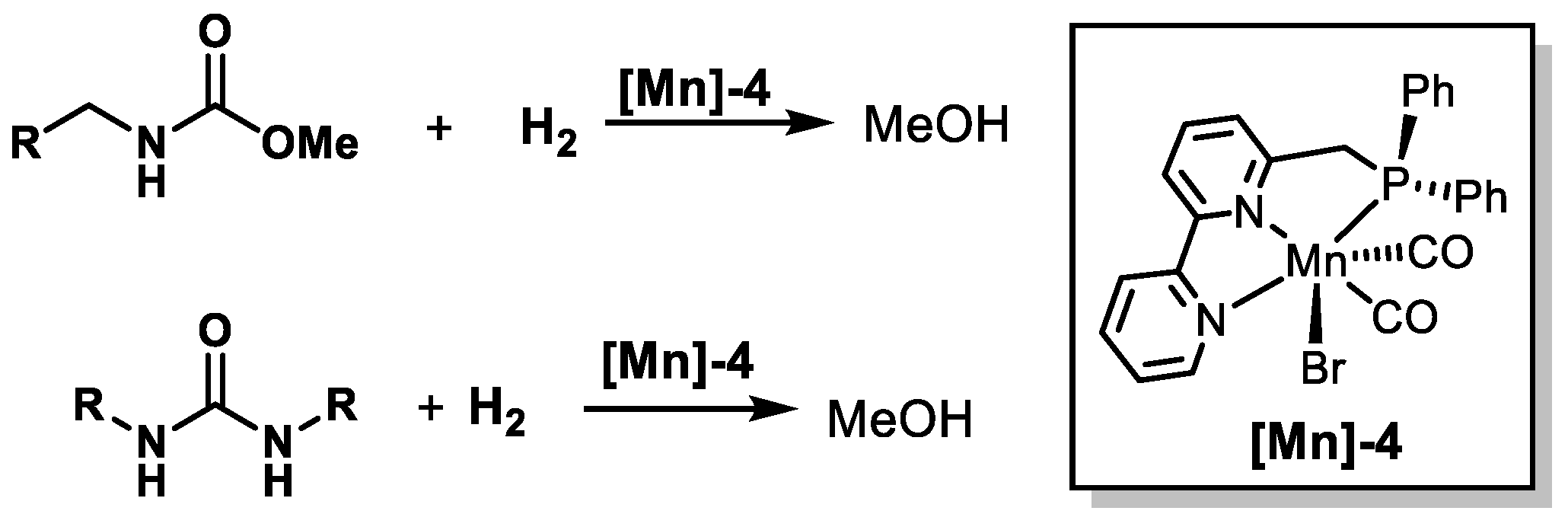
2.3. Mn-Based Catalysts
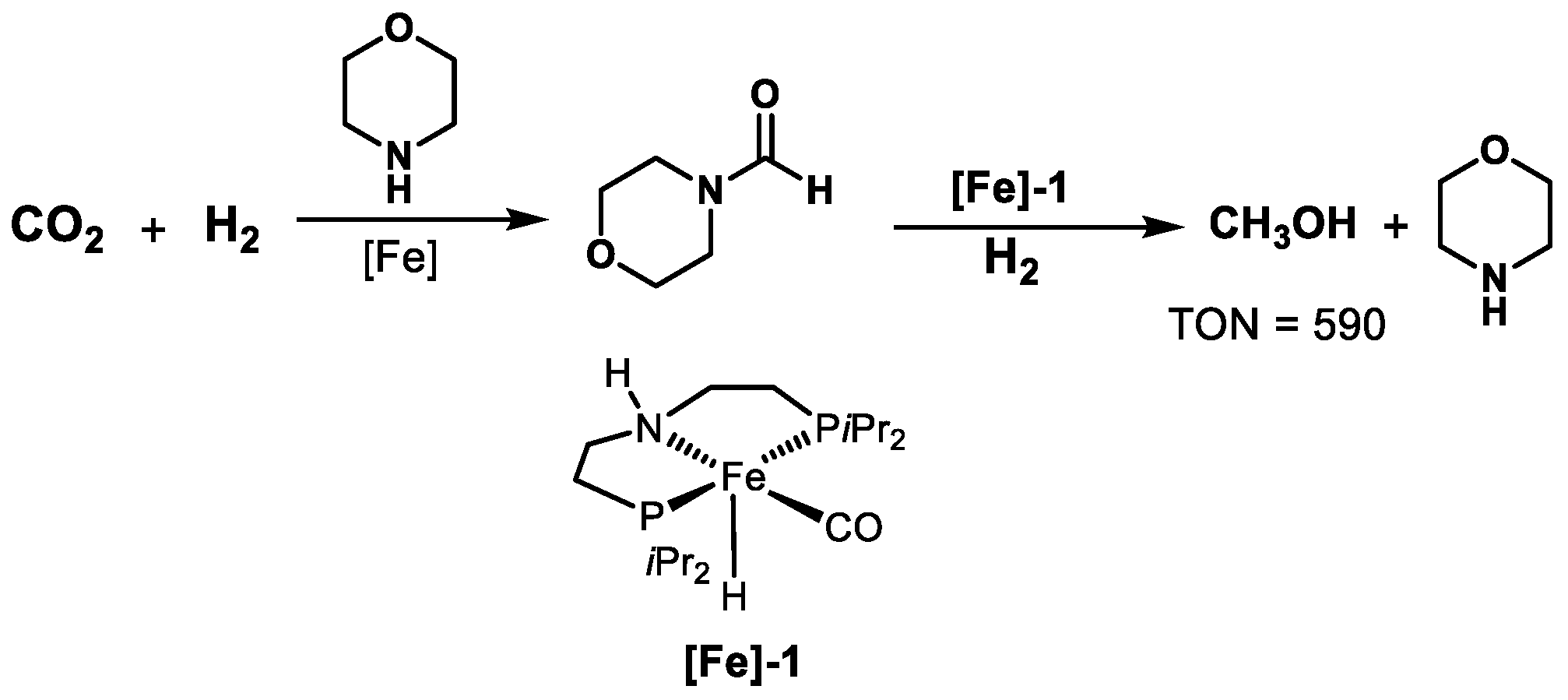
2.4. Fe-Based Catalysts
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Schneider, S.H. The Greenhouse Effect: Science and Policy. Science 1989, 243, 771–781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aresta, M.; Dibenedetto, A.; Angelini, A. Catalysis for the Valorization of Exhaust Carbon: From CO2 to Chemicals, Materials, and Fuels. Technological Use of CO2. Chem. Rev. 2014, 114, 1709–1742. [Google Scholar] [CrossRef] [PubMed]
- Peters, M.; Kohler, B.; Kuckshinrichs, W.; Leitner, W.; Markewitz, P.; Muller, T.E. Chemical Technologies for Exploiting and Recycling Carbon Dioxide into the Value Chain. ChemSusChem 2011, 4, 1216–1240. [Google Scholar] [CrossRef] [PubMed]
- Markewitz, P.; Kuckshinrichs, W.; Leitner, W.; Linssen, J.; Zapp, P.; Bongartz, R.; Schreiber, A.; Muller, T.E. Worldwide innovations in the development of carbon capture technologies and the utilization of CO2. Energy Environ. Sci. 2012, 5, 7281–7305. [Google Scholar] [CrossRef] [Green Version]
- Olah, G.A. The methanol economy. Chem. Eng. News 2003, 81, 5. [Google Scholar] [CrossRef]
- Olah, G.A. Beyond oil and gas: The methanol economy. Angew. Chem. Int. Ed. 2005, 44, 2636–2639. [Google Scholar] [CrossRef] [PubMed]
- Olah, G.A.; Prakash, G.K.S.; Goeppert, A. Anthropogenic Chemical Carbon Cycle for a Sustainable Future. J. Am. Chem. Soc. 2011, 133, 12881–12898. [Google Scholar] [CrossRef]
- Olah, G.A. Towards Oil Independence Through Renewable Methanol Chemistry. Angew. Chem. Int. Ed. 2013, 52, 104–107. [Google Scholar] [CrossRef]
- Ganesh, I. Conversion of carbon dioxide into methanol—A potential liquid fuel: Fundamental challenges and opportunities (a review). Renew. Sustain. Energy Rev. 2014, 31, 221–257. [Google Scholar] [CrossRef]
- Garg, N.; Paira, S.; Sundararaju, B. Efficient Transfer Hydrogenation of Ketones using Methanol as Liquid Organic Hydrogen Carrier. ChemCatChem 2020, 12, 3472–3476. [Google Scholar] [CrossRef]
- Xie, Y.; Hu, P.; Ben-David, Y.; Milstein, D. A Reversible Liquid Organic Hydrogen Carrier System Based on Methanol-Ethylenediamine and Ethylene Urea. Angew. Chem. Int. Ed. 2019, 58, 5105–5109. [Google Scholar] [CrossRef] [PubMed]
