
An Overview on Transport Phenomena within Solid Electrolyte Interphase and Their Impact on the Performance and Durability of Lithium-Ion Batteries
 , , , and
, , , and
Abstract
:
1. Introduction
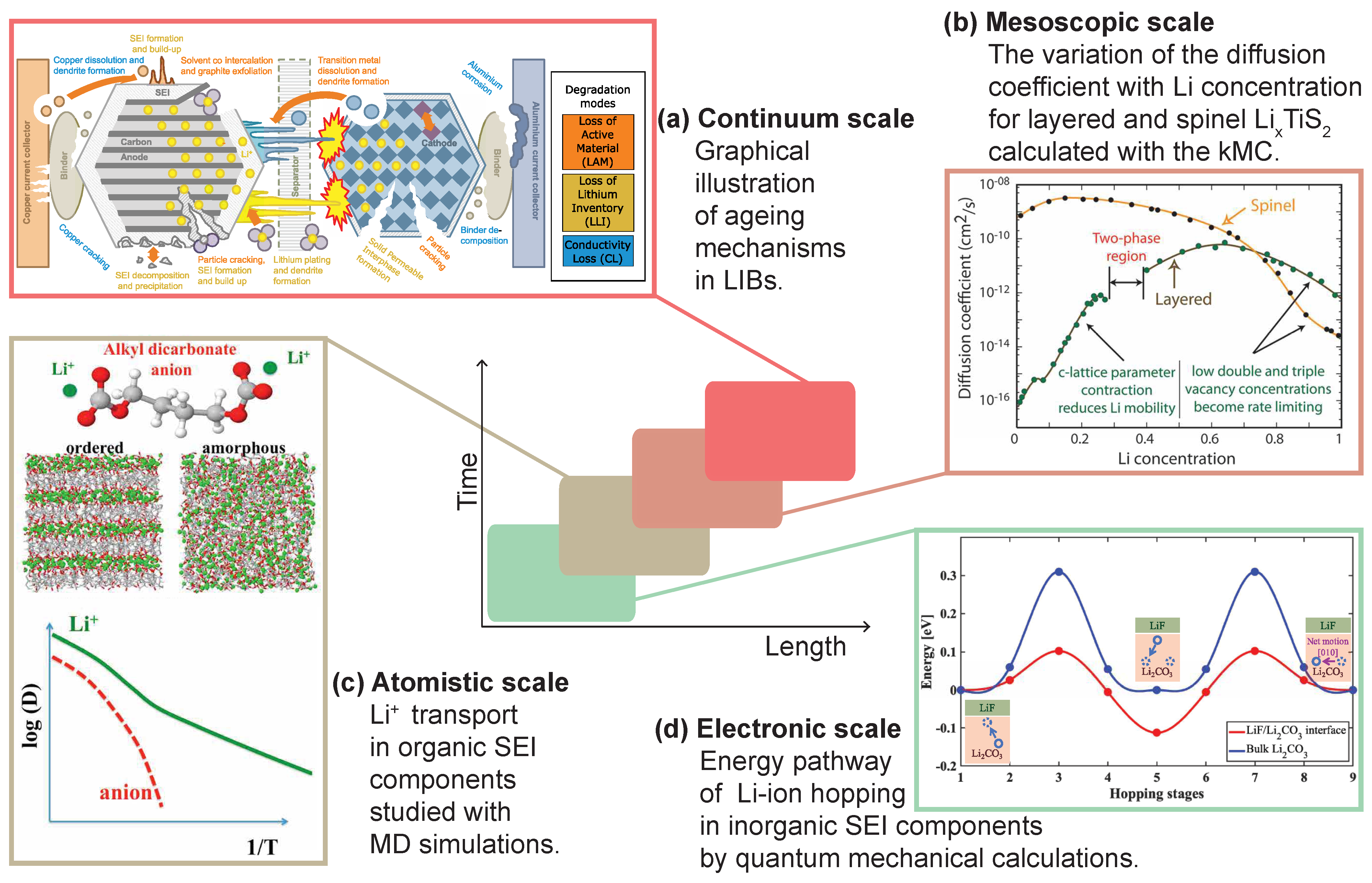
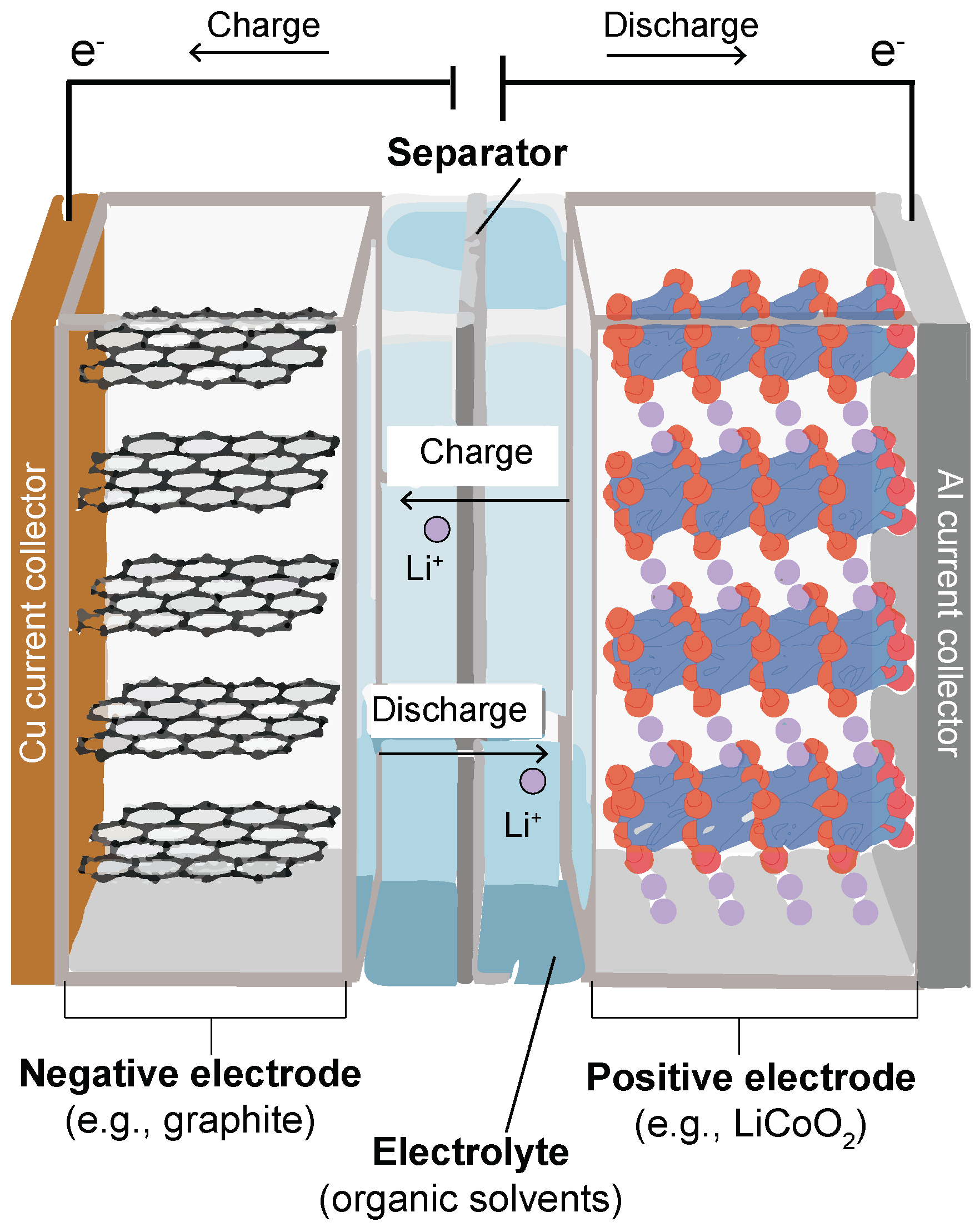
2. Modeling of Li-Ion Diffusion
- Interstitial diffusion occurs when lithium ions in interstitial positions in the crystal lattice move through interstitial sites or gaps between atoms.
- Substitutional diffusion occurs when lithium ions replace other ions in the crystal lattice, resulting in the migration of Li ions within the lattice typically into vacant lattice sites. It is also known as the “knock-off” mechanism.
- Vacancy diffusion can happen when a moves into a vacant site, leaving a vacancy behind, and another lithium ion moves into the empty site, resulting in net lithium diffusion.
- Hopping diffusion can occur when lithium ions jump from one lattice site to another through a series of intermediate sites, facilitated by thermal energy.
2.1. Electronic Scale Models
2.2. Atomistic Scale Models
2.3. Bridging Micro- to Macro-Properties
3. Experimental Measure of Li-Ion Diffusion
4. Challenges and Perspectives
- Investigating the diffusion of lithium ions in an SEI that is as realistic as possible, rather than just in the components of an ideal SEI that do not fully capture the complexity of the true SEI structure.
- Employing a multiscale approach that transfers information from electronic/atomic scale to macro-scale, defined, bottom-up approach. This approach is advantageous for designing battery materials because it allows for a more comprehensive understanding of underlying physics and chemistry at each length scale. In contrast, a top-down approach relies more on empirical or phenomenological relationships and does not capture the full complexity of the system at each scale.
- We also suggest the investigation “in vitro” and “in operando” of the SEI components to obtain a real measurement of the diffusion of Li atoms. These measures are crucial to validate all the past and future SEI models.
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kato, Y.; Ogumi, Z.; Martín, J.M.P. (Eds.) Lithium-Ion Batteries: Overview, Simulation, and Diagnostics; Pan Stanford Publishing: Singapore, 2019. [Google Scholar] [CrossRef]
- Pistoia, G. (Ed.) Lithium-Ion Batteries: Advances and Applications, 1st ed.; Elsevier: Amsterdam, The Netherlands, 2014. [Google Scholar] [CrossRef]
- Hannan, M.; Hoque, M.M.; Mohamed, A.; Ayob, A. Review of energy storage systems for electric vehicle applications: Issues and challenges. Renew. Sustain. Energy Rev. 2017, 69, 771–789. [Google Scholar] [CrossRef]
- Dinger, A.; Martin, R.; Mosquet, X.; Rabl, M.; Rizoulis, D.; Russo, M.; Sticher, G. Batteries for electric cars: Challenges, opportunities, and the outlook to 2020. Boston Consult. Group 2010, 7, 2017. [Google Scholar]
- De Angelis, P.; Tuninetti, M.; Bergamasco, L.; Calianno, L.; Asinari, P.; Laio, F.; Fasano, M. Data-driven appraisal of renewable energy potentials for sustainable freshwater production in Africa. Renew. Sustain. Energy Rev. 2021, 149, 111414. [Google Scholar] [CrossRef]
- Dunn, B.; Kamath, H.; Tarascon, J.M. Electrical energy storage for the grid: A battery of choices. Science 2011, 334, 928–935. [Google Scholar] [CrossRef] [Green Version]
- Eftekhari, A. (Ed.) Future Lithium-Ion Batteries; The Royal Society of Chemistry: London, UK, 2019. [Google Scholar] [CrossRef]
- Amici, J.; Asinari, P.; Ayerbe, E.; Barboux, P.; Bayle-Guillemaud, P.; Behm, R.J.; Berecibar, M.; Berg, E.; Bhowmik, A.; Bodoardo, S.; et al. A roadmap for transforming research to invent the batteries of the future designed within the European large scale research initiative BATTERY 2030+. Adv. Energy Mater. 2022, 12, 2102785. [Google Scholar] [CrossRef]
- Tarascon, J.M.; Armand, M. Issues and challenges facing rechargeable lithium batteries. Nature 2001, 414, 359–367. [Google Scholar] [CrossRef]
- Xu, K. Nonaqueous liquid electrolytes for lithium-based rechargeable batteries. Chem. Rev. 2004, 104, 4303–4418. [Google Scholar] [CrossRef] [PubMed]
- Latz, A.; Zausch, J. Thermodynamic consistent transport theory of Li-ion batteries. J. Power Sources 2011, 196, 3296–3302. [Google Scholar] [CrossRef] [Green Version]
- Latz, A.; Zausch, J. Thermodynamic derivation of a Butler–Volmer model for intercalation in Li-ion batteries. Electrochim. Acta 2013, 110, 358–362. [Google Scholar] [CrossRef]
- Xie, J.; Lu, Y.C. A retrospective on lithium-ion batteries. Nat. Commun. 2020, 11, 1–4. [Google Scholar] [CrossRef]
- Goodenough, J.B.; Kim, Y. Challenges for rechargeable Li batteries. Chem. Mater. 2010, 22, 587–603. [Google Scholar] [CrossRef]
- Li, F.; Bashir, S.; Liu, J.L. Nanostructured Materials for Next-Generation Energy Storage and Conversion. Fuel Cells 2018. [Google Scholar] [CrossRef]
- Xu, X.; Li, F.; Zhang, D.; Ji, S.; Huo, Y.; Liu, J. Facile construction of CoSn/Co3Sn2@ C nanocages as anode for superior lithium-/sodium-ion storage. Carbon Neutralization 2023, 2, 54–62. [Google Scholar] [CrossRef]
- Yang, T.; Fan, W.; Wang, C.; Lei, Q.; Ma, Z.; Yu, L.; Zuo, X.; Nan, J. 2,3,4,5,6-Pentafluorophenyl methanesulfonate as a versatile electrolyte additive matches LiNi0.5Co0.2Mn0.3O2/graphite batteries working in a wide-temperature range. ACS Appl. Mater. Interfaces 2018, 10, 31735–31744. [Google Scholar] [CrossRef]
- Wang, C.; Yu, L.; Fan, W.; Liu, J.; Ouyang, L.; Yang, L.; Zhu, M. Lithium difluorophosphate as a promising electrolyte lithium additive for high-voltage lithium-ion batteries. ACS Appl. Energy Mater. 2018, 1, 2647–2656. [Google Scholar] [CrossRef]
- Jiang, Y.; Wu, Y.; Rui, X.; Peng, Y.; Xu, X.; Li, J.; Zhang, S.; Feng, X. In Situ Inorganic-Rich Electrode–Electrolyte Interphases for Safer 4.5 V Gr|| NCM811 Batteries Enabled by an Ethylene Carbonate-Free Electrolyte. ACS Appl. Energy Mater. 2022, 5, 11748–11755. [Google Scholar] [CrossRef]
- Schultz, C.; Vedder, S.; Winter, M.; Nowak, S. Investigation of the decomposition of organic solvent-based lithium ion battery electrolytes with liquid chromatography-mass spectrometry. Spectrosc. Eur 2016, 28, 21–24. [Google Scholar]
- Aurbach, D.; Markovsky, B.; Levi, M.; Levi, E.; Schechter, A.; Moshkovich, M.; Cohen, Y. New insights into the interactions between electrode materials and electrolyte solutions for advanced nonaqueous batteries. J. Power Sources 1999, 81, 95–111. [Google Scholar] [CrossRef]
- Aurbach, D. Review of selected electrode–solution interactions which determine the performance of Li and Li ion batteries. J. Power Sources 2000, 89, 206–218. [Google Scholar] [CrossRef]
- Yao, Y.X.; Yan, C.; Zhang, Q. Emerging interfacial chemistry of graphite anodes in lithium-ion batteries. Chem. Commun. 2020, 56, 14570–14584. [Google Scholar] [CrossRef]
- Vetter, J.; Novák, P.; Wagner, M.R.; Veit, C.; Möller, K.C.; Besenhard, J.; Winter, M.; Wohlfahrt-Mehrens, M.; Vogler, C.; Hammouche, A. Ageing mechanisms in lithium-ion batteries. J. Power Sources 2005, 147, 269–281. [Google Scholar] [CrossRef]
- Pastor-Fernández, C.; Yu, T.F.; Widanage, W.D.; Marco, J. Critical review of non-invasive diagnosis techniques for quantification of degradation modes in lithium-ion batteries. Renew. Sustain. Energy Rev. 2019, 109, 138–159. [Google Scholar] [CrossRef]
- Reniers, J.M.; Mulder, G.; Howey, D.A. Review and performance comparison of mechanical-chemical degradation models for lithium-ion batteries. J. Electrochem. Soc. 2019, 166, A3189. [Google Scholar] [CrossRef] [Green Version]
- Pinson, M.B.; Bazant, M.Z. Theory of SEI formation in rechargeable batteries: Capacity fade, accelerated aging and lifetime prediction. J. Electrochem. Soc. 2012, 160, A243. [Google Scholar] [CrossRef]