- Johnson, T.C.; Morris, D.J.; Wills, M. Hydrogen generation from formic acid and alcohols using homogeneous catalysts. Chem. Soc. Rev. 2010, 39, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Trincado, M.; Bösken, J.; Grützmacher, H. Homogeneously catalyzed acceptorless dehydrogenation of alcohols: A progress report. Coordin. Chem. Rev. 2021, 443, 213967. [Google Scholar] [CrossRef]
- Nielsen, M.; Alberico, E.; Baumann, W.; Drexler, H.-J.; Junge, H.; Gladiali, S.; Beller, M. Low-temperature aqueous-phase methanol dehydrogenation to hydrogen and carbon dioxide. Nature 2013, 495, 85–89. [Google Scholar] [CrossRef]
- Rodríguez-Lugo, R.E.; Trincado, M.; Vogt, M.; Tewes, F.; Santiso-Quinones, G.; Grützmacher, H. A homogeneous transition metal complex for clean hydrogen production from methanol–water mixtures. Nat. Chem. 2013, 5, 342–347. [Google Scholar] [CrossRef]
- Cortright, R.D.; Davda, R.R.; Dumesic, J.A. Hydrogen from catalytic reforming of biomass-derived hydrocarbons in liquid water. Nature 2002, 418, 964–967. [Google Scholar] [CrossRef]
- Palo, D.R.; Dagle, R.A.; Holladay, J.D. Methanol steam reforming for hydrogen production. Chem. Rev. 2007, 107, 3992–4021. [Google Scholar] [CrossRef]
- Sa, S.; Silva, H.; Brandao, L.; Sousa, J.M.; Mendes, A. Catalysts for methanol steam reforming—A review. Appl. Catal. B Environ. 2010, 99, 43–57. [Google Scholar] [CrossRef]
- Behrens, M.; Studt, F.; Kasatkin, I.; Kühl, S.; Hävecker, M.; Abild-Pedersen, F.; Zander, S.; Girgsdies, F.; Kurr, P.; Kniep, B.-L.; et al. The Active Site of Methanol Synthesis over Cu/ZnO/Al2O3 Industrial Catalysts. Science 2012, 336, 893–897. [Google Scholar] [CrossRef]
- Bai, S.-T.; De Smet, G.; Liao, Y.; Sun, R.; Zhou, C.; Beller, M.; Maes, B.U.W.; Sels, B.F. Homogeneous and heterogeneous catalysts for hydrogenation of CO2 to methanol under mild conditions. Chem. Soc. Rev. 2021, 50, 4259–4298. [Google Scholar] [CrossRef]
- Li, Y.-N.; Ma, R.; He, L.-N.; Diao, Z.-F. Homogeneous hydrogenation of carbon dioxide to methanol. Catal. Sci. Technol. 2014, 4, 1498–1512. [Google Scholar] [CrossRef]