- Gauthier, M.; Carney, T.J.; Grimaud, A.; Giordano, L.; Pour, N.; Chang, H.H.; Fenning, D.P.; Lux, S.F.; Paschos, O.; Bauer, C.; et al. Electrode–electrolyte interface in Li-ion batteries: Current understanding and new insights. J. Phys. Chem. Lett. 2015, 6, 4653–4672. [Google Scholar] [CrossRef]
- An, S.J.; Li, J.; Daniel, C.; Mohanty, D.; Nagpure, S.; Wood III, D.L. The state of understanding of the lithium-ion-battery graphite solid electrolyte interphase (SEI) and its relationship to formation cycling. Carbon 2016, 105, 52–76. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Chae, O.B.; Lucht, B.L. Perspective—structure and stability of the solid electrolyte interphase on silicon anodes of lithium-ion batteries. J. Electrochem. Soc. 2021, 168, 030521. [Google Scholar] [CrossRef]
- Rago, N.D.; Bareno, J.; Li, J.; Du, Z.; Wood III, D.L.; Steele, L.A.; Lamb, J.; Spangler, S.; Grosso, C.; Fenton, K.; et al. Effect of overcharge on Li (Ni0.5Mn0.3Co0.2) O2/Graphite lithium ion cells with poly (vinylidene fluoride) binder. I-Microstructural changes in the anode. J. Power Sources 2018, 385, 148–155. [Google Scholar] [CrossRef]
- Wang, A.; Kadam, S.; Li, H.; Shi, S.; Qi, Y. Review on modeling of the anode solid electrolyte interphase (SEI) for lithium-ion batteries. Npj Comput. Mater. 2018, 4, 1–26. [Google Scholar] [CrossRef] [Green Version]
- Malmgren, S.; Ciosek, K.; Hahlin, M.; Gustafsson, T.; Gorgoi, M.; Rensmo, H.; Edström, K. Comparing anode and cathode electrode/electrolyte interface composition and morphology using soft and hard X-ray photoelectron spectroscopy. Electrochim. Acta 2013, 97, 23–32. [Google Scholar] [CrossRef]
- Peled, E. The electrochemical behavior of alkali and alkaline earth metals in nonaqueous battery systems—The solid electrolyte interphase model. J. Electrochem. Soc. 1979, 126, 2047. [Google Scholar] [CrossRef]
- Peled, E.; Golodnitsky, D.; Ardel, G. Advanced model for solid electrolyte interphase electrodes in liquid and polymer electrolytes. J. Electrochem. Soc. 1997, 144, L208. [Google Scholar] [CrossRef]
- Villevieille, C. Interfaces and Interphases in Batteries: How to Identify and Monitor Them Properly Using Surface Sensitive Characterization Techniques. Adv. Mater. Interfaces 2022, 9, 2101865. [Google Scholar] [CrossRef]
- Wang, Y.; Nakamura, S.; Ue, M.; Balbuena, P.B. Theoretical studies to understand surface chemistry on carbon anodes for lithium-ion batteries: Reduction mechanisms of ethylene carbonate. J. Am. Chem. Soc. 2001, 123, 11708–11718. [Google Scholar] [CrossRef] [PubMed]
- Leung, K. Electronic structure modeling of electrochemical reactions at electrode/electrolyte interfaces in lithium ion batteries. J. Phys. Chem. 2013, 117, 1539–1547. [Google Scholar] [CrossRef] [Green Version]
- Aurbach, D.; Zaban, A.; Ein-Eli, Y.; Weissman, I.; Chusid, O.; Markovsky, B.; Levi, M.; Levi, E.; Schechter, A.; Granot, E. Recent studies on the correlation between surface chemistry, morphology, three-dimensional structures and performance of Li and Li-C intercalation anodes in several important electrolyte systems. J. Power Sources 1997, 68, 91–98. [Google Scholar] [CrossRef]
- Schroder, K.W.; Celio, H.; Webb, L.J.; Stevenson, K.J. Examining solid electrolyte interphase formation on crystalline silicon electrodes: Influence of electrochemical preparation and ambient exposure conditions. J. Phys. Chem. C 2012, 116, 19737–19747. [Google Scholar] [CrossRef]
- Yun, K.S.; Pai, S.J.; Yeo, B.C.; Lee, K.R.; Kim, S.J.; Han, S.S. Simulation protocol for prediction of a solid-electrolyte interphase on the silicon-based anodes of a lithium-ion battery: ReaxFF reactive force field. J. Phys. Chem. Lett. 2017, 8, 2812–2818. [Google Scholar] [CrossRef]
- Lu, P.; Li, C.; Schneider, E.W.; Harris, S.J. Chemistry, impedance, and morphology evolution in solid electrolyte interphase films during formation in lithium ion batteries. J. Phys. Chem. C 2014, 118, 896–903. [Google Scholar] [CrossRef]
- Winter, M. The solid electrolyte interphase–the most important and the least understood solid electrolyte in rechargeable Li batteries. Z. für Phys. Chem. 2009, 223, 1395–1406. [Google Scholar] [CrossRef]
- Meda, U.S.; Lal, L.; Sushantha, M.; Garg, P. Solid Electrolyte Interphase (SEI), a boon or a bane for lithium batteries: A review on the recent advances. J. Energy Storage 2021, 47, 103564. [Google Scholar] [CrossRef]
- An, S.J.; Li, J.; Du, Z.; Daniel, C.; Wood III, D.L. Fast formation cycling for lithium ion batteries. J. Power Sources 2017, 342, 846–852. [Google Scholar] [CrossRef]
- Wood, D.L.; Li, J.; An, S.J. Formation challenges of lithium-ion battery manufacturing. Joule 2019, 3, 2884–2888. [Google Scholar] [CrossRef]
- Shi, S.; Lu, P.; Liu, Z.; Qi, Y.; Hector Jr, L.G.; Li, H.; Harris, S.J. Direct calculation of Li-ion transport in the solid electrolyte interphase. J. Am. Chem. Soc. 2012, 134, 15476–15487. [Google Scholar] [CrossRef]
- Ahmad, Z.; Venturi, V.; Hafiz, H.; Viswanathan, V. Interfaces in solid electrolyte interphase: Implications for lithium-ion batteries. J. Phys. Chem. C 2021, 125, 11301–11309. [Google Scholar] [CrossRef]
- Chattopadhyay, S.; Lipson, A.L.; Karmel, H.J.; Emery, J.D.; Fister, T.T.; Fenter, P.A.; Hersam, M.C.; Bedzyk, M.J. In situ X-ray study of the solid electrolyte interphase (SEI) formation on graphene as a model Li-ion battery anode. Chem. Mater. 2012, 24, 3038–3043. [Google Scholar] [CrossRef]
- Seidl, L.; Martens, S.; Ma, J.; Stimming, U.; Schneider, O. In situ scanning tunneling microscopy studies of the SEI formation on graphite electrodes for Li+-ion batteries. Nanoscale 2016, 8, 14004–14014. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, Y.; Pei, A.; Yan, K.; Sun, Y.; Wu, C.L.; Joubert, L.M.; Chin, R.; Koh, A.L.; Yu, Y.; et al. Atomic structure of sensitive battery materials and interfaces revealed by cryo–electron microscopy. Science 2017, 358, 506–510. [Google Scholar] [CrossRef] [Green Version]