- Alberico, E.; Nielsen, M. Towards a methanol economy based on homogeneous catalysis: Methanol to H2 and CO2 to methanol. Chem. Commun. 2015, 51, 6714–6725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, K.; Razzaq, R.; Hu, Y.; Ding, K. Homogeneous Reduction of Carbon Dioxide with Hydrogen. Top. Curr. Chem. 2017, 375, 1–26. [Google Scholar]
- Sordakis, K.; Tang, C.; Vogt, L.K.; Junge, H.; Dyson, P.J.; Beller, M.; Laurenczy, G. Homogeneous Catalysis for Sustainable Hydrogen Storage in Formic Acid and Alcohols. Chem. Rev. 2018, 118, 372–433. [Google Scholar] [CrossRef] [PubMed]
- Bernskoetter, W.H.; Hazari, N. Reversible Hydrogenation of Carbon Dioxide to Formic Acid and Methanol: Lewis Acid Enhancement of Base Metal Catalysts. Acc. Chem. Res. 2017, 50, 1049–1058. [Google Scholar] [CrossRef]
- Kar, S.; Goeppert, A.; Prakash, G.K.S. Integrated CO2 Capture and Conversion to Formate and Methanol: Connecting Two Threads. Acc. Chem. Res. 2019, 52, 2892–2903. [Google Scholar] [CrossRef]
- Kar, S.; Kothandaraman, J.; Goeppert, A.; Prakash, G.K.S. Advances in catalytic homogeneous hydrogenation of carbon dioxide to methanol. J. CO2 Util. 2018, 23, 212–218. [Google Scholar] [CrossRef]
- Xie, S.; Zhang, W.; Lan, X.; Lin, H. CO2 Reduction to Methanol in the Liquid Phase: A Review. ChemSusChem 2020, 13, 6141–6159. [Google Scholar]
- Wang, W.-H.; Himeda, Y.; Muckerman, J.T.; Manbeck, G.F.; Fujita, E. CO2 Hydrogenation to Formate and Methanol as an Alternative to Photo- and Electrochemical CO2 Reduction. Chem. Rev. 2015, 115, 12936–12973. [Google Scholar] [CrossRef]
- Ashley, A.E.; Thompson, A.L.; O’Hare, D. Non-Metal-Mediated Homogeneous Hydrogenation of CO2 to CH3OH. Angew. Chem. Int. Ed. 2009, 48, 9839–9843. [Google Scholar] [CrossRef] [Green Version]
- Courtemanche, M.A.; Legare, M.A.; Maron, L.; Fontaine, F.G. Reducing CO2 to Methanol Using Frustrated Lewis Pairs: On the Mechanism of Phosphine-Borane-Mediated Hydroboration of CO2. J. Am. Chem. Soc. 2014, 136, 10708–10717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fontaine, F.G.; Courtemanche, M.A.; Legare, M.A. Transition-Metal-Free Catalytic Reduction of Carbon Dioxide. Chem. Eur. J. 2014, 20, 2990–2996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sgro, M.J.; Stephan, D.W. Synthesis and reactivity of ruthenium tridentate bis-phosphinite ligand complexes. Dalton Trans. 2013, 42, 10460–10472. [Google Scholar] [CrossRef] [PubMed]
- Riduan, S.N.; Zhang, Y.G.; Ying, J.Y. Conversion of Carbon Dioxide into Methanol with Silanes over N-Heterocyclic Carbene Catalysts. Angew. Chem. Int. Ed. 2009, 48, 3322–3325. [Google Scholar] [CrossRef]
- Riduan, S.N.; Ying, J.Y.; Zhang, Y.G. Mechanistic Insights into the Reduction of Carbon Dioxide with Silanes over N-Heterocyclic Carbene Catalysts. ChemCatChem 2013, 5, 1490–1496. [Google Scholar] [CrossRef]
- Huang, F.; Lu, G.; Zhao, L.L.; Li, H.X.; Wang, Z.X. The Catalytic Role of N-Heterocyclic Carbene in a Metal-Free Conversion of Carbon Dioxide into Methanol: A Computational Mechanism Study. J. Am. Chem. Soc. 2010, 132, 12388–12396. [Google Scholar] [CrossRef]
- Chakraborty, S.; Zhang, J.; Krause, J.A.; Guan, H.R. An Efficient Nickel Catalyst for the Reduction of Carbon Dioxide with a Borane. J. Am. Chem. Soc. 2010, 132, 8872–8873. [Google Scholar] [CrossRef]
- Gomes, C.D.; Blondiaux, E.; Thuery, P.; Cantat, T. Metal-Free Reduction of CO2 with Hydroboranes: Two Efficient Pathways at Play for the Reduction of CO2 to Methanol. Chem. Eur. J. 2014, 20, 7098–7106. [Google Scholar] [CrossRef]
- Tominaga, K.-i.; Sasaki, Y.; Kawai, M.; Watanabe, T.; Saito, M. Ruthenium complex catalysed hydrogenation of carbon dioxide to carbon monoxide, methanol and methane. J. Chem. Soc. Chem. Commun. 1993, 629–631. [Google Scholar] [CrossRef]
- Tominaga, K.-i.; Sasaki, Y.; Watanabe, T.; Saito, M. Homogeneous Hydrogenation of Carbon Dioxide to Methanol Catalyzed by Ruthenium Cluster Anions in the Presence of Halide Anions. Bull. Chem. Soc. Jpn. 1995, 68, 2837–2842. [Google Scholar] [CrossRef]
- Huff, C.A.; Sanford, M.S. Cascade Catalysis for the Homogeneous Hydrogenation of CO2 to Methanol. J. Am. Chem. Soc. 2011, 133, 18122–18125. [Google Scholar] [CrossRef] [PubMed]
- Chu, W.-Y.; Culakova, Z.; Wang, B.T.; Goldberg, K.I. Acid-Assisted Hydrogenation of CO2 to Methanol in a Homogeneous Catalytic Cascade System. ACS Catal. 2019, 9, 9317–9326. [Google Scholar] [CrossRef]