- Shinagawa, C.; Ushiyama, H.; Yamashita, K. Multiscale simulations for lithium-ion batteries: SEI film growth and capacity fading. J. Electrochem. Soc. 2017, 164, A3018. [Google Scholar] [CrossRef]
- Horstmann, B.; Single, F.; Latz, A. Review on multi-scale models of solid-electrolyte interphase formation. Curr. Opin. Electrochem. 2019, 13, 61–69. [Google Scholar] [CrossRef] [Green Version]
- Franco, A.A.; Rucci, A.; Brandell, D.; Frayret, C.; Gaberscek, M.; Jankowski, P.; Johansson, P. Boosting rechargeable batteries R&D by multiscale modeling: Myth or reality? Chem. Rev. 2019, 119, 4569–4627. [Google Scholar] [CrossRef] [Green Version]
- Fong, R.; Von Sacken, U.; Dahn, J.R. Studies of lithium intercalation into carbons using nonaqueous electrochemical cells. J. Electrochem. Soc. 1990, 137, 2009. [Google Scholar] [CrossRef]
- Xu, K.; von Cresce, A.; Lee, U. Differentiating contributions to “ion transfer” barrier from interphasial resistance and Li+ desolvation at electrolyte/graphite interface. Langmuir 2010, 26, 11538–11543. [Google Scholar] [CrossRef]
- Single, F.; Horstmann, B.; Latz, A. Dynamics and morphology of solid electrolyte interphase (SEI). Phys. Chem. Chem. Phys. 2016, 18, 17810–17814. [Google Scholar] [CrossRef] [PubMed]
- von Kolzenberg, L.; Latz, A.; Horstmann, B. Solid–electrolyte interphase during battery cycling: Theory of growth regimes. ChemSusChem 2020, 13, 3901–3910. [Google Scholar] [CrossRef] [PubMed]
- Van der Ven, A.; Bhattacharya, J.; Belak, A.A. Understanding Li diffusion in Li-intercalation compounds. Accounts Chem. Res. 2013, 46, 1216–1225. [Google Scholar] [CrossRef]
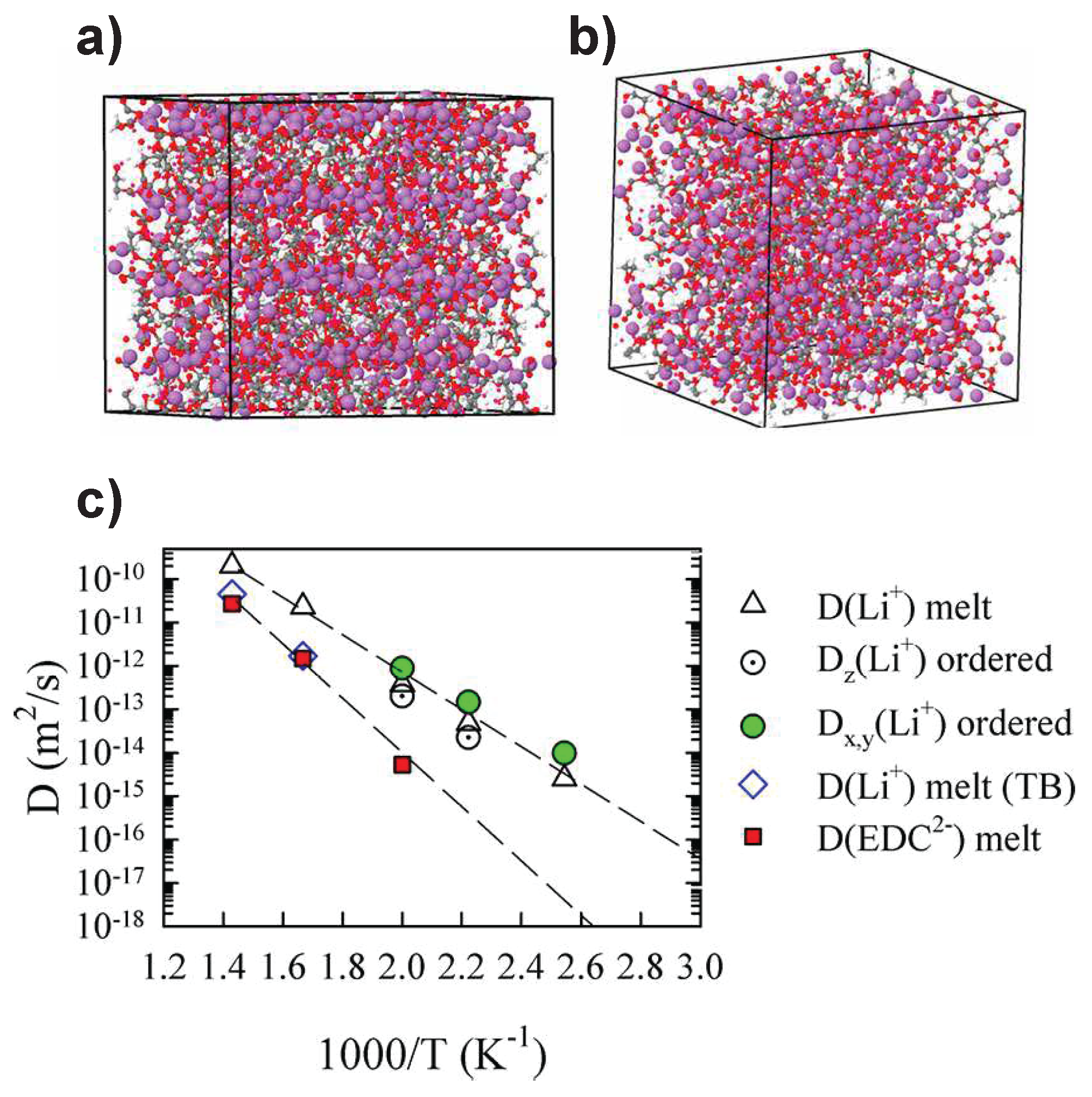
- Bedrov, D.; Borodin, O.; Hooper, J.B. Li+ transport and mechanical properties of model solid electrolyte interphases (SEI): Insight from atomistic molecular dynamics simulations. J. Phys. Chem. C 2017, 121, 16098–16109. [Google Scholar] [CrossRef]
- Moosbauer, D.; Zugmann, S.; Amereller, M.; Gores, H.J. Effect of ionic liquids as additives on lithium electrolytes: Conductivity, electrochemical stability, and aluminum corrosion. J. Chem. Eng. Data 2010, 55, 1794–1798. [Google Scholar] [CrossRef]
- Adenusi, H.; Chass, G.A.; Passerini, S.; Tian, K.V.; Chen, G. Lithium Batteries and the Solid Electrolyte Interphase (SEI)—Progress and Outlook. Adv. Energy Mater. 2023, 13, 2203307. [Google Scholar] [CrossRef]
- Soto, F.A.; Marzouk, A.; El-Mellouhi, F.; Balbuena, P.B. Understanding ionic diffusion through SEI components for lithium-ion and sodium-ion batteries: Insights from first-principles calculations. Chem. Mater. 2018, 30, 3315–3322. [Google Scholar] [CrossRef]
- Ma, X.X.; Shen, X.; Chen, X.; Fu, Z.H.; Yao, N.; Zhang, R.; Zhang, Q. The Origin of Fast Lithium-Ion Transport in the Inorganic Solid Electrolyte Interphase on Lithium Metal Anodes. Small Struct. 2022, 3, 2200071. [Google Scholar] [CrossRef]
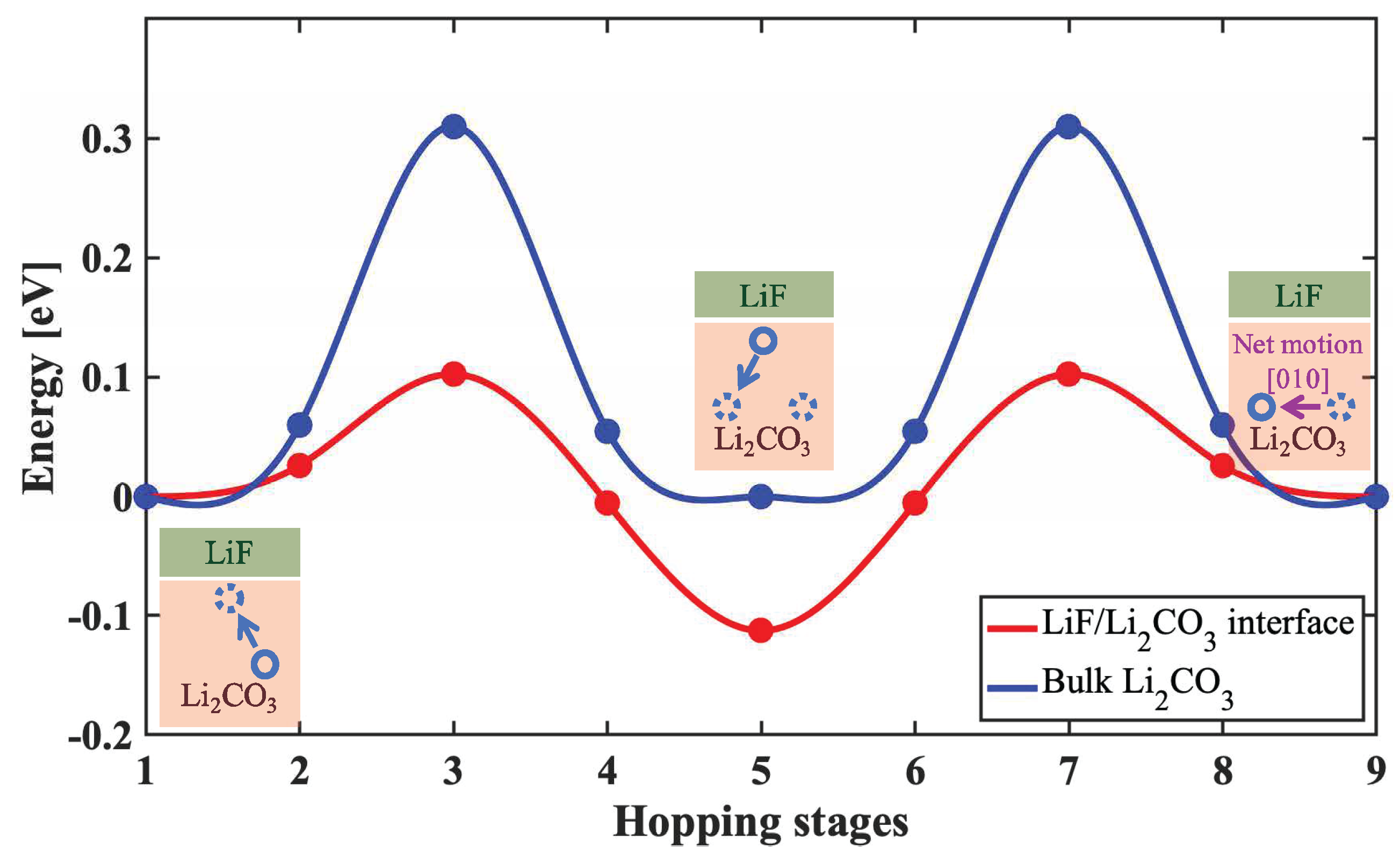
- Shi, S.; Qi, Y.; Li, H.; Hector Jr, L.G. Defect thermodynamics and diffusion mechanisms in Li2CO3 and implications for the solid electrolyte interphase in Li-ion batteries. J. Phys. Chem. C 2013, 117, 8579–8593. [Google Scholar] [CrossRef]
- Alzate-Vargas, L.; Vikrant, K.; Allu, S.; Fattebert, J.L. Atomistic modeling of LiF microstructure ionic conductivity and its influence on nucleation and plating. Phys. Rev. Mater. 2022, 6, 095402. [Google Scholar] [CrossRef]
- Benitez, L.; Seminario, J.M. Ion diffusivity through the solid electrolyte interphase in lithium-ion batteries. J. Electrochem. Soc. 2017, 164, E3159. [Google Scholar] [CrossRef]
- Safari, M.; Morcrette, M.; Teyssot, A.; Delacourt, C. Multimodal physics-based aging model for life prediction of Li-ion batteries. J. Electrochem. Soc. 2008, 156, A145. [Google Scholar] [CrossRef]
- Santhanagopalan, S.; Guo, Q.; Ramadass, P.; White, R.E. Review of models for predicting the cycling performance of lithium ion batteries. J. Power Sources 2006, 156, 620–628. [Google Scholar] [CrossRef]
- Ning, G.; Popov, B.N. Cycle life modeling of lithium-ion batteries. J. Electrochem. Soc. 2004, 151, A1584. [Google Scholar] [CrossRef] [Green Version]
- Maier, J. Physical Chemistry of Ionic Materials: Ions and Electrons in Solids; John Wiley & Sons: Hoboken, NJ, USA, 2023. [Google Scholar]
- Pan, J.; Cheng, Y.T.; Qi, Y. General method to predict voltage-dependent ionic conduction in a solid electrolyte coating on electrodes. Phys. Rev. B 2015, 91, 134116. [Google Scholar] [CrossRef] [Green Version]
- Pan, J.; Zhang, Q.; Xiao, X.; Cheng, Y.T.; Qi, Y. Design of nanostructured heterogeneous solid ionic coatings through a multiscale defect model. ACS Appl. Mater. Interfaces 2016, 8, 5687–5693. [Google Scholar] [CrossRef]
- Iddir, H.; Curtiss, L.A. Li ion diffusion mechanisms in bulk monoclinic Li2CO3 crystals from density functional studies. J. Phys. Chem. C 2010, 114, 20903–20906. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 1965, 140, A1133. [Google Scholar] [CrossRef] [Green Version]
- Nazri, G.; Muller, R.H. Composition of surface layers on Li electrodes in PC, LiClO4 of very low water content. J. Electrochem. Soc. 1985, 132, 2050. [Google Scholar] [CrossRef]
- Jónsson, H.; Mills, G.; Jacobsen, K.W. Nudged elastic band method for finding minimum energy paths of transitions. In Classical and Quantum Dynamics in Condensed Phase Simulations; World Scientific: London, UK, 1998; pp. 385–404. [Google Scholar] [CrossRef] [Green Version]
- Toyoura, K.; Koyama, Y.; Kuwabara, A.; Tanaka, I. Effects of off-stoichiometry of LiC6 on the lithium diffusion mechanism and diffusivity by first principles calculations. J. Phys. Chem. C 2010, 114, 2375–2379. [Google Scholar] [CrossRef]