- Wesselbaum, S.; Vom Stein, T.; Klankermayer, J.; Leitner, W. Hydrogenation of Carbon Dioxide to Methanol by Using a Homogeneous Ruthenium–Phosphine Catalyst. Angew. Chem. Int. Ed. 2012, 51, 7499–7502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schieweck, B.G.; Juerling-Will, P.; Klankermayer, J. Structurally Versatile Ligand System for the Ruthenium Catalyzed One-Pot Hydrogenation of CO2 to Methanol. ACS Catal. 2020, 10, 3890–3894. [Google Scholar] [CrossRef]
- Rezayee, N.M.; Huff, C.A.; Sanford, M.S. Tandem Amine and Ruthenium-Catalyzed Hydrogenation of CO2 to Methanol. J. Am. Chem. Soc. 2015, 137, 1028–1031. [Google Scholar] [CrossRef] [PubMed]
- Khusnutdinova, J.R.; Garg, J.A.; Milstein, D. Combining Low-Pressure CO2 Capture and Hydrogenation to Form Methanol. ACS Catal. 2015, 5, 2416–2422. [Google Scholar] [CrossRef]
- Zhang, L.; Han, Z.; Zhao, X.; Wang, Z.; Ding, K. Highly Efficient Ruthenium-Catalyzed N-Formylation of Amines with H2 and CO2. Angew. Chem. Int. Ed. 2015, 54, 6186–6189. [Google Scholar] [CrossRef]
- Kar, S.; Goeppert, A.; Prakash, G.K.S. Catalytic Homogeneous Hydrogenation of CO to Methanol via Formamide. J. Am. Chem. Soc. 2019, 141, 12518–12521. [Google Scholar] [CrossRef]
- Kothandaraman, J.; Goeppert, A.; Czaun, M.; Olah, G.A.; Prakash, G.K.S. Conversion of CO2 from Air into Methanol Using a Polyamine and a Homogeneous Ruthenium Catalyst. J. Am. Chem. Soc. 2016, 138, 778–781. [Google Scholar] [CrossRef]
- Kar, S.; Sen, R.; Goeppert, A.; Prakash, G.K.S. Integrative CO2 Capture and Hydrogenation to Methanol with Reusable Catalyst and Amine: Toward a Carbon Neutral Methanol Economy. J. Am. Chem. Soc. 2018, 140, 1580–1583. [Google Scholar] [CrossRef]
- Sen, R.; Goeppert, A.; Kar, S.; Prakash, G.K.S. Hydroxide Based Integrated CO2 Capture from Air and Conversion to Methanol. J. Am. Chem. Soc. 2020, 142, 4544–4549. [Google Scholar] [CrossRef] [PubMed]
- Kar, S.; Goeppert, A.; Prakash, G.K.S. Combined CO2 Capture and Hydrogenation to Methanol: Amine Immobilization Enables Easy Recycling of Active Elements. ChemSusChem 2019, 12, 3172–3177. [Google Scholar] [CrossRef] [PubMed]