- Chen, Y.; Ouyang, C.; Song, L.; Sun, Z. Electrical and lithium ion dynamics in three main components of solid electrolyte interphase from density functional theory study. J. Phys. Chem. C 2011, 115, 7044–7049. [Google Scholar] [CrossRef]
- Persson, K.; Sethuraman, V.A.; Hardwick, L.J.; Hinuma, Y.; Meng, Y.S.; Van Der Ven, A.; Srinivasan, V.; Kostecki, R.; Ceder, G. Lithium diffusion in graphitic carbon. J. Phys. Chem. Lett. 2010, 1, 1176–1180. [Google Scholar] [CrossRef] [Green Version]
- Ramasubramanian, A.; Yurkiv, V.; Foroozan, T.; Ragone, M.; Shahbazian-Yassar, R.; Mashayek, F. Lithium diffusion mechanism through solid–electrolyte interphase in rechargeable lithium batteries. J. Phys. Chem. C 2019, 123, 10237–10245. [Google Scholar] [CrossRef]
- Zheng, J.; Ju, Z.; Zhang, B.; Nai, J.; Liu, T.; Liu, Y.; Xie, Q.; Zhang, W.; Wang, Y.; Tao, X. Lithium ion diffusion mechanism on the inorganic components of the solid–electrolyte interphase. J. Mater. Chem. A 2021, 9, 10251–10259. [Google Scholar] [CrossRef]
- Car, R.; Parrinello, M. Unified approach for molecular dynamics and density-functional theory. Phys. Rev. Lett. 1985, 55, 2471. [Google Scholar] [CrossRef] [Green Version]
- Frenkel, D.; Smit, B. Understanding Molecular Simulation: From Algorithms to Applications; Elsevier: Amsterdam, The Netherlands, 2001; Volume 1. [Google Scholar] [CrossRef]
- Satoh, A. Introduction to Practice of Molecular Simulation: Molecular Dynamics, Monte Carlo, Brownian Dynamics, Lattice Boltzmann and Dissipative Particle Dynamics; Elsevier: Amsterdam, The Netherlands, 2010. [Google Scholar] [CrossRef]
- Allen, M.P.; Tildesley, D.J. Computer Simulation of Liquids, 2nd ed.; Oxford University Press: Oxford, UK, 2017. [Google Scholar] [CrossRef]
- Senftle, T.P.; Hong, S.; Islam, M.M.; Kylasa, S.B.; Zheng, Y.; Shin, Y.K.; Junkermeier, C.; Engel-Herbert, R.; Janik, M.J.; Aktulga, H.M.; et al. The ReaxFF reactive force-field: Development, applications and future directions. Npj Comput. Mater. 2016, 2, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Harrison, J.A.; Schall, J.D.; Maskey, S.; Mikulski, P.T.; Knippenberg, M.T.; Morrow, B.H. Review of force fields and intermolecular potentials used in atomistic computational materials research. Appl. Phys. Rev. 2018, 5, 031104. [Google Scholar] [CrossRef]
- Unke, O.T.; Chmiela, S.; Sauceda, H.E.; Gastegger, M.; Poltavsky, I.; Schütt, K.T.; Tkatchenko, A.; Muüller, K.R. Machine learning force fields. Chem. Rev. 2021, 121, 10142–10186. [Google Scholar] [CrossRef] [PubMed]
- Pauling, L. The nature of the chemical bond. II. The one-electron bond and the three-electron bond. J. Am. Chem. Soc. 1931, 53, 3225–3237. [Google Scholar] [CrossRef]
- Brenner, D.W. Empirical potential for hydrocarbons for use in simulating the chemical vapor deposition of diamond films. Phys. Rev. B 1990, 42, 9458. [Google Scholar] [CrossRef] [PubMed]
- Stuart, S.J.; Tutein, A.B.; Harrison, J.A. A reactive potential for hydrocarbons with intermolecular interactions. J. Chem. Phys. 2000, 112, 6472–6486. [Google Scholar] [CrossRef] [Green Version]
- Tersoff, J. New empirical approach for the structure and energy of covalent systems. Phys. Rev. B 1988, 37, 6991. [Google Scholar] [CrossRef] [PubMed]
- Van Duin, A.C.; Dasgupta, S.; Lorant, F.; Goddard, W.A. ReaxFF: A reactive force field for hydrocarbons. J. Phys. Chem. A 2001, 105, 9396–9409. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Canongia Lopes, J.N.; Pádua, A.A. Molecular force field for ionic liquids composed of triflate or bistriflylimide anions. J. Phys. Chem. B 2004, 108, 16893–16898. [Google Scholar] [CrossRef]
- Borodin, O.; Smith, G.D. Development of many- body polarizable force fields for Li-battery components: 1. Ether, Alkane, and carbonate-based solvents. J. Phys. Chem. B 2006, 110, 6279–6292. [Google Scholar] [CrossRef]
- Borodin, O.; Smith, G.D.; Fan, P. Molecular dynamics simulations of lithium alkyl carbonates. J. Phys. Chem. B 2006, 110, 22773–22779. [Google Scholar] [CrossRef]
- Starovoytov, O.N.; Borodin, O.; Bedrov, D.; Smith, G.D. Development of a polarizable force field for molecular dynamics simulations of poly (ethylene oxide) in aqueous solution. J. Chem. Theory Comput. 2011, 7, 1902–1915. [Google Scholar] [CrossRef] [PubMed]
- Chiavazzo, E.; Fasano, M.; Asinari, P.; Decuzzi, P. Scaling behaviour for the water transport in nanoconfined geometries. Nat. Commun. 2014, 5, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Fasano, M.; Bevilacqua, A.; Chiavazzo, E.; Humplik, T.; Asinari, P. Mechanistic correlation between water infiltration and framework hydrophilicity in MFI zeolites. Sci. Rep. 2019, 9, 18429. [Google Scholar] [CrossRef] [Green Version]
- Márquez, A.; Balbuena, P.B. Molecular dynamics study of Graphite/Electrolyte interfaces. J. Electrochem. Soc. 2001, 148, A624. [Google Scholar] [CrossRef]
- Zhuang, G.V.; Xu, K.; Yang, H.; Jow, T.R.; Ross, P.N. Lithium ethylene dicarbonate identified as the primary product of chemical and electrochemical reduction of EC in 1.2 M LiPF6/EC: EMC electrolyte. J. Phys. Chem. B 2005, 109, 17567–17573. [Google Scholar] [CrossRef]
- Nie, M.; Chalasani, D.; Abraham, D.P.; Chen, Y.; Bose, A.; Lucht, B.L. Lithium ion battery graphite solid electrolyte interphase revealed by microscopy and spectroscopy. J. Phys. Chem. C 2013, 117, 1257–1267. [Google Scholar] [CrossRef]
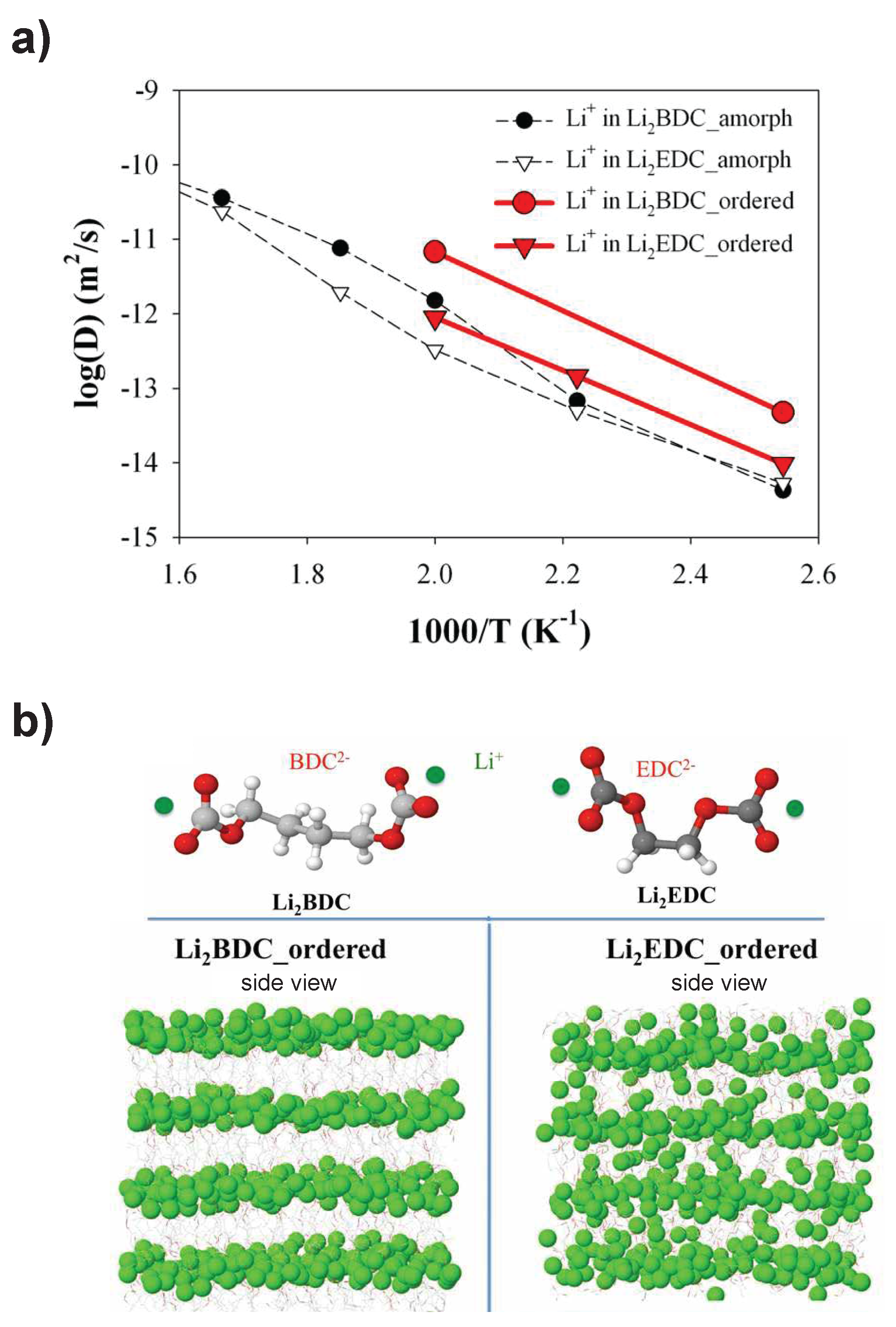
- Borodin, O.; Zhuang, G.V.; Ross, P.N.; Xu, K. Molecular dynamics simulations and experimental study of lithium ion transport in dilithium ethylene dicarbonate. J. Phys. Chem. C 2013, 117, 7433–7444. [Google Scholar] [CrossRef]