- Everett, M.; Wass, D.F. Highly productive CO2 hydrogenation to methanol—A tandem catalytic approach via amide intermediates. Chem. Commun. 2017, 53, 9502–9504. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, A.; Watari, R.; Kuwata, S.; Kayaki, Y. Poly(ethyleneimine)-Mediated Consecutive Hydrogenation of Carbon Dioxide to Methanol with Ru Catalysts. Eur. J. Inorg. Chem. 2019, 2019, 2375–2380. [Google Scholar] [CrossRef]
- Balaraman, E.; Gunanathan, C.; Zhang, J.; Shimon, L.J.W.; Milstein, D. Efficient hydrogenation of organic carbonates, carbamates and formates indicates alternative routes to methanol based on CO2 and CO. Nat. Chem. 2011, 3, 609–614. [Google Scholar] [CrossRef]
- Balaraman, E.; Ben-David, Y.; Milstein, D. Unprecedented Catalytic Hydrogenation of Urea Derivatives to Amines and Methanol. Angew. Chem. Int. Ed. 2011, 50, 11702–11705. [Google Scholar] [CrossRef]
- Han, Z.; Rong, L.; Wu, J.; Zhang, L.; Wang, Z.; Ding, K. Catalytic Hydrogenation of Cyclic Carbonates: A Practical Approach from CO2 and Epoxides to Methanol and Diols. Angew. Chem. Int. Ed. 2012, 51, 13041–13045. [Google Scholar] [CrossRef]
- van der Vlugt, J.I. Cooperative Catalysis with First-Row Late Transition Metals. Eur. J. Inorg. Chem. 2012, 2012, 363–375. [Google Scholar] [CrossRef]
- Dalle, K.E.; Warnan, J.; Leung, J.J.; Reuillard, B.; Karmel, I.S.; Reisner, E. Electro- and Solar-Driven Fuel Synthesis with First Row Transition Metal Complexes. Chem. Rev. 2019, 119, 2752–2875. [Google Scholar] [CrossRef]
- Taylor, L.J.; Kays, D.L. Low-coordinate first-row transition metal complexes in catalysis and small molecule activation. Dalton Trans. 2019, 48, 12365–12381. [Google Scholar] [CrossRef] [Green Version]
- Schneidewind, J.; Adam, R.; Baumann, W.; Jackstell, R.; Beller, M. Low-Temperature Hydrogenation of Carbon Dioxide to Methanol with a Homogeneous Cobalt Catalyst. Angew. Chem. Int. Ed. 2017, 56, 1890–1893. [Google Scholar] [CrossRef] [PubMed]
- Scharnagl, F.K.; Hertrich, M.F.; Neitzel, G.; Jackstell, R.; Beller, M. Homogeneous Catalytic Hydrogenation of CO2 to Methanol—Improvements with Tailored Ligands. Adv. Synth. Catal. 2019, 361, 374–379. [Google Scholar]