- Jow, T.R.; Marx, M.B.; Allen, J.L. Distinguishing Li+ charge transfer kinetics at NCA/electrolyte and graphite/electrolyte interfaces, and NCA/electrolyte and LFP/electrolyte interfaces in Li-ion cells. J. Electrochem. Soc. 2012, 159, A604. [Google Scholar] [CrossRef]
- Jow, T.R.; Allen, J.; Marx, M.; Nechev, K.; Deveney, B.; Rickman, S. Electrolytes, SEI and charge discharge kinetics in Li-ion batteries. ECS Trans. 2010, 25, 3. [Google Scholar] [CrossRef]
- Xu, K.; Lam, Y.; Zhang, S.S.; Jow, T.R.; Curtis, T.B. Solvation sheath of Li+ in nonaqueous electrolytes and its implication of graphite/electrolyte interface chemistry. J. Phys. Chem. C 2007, 111, 7411–7421. [Google Scholar] [CrossRef]
- Borodin, O. Polarizable force field development and molecular dynamics simulations of ionic liquids. J. Phys. Chem. B 2009, 113, 11463–11478. [Google Scholar] [CrossRef]
- Muralidharan, A.; Chaudhari, M.I.; Pratt, L.R.; Rempe, S.B. Molecular dynamics of lithium ion transport in a model solid electrolyte interphase. Sci. Rep. 2018, 8, 10736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muralidharan, A.; Chaudhari, M.; Rempe, S.; Pratt, L.R. Molecular dynamics simulations of lithium ion transport through a model solid electrolyte interphase (sei) layer. ECS Trans. 2017, 77, 1155. [Google Scholar] [CrossRef]
- Kim, S.Y.; Ostadhossein, A.; Van Duin, A.C.; Xiao, X.; Gao, H.; Qi, Y. Self-generated concentration and modulus gradient coating design to protect Si nano-wire electrodes during lithiation. Phys. Chem. Chem. Phys. 2016, 18, 3706–3715. [Google Scholar] [CrossRef] [PubMed]
- Bedrov, D.; Smith, G.D.; van Duin, A.C. Reactions of singly-reduced ethylene carbonate in lithium battery electrolytes: A molecular dynamics simulation study using the ReaxFF. J. Phys. Chem. A 2012, 116, 2978–2985. [Google Scholar] [CrossRef]
- Islam, M.M.; Van Duin, A.C. Reductive decomposition reactions of ethylene carbonate by explicit electron transfer from lithium: An eReaxFF molecular dynamics study. J. Phys. Chem. C 2016, 120, 27128–27134. [Google Scholar] [CrossRef]
- Kim, S.P.; Van Duin, A.C.; Shenoy, V.B. Effect of electrolytes on the structure and evolution of the solid electrolyte interphase (SEI) in Li-ion batteries: A molecular dynamics study. J. Power Sources 2011, 196, 8590–8597. [Google Scholar] [CrossRef]
- De Angelis, P. Reactive and Non-Reactive Interface Modelling for Energy Materials. Ph.D. Thesis, Politecnico di Torino, Torino, IT, USA, 2022. [Google Scholar]
- Chenoweth, K.; Van Duin, A.C.; Goddard, W.A. ReaxFF reactive force field for molecular dynamics simulations of hydrocarbon oxidation. J. Phys. Chem. A 2008, 112, 1040–1053. [Google Scholar] [CrossRef] [Green Version]
- Castro-Marcano, F.; Kamat, A.M.; Russo, M.F., Jr.; van Duin, A.C.; Mathews, J.P. Combustion of an Illinois No. 6 coal char simulated using an atomistic char representation and the ReaxFF reactive force field. Combust. Flame 2012, 159, 1272–1285. [Google Scholar] [CrossRef]
- Strachan, A.; van Duin, A.C.; Chakraborty, D.; Dasgupta, S.; Goddard, W.A., III. Shock waves in high-energy materials: The initial chemical events in nitramine RDX. Phys. Rev. Lett. 2003, 91, 098301. [Google Scholar] [CrossRef] [Green Version]
- Weismiller, M.R.; Van Duin, A.C.; Lee, J.; Yetter, R.A. ReaxFF reactive force field development and applications for molecular dynamics simulations of ammonia borane dehydrogenation and combustion. J. Phys. Chem. A 2010, 114, 5485–5492. [Google Scholar] [CrossRef] [PubMed]
- Agrawalla, S.; Van Duin, A.C. Development and application of a ReaxFF reactive force field for hydrogen combustion. J. Phys. Chem. A 2011, 115, 960–972. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Bonati, L.; Polino, D.; Parrinello, M. Using metadynamics to build neural network potentials for reactive events: The case of urea decomposition in water. Catal. Today 2022, 387, 143–149. [Google Scholar] [CrossRef]
- Andersen, M.; Panosetti, C.; Reuter, K. A practical guide to surface kinetic Monte Carlo simulations. Front. Chem. 2019, 7, 202. [Google Scholar] [CrossRef]
- Methekar, R.N.; Northrop, P.W.; Chen, K.; Braatz, R.D.; Subramanian, V.R. Kinetic Monte Carlo simulation of surface heterogeneity in graphite anodes for lithium-ion batteries: Passive layer formation. J. Electrochem. Soc. 2011, 158, A363. [Google Scholar] [CrossRef]
- Fitzgerald, M.; Picard, R.; Silver, R. Canonical transition probabilities for adaptive Metropolis simulation. Europhys. Lett. 1999, 46, 282. [Google Scholar] [CrossRef]
- Esmaeilpour, M.; Jana, S.; Li, H.; Soleymanibrojeni, M.; Wenzel, W. A Solution-Mediated Pathway for the Growth of the Solid Electrolyte Interphase in Lithium-Ion Batteries. Adv. Energy Mater. 2023, 2203966. [Google Scholar] [CrossRef]
- Colclasure, A.M.; Smith, K.A.; Kee, R.J. Modeling detailed chemistry and transport for solid-electrolyte-interface (SEI) films in Li–ion batteries. Electrochim. Acta 2011, 58, 33–43. [Google Scholar] [CrossRef]
- Christensen, J.; Newman, J. A mathematical model for the lithium-ion negative electrode solid electrolyte interphase. J. Electrochem. Soc. 2004, 151, A1977. [Google Scholar] [CrossRef] [Green Version]
- Ning, G.; White, R.E.; Popov, B.N. A generalized cycle life model of rechargeable Li-ion batteries. Electrochim. Acta 2006, 51, 2012–2022. [Google Scholar] [CrossRef]
- Doyle, M.; Fuller, T.F.; Newman, J. Modeling of galvanostatic charge and discharge of the lithium/polymer/insertion cell. J. Electrochem. Soc. 1993, 140, 1526. [Google Scholar] [CrossRef]
- Ramadass, P.; Haran, B.; Gomadam, P.M.; White, R.; Popov, B.N. Development of first principles capacity fade model for Li-ion cells. J. Electrochem. Soc. 2004, 151, A196. [Google Scholar] [CrossRef]