- Kar, S.; Goeppert, A.; Kothandaraman, J.; Prakash, G.K.S. Manganese-Catalyzed Sequential Hydrogenation of CO2 to Methanol via Formamide. ACS Catal. 2017, 7, 6347–6351. [Google Scholar] [CrossRef]
- Kumar, A.; Janes, T.; Espinosa-Jalapa, N.A.; Milstein, D. Manganese Catalyzed Hydrogenation of Organic Carbonates to Methanol and Alcohols. Angew. Chem. Int. Ed. 2018, 57, 12076–12080. [Google Scholar] [CrossRef] [PubMed]
- Zubar, V.; Lebedev, Y.; Azofra, L.M.; Cavallo, L.; El-Sepelgy, O.; Rueping, M. Hydrogenation of CO2-Derived Carbonates and Polycarbonates to Methanol and Diols by Metal–Ligand Cooperative Manganese Catalysis. Angew. Chem. Int. Ed. 2018, 57, 13439–13443. [Google Scholar] [CrossRef] [PubMed]
- Kaithal, A.; Hölscher, M.; Leitner, W. Catalytic Hydrogenation of Cyclic Carbonates using Manganese Complexes. Angew. Chem. Int. Ed. 2018, 57, 13449–13453. [Google Scholar] [CrossRef] [Green Version]
- Das, U.K.; Kumar, A.; Ben-David, Y.; Iron, M.A.; Milstein, D. Manganese Catalyzed Hydrogenation of Carbamates and Urea Derivatives. J. Am. Chem. Soc. 2019, 141, 12962–12966. [Google Scholar] [CrossRef]
- Lane, E.M.; Zhang, Y.; Hazari, N.; Bernskoetter, W.H. Sequential Hydrogenation of CO2 to Methanol Using a Pincer Iron Catalyst. Organometallics 2019, 38, 3084–3091. [Google Scholar] [CrossRef]
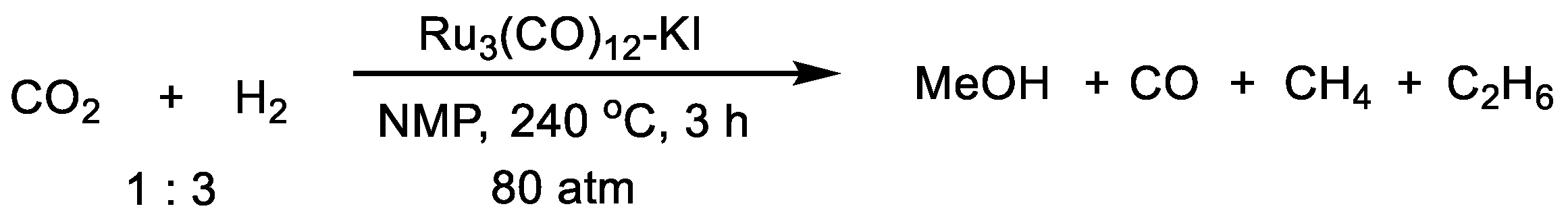







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | Auxiliary | T (°C) | P (bar) | Solvent | Time (h) | TON | Ref. |
|---|---|---|---|---|---|---|---|
| Ru3(CO)12 | KI | 240 | 80 | NMP | 3 | 32 | [39] |
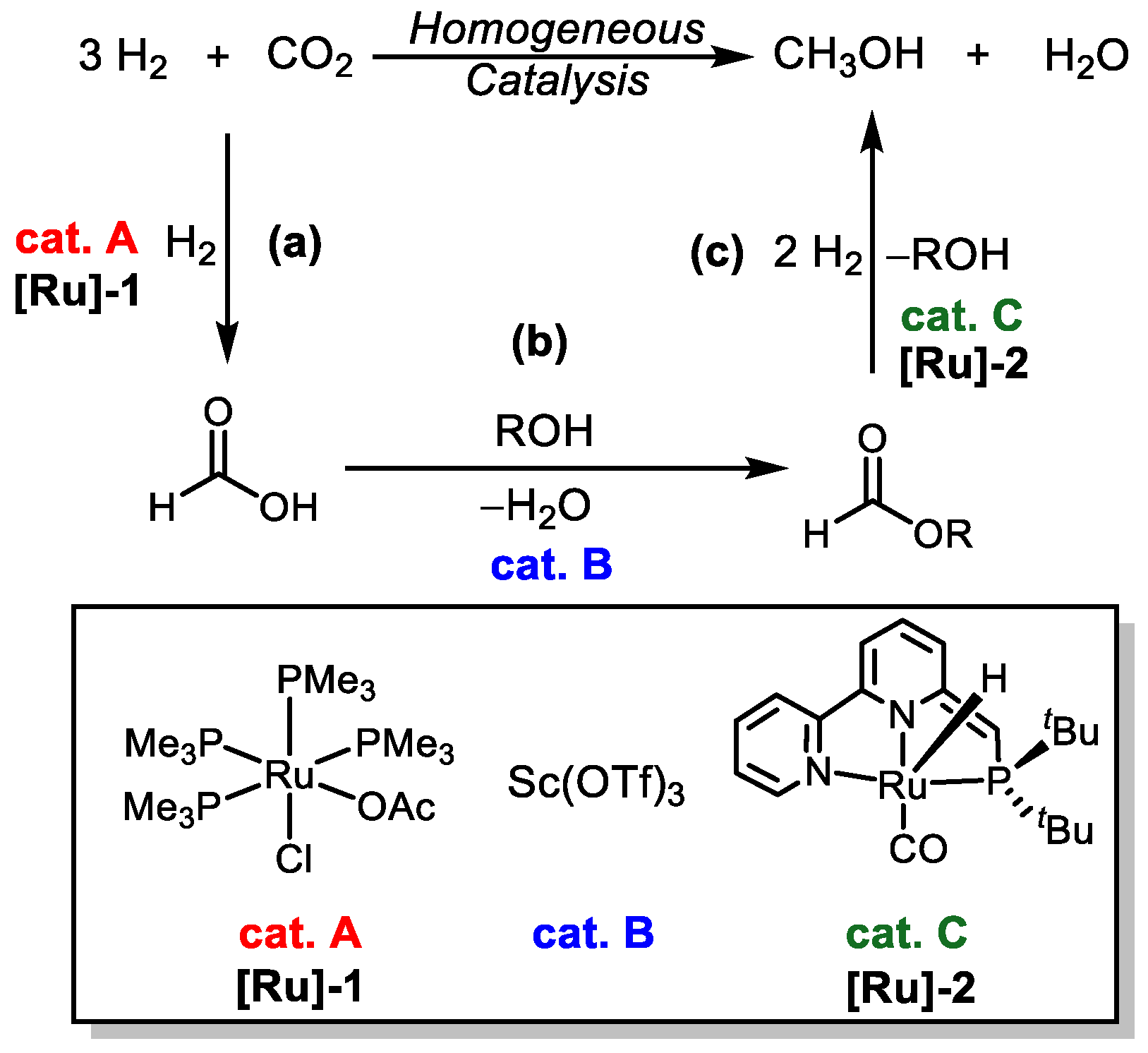
| [Ru]-1/[Ru]-3 | Sc(OTf)3 | 135 | 40 | Dioxane | 16 | 21 | [41] |
| Ru(H)2[P(CH2CH2PPh2)3] | Sc(OTf)3 | 90 | 62 | Dioxane EtOH | 16 | 428 | [42] |
| Ir-(tBuPCP)(CO) | |||||||
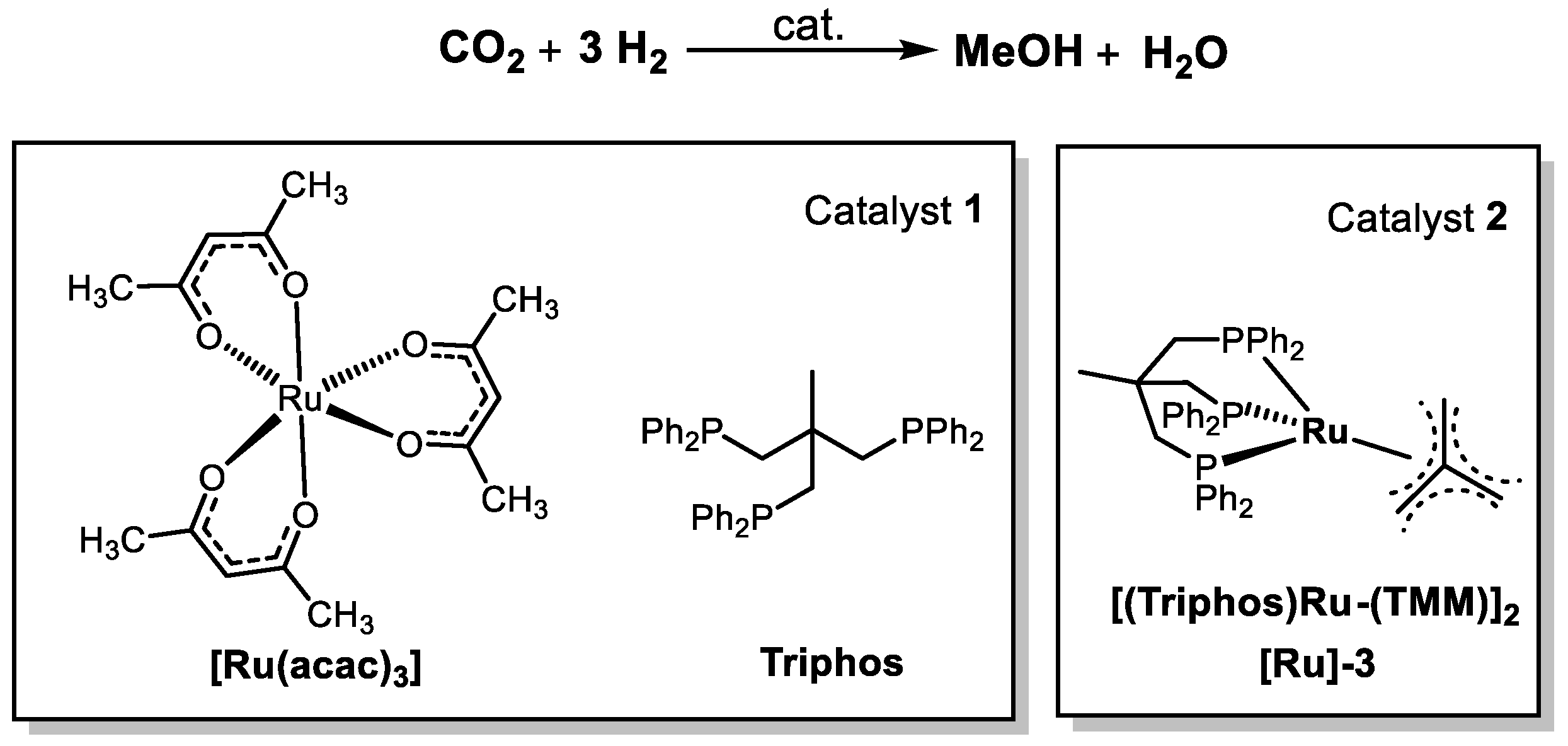
| [Ru(acac)3] | Triphos MSA | 140 | 80 | EtOH | 24 | 135 | [43] |
| [Ru]-3 | HNTf2 | 140 | 80 | EtOH | 24 | 221 | [43] |
| (cod)Ru(methallyl)2 | Tdppcy | 120 | 120 | THF | 20 | 1100 | [44] |
| Al(OTf)3 | |||||||
| (cod)Ru(methallyl)2 | Tdppcy Al(OTf)3 | 120 | 120 | EtOH | 20 | 2100 | [44] |
| Ru-MACHO-BH | Dimethylamine | 95~155 | 50 | THF | 54 | 550 | [45] |
| [Ru]-5 | Valinol | 135 | 60 | DMSO/THF | 19 | 322 | [46] |