- Single, F.; Latz, A.; Horstmann, B. Identifying the mechanism of continued growth of the solid–electrolyte interphase. ChemSusChem 2018, 11, 1950–1955. [Google Scholar] [CrossRef] [Green Version]
- Cheng, X.B.; Zhang, R.; Zhao, C.Z.; Wei, F.; Zhang, J.G.; Zhang, Q. A review of solid electrolyte interphases on lithium metal anode. Adv. Sci. 2016, 3, 1500213. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Hu, R.; Zhang, D.; Liu, J.; Liu, F.; Cui, J.; Lin, Z.; Wu, J.; Zhu, M. Constructing Li-Rich Artificial SEI Layer in Alloy–Polymer Composite Electrolyte to Achieve High Ionic Conductivity for All-Solid-State Lithium Metal Batteries. Adv. Mater. 2021, 33, 2004711. [Google Scholar] [CrossRef]
- Lowe, J.S.; Siegel, D.J. Modeling the interface between lithium metal and its native oxide. ACS Appl. Mater. Interfaces 2020, 12, 46015–46026. [Google Scholar] [CrossRef]
- Lorger, S.; Usiskin, R.; Maier, J. Transport and charge carrier chemistry in lithium oxide. J. Electrochem. Soc. 2019, 166, A2215. [Google Scholar] [CrossRef]
- Guo, R.; Gallant, B.M. Li2O solid electrolyte interphase: Probing transport properties at the chemical potential of lithium. Chem. Mater. 2020, 32, 5525–5533. [Google Scholar] [CrossRef]
- Less, G.B.; Seo, J.H.; Han, S.; Sastry, A.M.; Zausch, J.; Latz, A.; Schmidt, S.; Wieser, C.; Kehrwald, D.; Fell, S. Micro-scale modeling of Li-ion batteries: Parameterization and validation. J. Electrochem. Soc. 2012, 159, A697. [Google Scholar] [CrossRef]
- Bhowmik, A.; Castelli, I.E.; Garcia-Lastra, J.M.; Jørgensen, P.B.; Winther, O.; Vegge, T. A perspective on inverse design of battery interphases using multi-scale modelling, experiments and generative deep learning. Energy Storage Mater. 2019, 21, 446–456. [Google Scholar] [CrossRef]
- Shi, S.; Gao, J.; Liu, Y.; Zhao, Y.; Wu, Q.; Ju, W.; Ouyang, C.; Xiao, R. Multi-scale computation methods: Their applications in lithium-ion battery research and development. Chin. Phys. B 2015, 25, 018212. [Google Scholar] [CrossRef]
- Single, F.; Horstmann, B.; Latz, A. Revealing SEI morphology: In-depth analysis of a modeling approach. J. Electrochem. Soc. 2017, 164, E3132. [Google Scholar] [CrossRef] [Green Version]
- Zeng, Z.; Liang, W.I.; Liao, H.G.; Xin, H.L.; Chu, Y.H.; Zheng, H. Visualization of electrode–electrolyte interfaces in LiPF6/EC/DEC electrolyte for lithium ion batteries via in situ TEM. Nano Lett. 2014, 14, 1745–1750. [Google Scholar] [CrossRef] [PubMed]
- An, S.J.; Li, J.; Daniel, C.; Meyer III, H.M.; Trask, S.E.; Polzin, B.J.; Wood III, D.L. Electrolyte volume effects on electrochemical performance and solid electrolyte interphase in Si-graphite/NMC lithium-ion pouch cells. ACS Appl. Mater. Interfaces 2017, 9, 18799–18808. [Google Scholar] [CrossRef]
- Merryweather, A.J.; Schnedermann, C.; Jacquet, Q.; Grey, C.P.; Rao, A. Operando optical tracking of single-particle ion dynamics in batteries. Nature 2021, 594, 522–528. [Google Scholar] [CrossRef]
- Clark, S.; Bleken, F.L.; Stier, S.; Flores, E.; Andersen, C.W.; Marcinek, M.; Szczesna-Chrzan, A.; Gaberscek, M.; Palacin, M.R.; Uhrin, M.; et al. Toward a unified description of battery data. Adv. Energy Mater. 2022, 12, 2102702. [Google Scholar] [CrossRef]
- Castelli, I.E.; Arismendi-Arrieta, D.J.; Bhowmik, A.; Cekic-Laskovic, I.; Clark, S.; Dominko, R.; Flores, E.; Flowers, J.; Ulvskov Frederiksen, K.; Friis, J.; et al. Data Management Plans: The Importance of Data Management in the BIG-MAP Project. Batter. Supercaps 2021, 4, 1803–1812. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, Y.; Tu, Y.; Wang, Q. Reductive decomposition of solvents and additives toward solid-electrolyte interphase formation in lithium-ion battery. J. Phys. Chem. C 2020, 124, 9099–9108. [Google Scholar] [CrossRef]
- Joraleechanchai, N.; Duangdangchote, S.; Sawangphruk, M. Machine Learning and Reactive Force Field Molecular Dynamics Investigation of Electrolytes for Ultra-fast Charging Li-ion Batteries. ECS Trans. 2020, 97, 45. [Google Scholar] [CrossRef]
- Barcaro, G.; Monti, S.; Sementa, L.; Carravetta, V. Parametrization of a reactive force field (ReaxFF) for molecular dynamics simulations of Si nanoparticles. J. Chem. Theory Comput. 2017, 13, 3854–3861. [Google Scholar] [CrossRef]
- Yao, N.; Chen, X.; Fu, Z.H.; Zhang, Q. Applying classical, ab initio, and machine-learning molecular dynamics simulations to the liquid electrolyte for rechargeable batteries. Chem. Rev. 2022, 122, 10970–11021. [Google Scholar] [CrossRef]
- Deringer, V.L. Modelling and understanding battery materials with machine-learning-driven atomistic simulations. J. Phys. Energy 2020, 2, 041003. [Google Scholar] [CrossRef]
- Lombardo, T.; Hoock, J.B.; Primo, E.N.; Ngandjong, A.C.; Duquesnoy, M.; Franco, A.A. Accelerated optimization methods for force-field parametrization in battery electrode manufacturing modeling. Batter. Supercaps 2020, 3, 721–730. [Google Scholar] [CrossRef]
- Chmiela, S.; Tkatchenko, A.; Sauceda, H.E.; Poltavsky, I.; Schütt, K.T.; Müller, K.R. Machine learning of accurate energy-conserving molecular force fields. Sci. Adv. 2017, 3, e1603015. [Google Scholar] [CrossRef] [Green Version]
- De Angelis, P.; Cappabianca, R.; Asinari, P.; Chiavazzo, E. SEI Builder-Jupyter notebooks to build inital SEI morphology. Zenodo 2022. [Google Scholar] [CrossRef]
- Edge, J.S.; O’Kane, S.; Prosser, R.; Kirkaldy, N.D.; Patel, A.N.; Hales, A.; Ghosh, A.; Ai, W.; Chen, J.; Yang, J.; et al. Lithium ion battery degradation: What you need to know. Phys. Chem. Chem. Phys. 2021, 23, 8200–8221. [Google Scholar] [CrossRef] [PubMed]
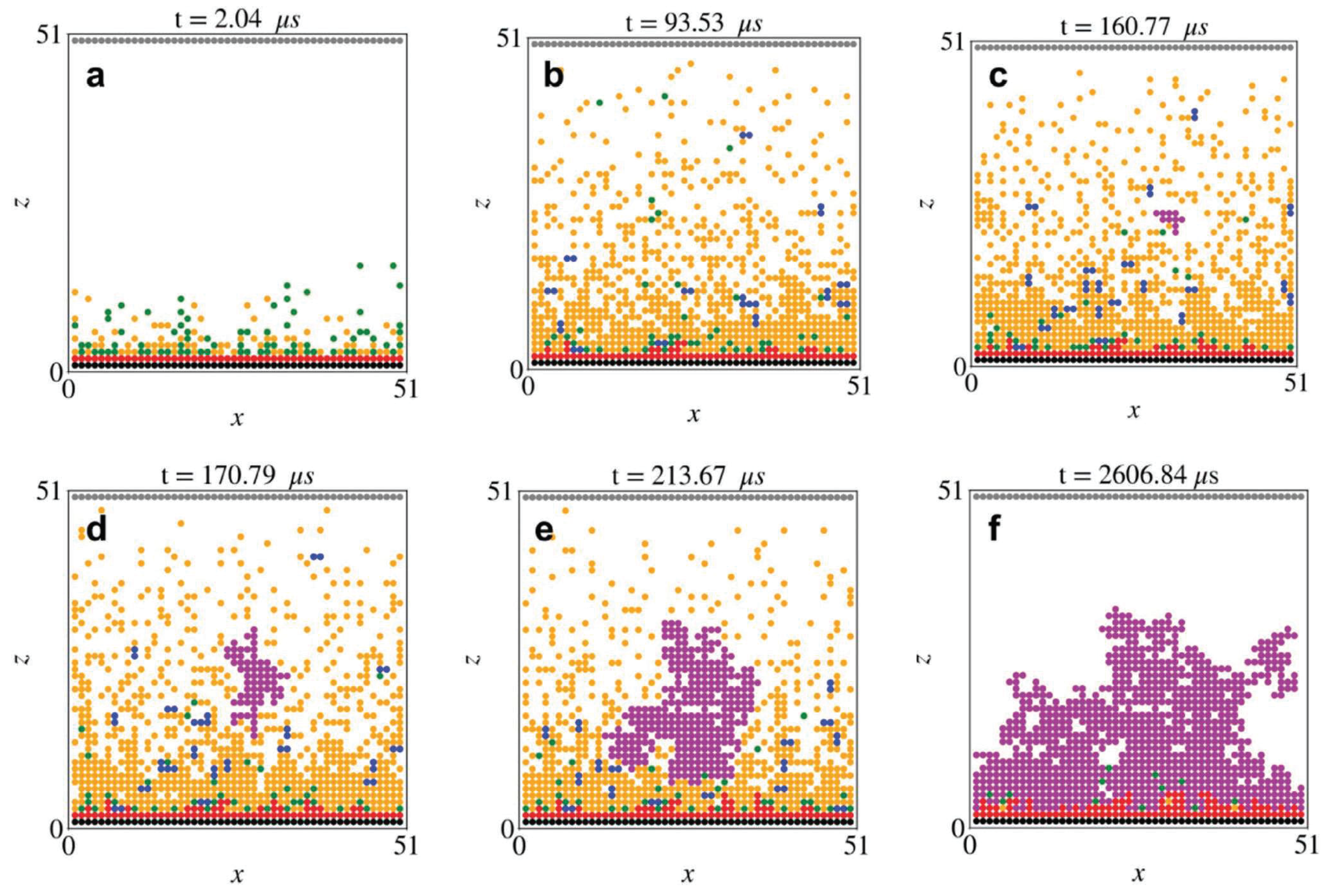
- Guan, P.; Liu, L.; Lin, X. Simulation and experiment on solid electrolyte interphase (SEI) morphology evolution and lithium-ion diffusion. J. Electrochem. Soc. 2015, 162, A1798. [Google Scholar] [CrossRef] [Green Version]
- Abbott, J.W.; Hanke, F. Kinetically Corrected Monte Carlo–Molecular Dynamics Simulations of Solid Electrolyte Interphase Growth. J. Chem. Theory Comput. 2022, 18, 925–934. [Google Scholar] [CrossRef]
- Gerasimov, M.; Soto, F.A.; Wagner, J.; Baakes, F.; Guo, N.; Ospina-Acevedo, F.; Roder, F.; Balbuena, P.B.; Krewer, U. Species Distribution During Solid Electrolyte Interphase Formation on Lithium Using MD/DFT-Parameterized Kinetic Monte Carlo Simulations. J. Phys. Chem. C 2023. [Google Scholar] [CrossRef]
- Kim, J.G.; Son, B.; Mukherjee, S.; Schuppert, N.; Bates, A.; Kwon, O.; Choi, M.J.; Chung, H.Y.; Park, S. A review of lithium and non-lithium based solid state batteries. J. Power Sources 2015, 282, 299–322. [Google Scholar] [CrossRef]
- Lee, D.; Lee, H.; Song, T.; Paik, U. Toward High Rate Performance Solid-State Batteries. Adv. Energy Mater. 2022, 12, 2200948. [Google Scholar] [CrossRef]
- Zhai, Y.; Yang, G.; Zeng, Z.; Song, S.; Li, S.; Hu, N.; Tang, W.; Wen, Z.; Lu, L.; Molenda, J. Composite hybrid quasi-solid electrolyte for high-energy lithium metal batteries. ACS Appl. Energy Mater. 2021, 4, 7973–7982. [Google Scholar] [CrossRef]
- Miara, L.; Windmüller, A.; Tsai, C.L.; Richards, W.D.; Ma, Q.; Uhlenbruck, S.; Guillon, O.; Ceder, G. About the compatibility between high voltage spinel cathode materials and solid oxide electrolytes as a function of temperature. ACS Appl. Mater. Interfaces 2016, 8, 26842–26850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.; Wan, J.; Liu, G.X.; Zuo, T.T.; Song, Y.X.; Liu, B.; Guo, Y.G.; Wen, R.; Wan, L.J. Interfacial Evolution of Lithium Dendrites and Their Solid Electrolyte Interphase Shells of Quasi-Solid-State Lithium-Metal Batteries. Angew. Chem. Int. Ed. 2020, 59, 18120–18125. [Google Scholar] [CrossRef] [PubMed]
- Thangadurai, V.; Weppner, W. Recent progress in solid oxide and lithium ion conducting electrolytes research. Ionics 2006, 12, 81–92. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.T.; Nachimuthu, S.; Brandell, D.; Jiang, J.C. Prediction of SEI Formation in All-Solid-State Batteries: Computational Insights from PCL-based Polymer Electrolyte Decomposition on Lithium-Metal. Batter. Supercaps 2022, 5, e202200088. [Google Scholar] [CrossRef]
- Wu, B.; Wang, S.; Lochala, J.; Desrochers, D.; Liu, B.; Zhang, W.; Yang, J.; Xiao, J. The role of the solid electrolyte interphase layer in preventing Li dendrite growth in solid-state batteries. Energy Environ. Sci. 2018, 11, 1803–1810. [Google Scholar] [CrossRef]
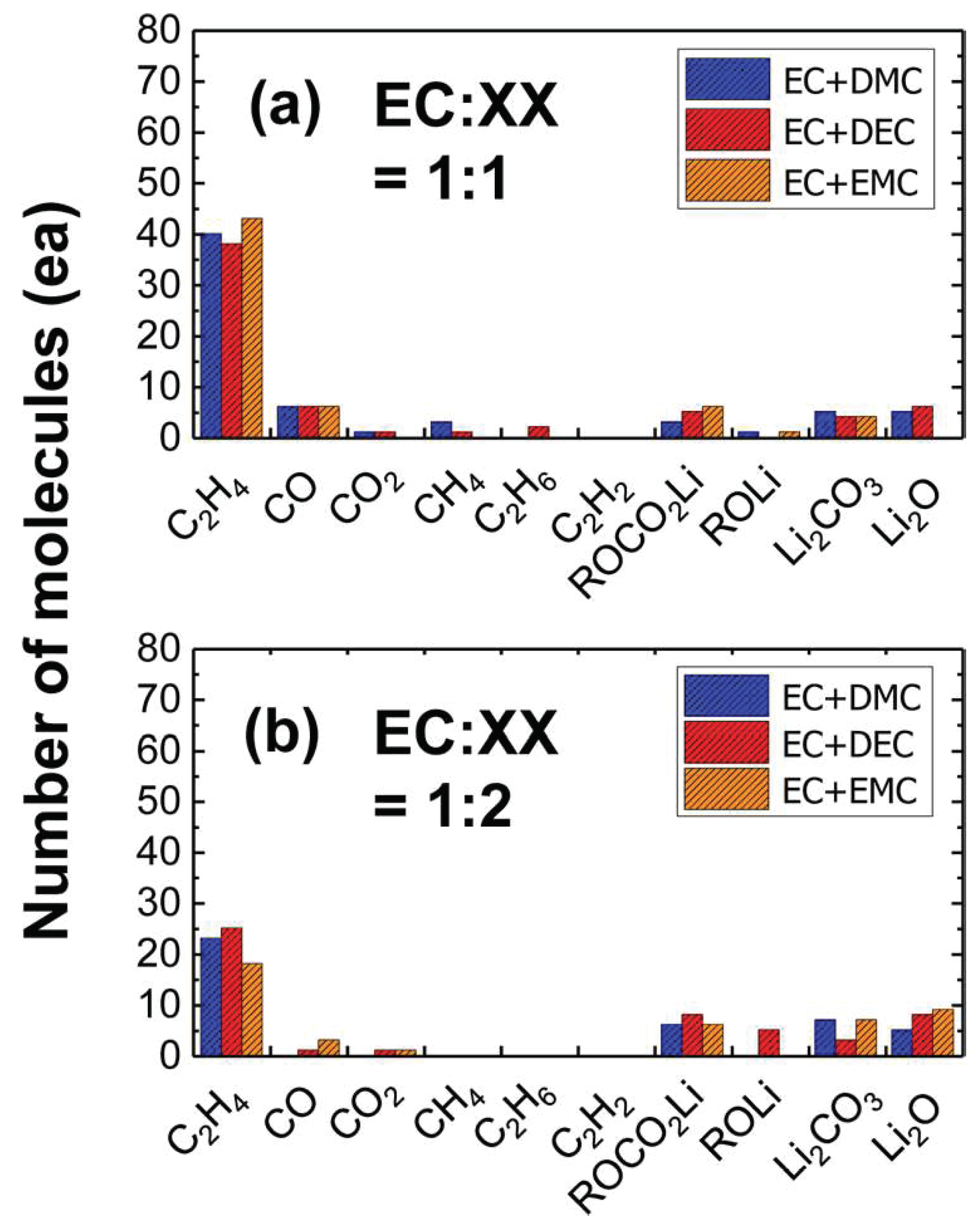
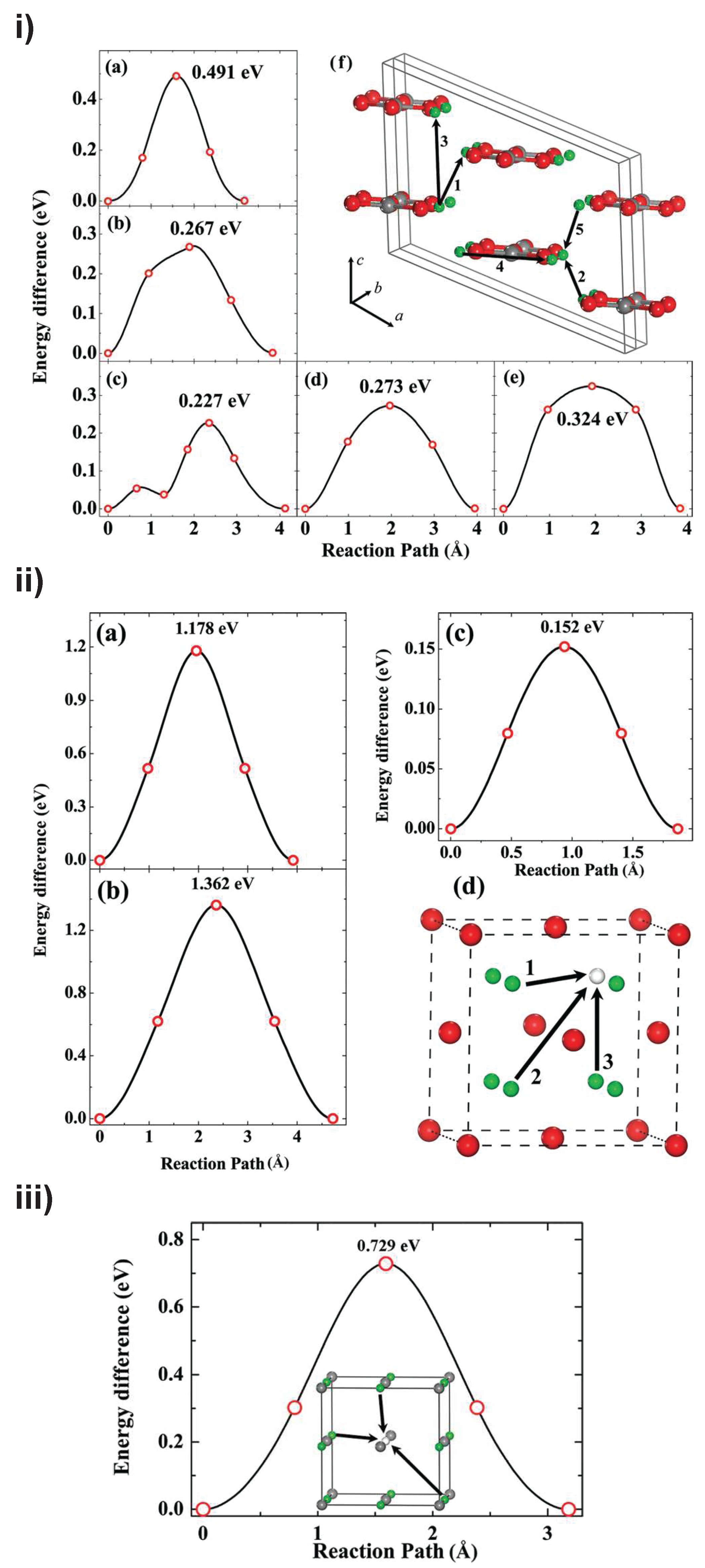



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Component | Li-Ion Diffusion Mechanism | ||||
|---|---|---|---|---|---|
 | Through the grain boundary | 0.68 | |||
 | Through the grain boundary | 0.78 | |||
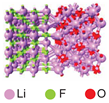 | Through the grain boundary | 0.45 | |||
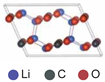 | Vacancy | 0.21 | |||
| Knock-off | 0.24 | ||||
| Direct-hopping | 0.48 | ||||
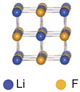 | Vacancy | 0.66 | |||
| Knock-off | 0.25 | ||||
| Direct-hopping | 0.54 | ||||
 | Vacancy | 0.24 | |||
| Knock-off | 0.49 | ||||
| Direct-hopping | 1.76 | ||||
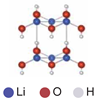 | Vacancy | 0.31 | |||
| Knock-off | 0.20 | ||||
| Direct-hopping | 0.29 | ||||
| System | Method | Calculated Parameters | |
|---|---|---|---|
| Iddir et al. [74] | 193 atoms (interstitial added to the supercell of 192 atoms of ) | DFT studies using NEB method for diffusion | Energy barriers for diffusion |
| Chen et al. [79] | 64 atoms for , 96 atoms for and for | DFT studies using NEB method for diffusion | Energy barriers for diffusion |
| Ahmad et al. [48] | 272 atoms (80 atoms of and 192 atoms of ) | NEB method and analysis of Car-Parrinello MD trajectories | Energy barriers and diffusion coefficient as a function of temperature (Arrhenius fit) |
| Bedrov et al. [60] | 4096 atoms (256 ) | Analysis of MSDs of obtained from MD simulations using polarizable APPLE&P FF | Diffusion coefficient as a function of temperature (Arrhenius fit) |
| Muralidhran et al. [110] | 4096 atoms (256 ) | Analysis of MSDs of obtained from MD simulations using non-polarizable OPLS-AA FF | Diffusion coefficient as a function of temperature (Arrhenius fit) |
| Van Der Ven et al. [59] | Layer and spinal crystal structure of electrode materials | kMC simulations to sample representative Li trajectories | Variation of the diffusion coefficients with Li concentration |
| Ramadass et al. [131] | Lithium-ion battery cell model | Diffusion coefficient is an input parameter of the mass transport equation | Capacity loss due to the side reaction over the negative electrode surface |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cappabianca, R.; De Angelis, P.; Fasano, M.; Chiavazzo, E.; Asinari, P. An Overview on Transport Phenomena within Solid Electrolyte Interphase and Their Impact on the Performance and Durability of Lithium-Ion Batteries. Energies 2023, 16, 5003. https://doi.org/10.3390/en16135003
Cappabianca R, De Angelis P, Fasano M, Chiavazzo E, Asinari P. An Overview on Transport Phenomena within Solid Electrolyte Interphase and Their Impact on the Performance and Durability of Lithium-Ion Batteries. Energies. 2023; 16(13):5003. https://doi.org/10.3390/en16135003
Chicago/Turabian StyleCappabianca, Roberta, Paolo De Angelis, Matteo Fasano, Eliodoro Chiavazzo, and Pietro Asinari. 2023. "An Overview on Transport Phenomena within Solid Electrolyte Interphase and Their Impact on the Performance and Durability of Lithium-Ion Batteries" Energies 16, no. 13: 5003. https://doi.org/10.3390/en16135003