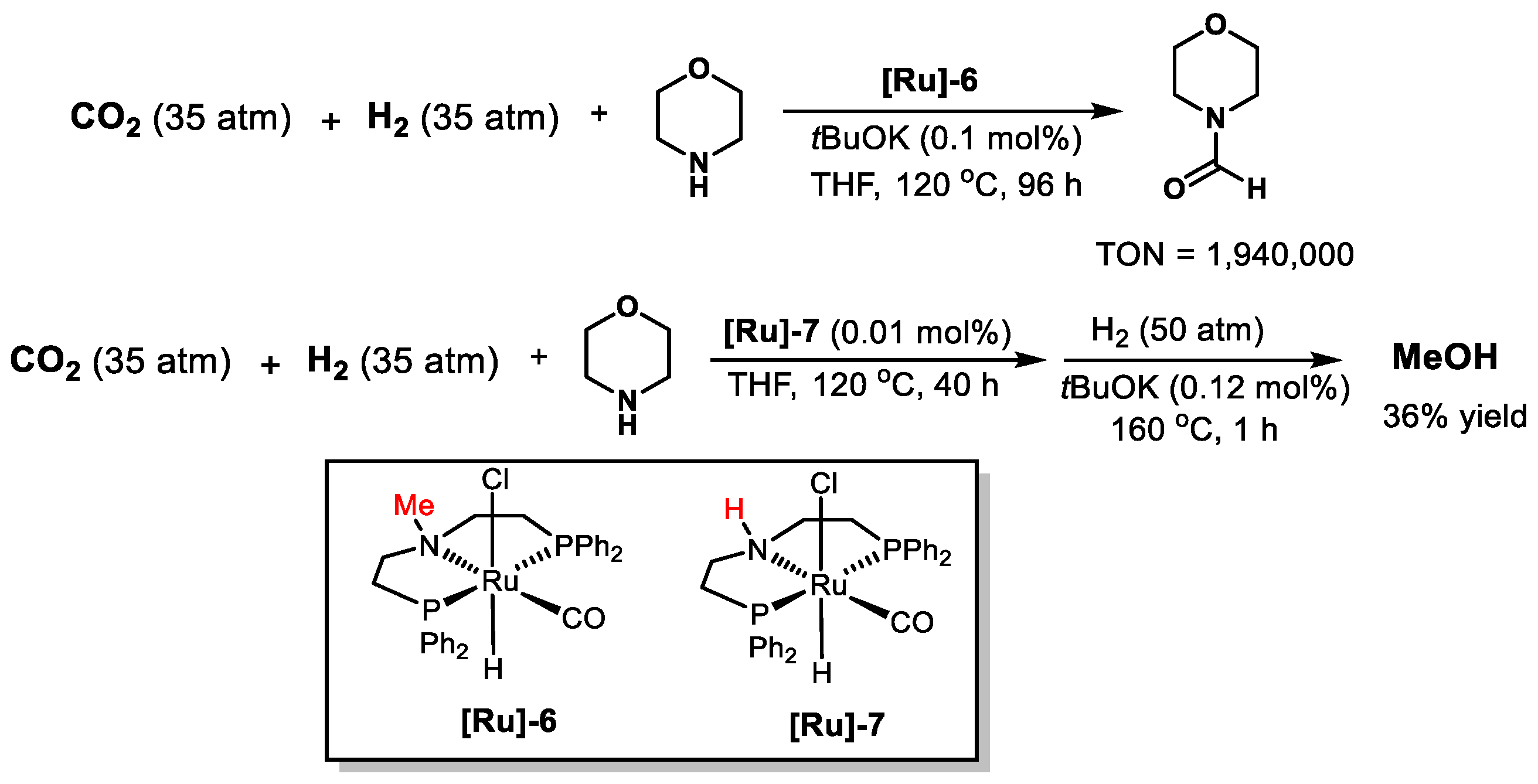
| [Ru]-6 | Morpholine | 120 | 70 | THF | 96 | 1,940,000 | [47] |
| [Ru]-4 | PEHA | 155 | 70 | THF | 40 | 1060 | [49] |
| [Ru]-4 | PEHA | 145 | 70 | 2-MeTHF/H2O | 72 | 520 | [50] |
| [Ru]-4 | Ethylene glycol | 140 | 70 | THF | 72 | 480 | [51] |
| [Ru]-8 | Amine | 100 | 40 | Toluene | 20 | 8900 | [53] |
| [Ru]-4 | PEIs | 150 | 80 | THF | 20 | 689 | [54] |
| Co(acac)3 | Triphos HNTf2 | 140 | 80 | THF/EtOH | 24 | 31 | [61] |
| Co(acac)3 | Triphos | 110 | 90 | THF/EtOH | 24 | 50 | [61] |
| Co(NTf2)2 | Triphos(p-tol) | 100 | 90 | THF/EtOH | 24 | 125 | [62] |
| [Mn]-1 | Morpholine | 110 | 80 | THF | 36 | 840 | [63] |
| [Fe]-1 | Morpholine | 120 | 69 | Dioxane | 16 | 590 | [68] |
| Catalyst | CO2 derivatives | T (°C) | P (bar) | Solvent | Time (h) | TON | Ref. |
|---|---|---|---|---|---|---|---|
| [Ru]-2 | Dimethyl carbonate | 145 | 60 | Dioxane | 1 | 2500 | [55] |
| [Ru]-9 | Dimethyl carbonate | 110 | 50 | THF | 14 | 4400 | [55] |
| [Ru]-2 | Methyl formate | 110 | 50 | THF | 14 | 4700 | [55] |
| [Ru]-2 | Urea | 100 | 13.6 | THF | 72 | 202 | [56] |
| [Ru]-7 | Ethylene carbonate | 140 | 60 | THF | 48 | 87,000 | [57] |
| [Mn]-2 | Carbonates | 110 | 50 | PhMe | 30 | 390 | [64] |
| [Mn]-3 | Cyclic carbonates | 140 | 50 | Dioxane | 16 | 920 | [65] |
| [Mn]-1 | Cyclic carbonates | 120 | 30 | THF | 40 | 175 | [66] |
| [Mn]-4 | Carbamates, urea | 130 | 20 | Toluene | 48 | 50 | [67] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xue, W.; Tang, C. Hydrogenation of CO2 or CO2 Derivatives to Methanol under Molecular Catalysis: A Review. Energies 2022, 15, 2011. https://doi.org/10.3390/en15062011
Xue W, Tang C. Hydrogenation of CO2 or CO2 Derivatives to Methanol under Molecular Catalysis: A Review. Energies. 2022; 15(6):2011. https://doi.org/10.3390/en15062011
Chicago/Turabian StyleXue, Wenxuan, and Conghui Tang. 2022. "Hydrogenation of CO2 or CO2 Derivatives to Methanol under Molecular Catalysis: A Review" Energies 15, no. 6: 2011. https://doi.org/10.3390/en15062011
APA StyleXue, W., & Tang, C. (2022). Hydrogenation of CO2 or CO2 Derivatives to Methanol under Molecular Catalysis: A Review. Energies, 15(6), 2011. https://doi.org/10.3390/en15062011