
Molecular Dynamics Simulation Studies of Properties, Preparation, and Performance of Silicon Carbide Materials: A Review

Abstract
:1. Introduction
2. Comparative Analysis of Potential Functions for MD Simulation of Silicon Carbide
2.1. Tersoff Potential
2.2. Tersoff/ZBL Potential
2.3. Vashishta Potential
2.4. EDIP
2.5. MEAM Potential
2.6. Gao-Weber Potential
2.7. Gao-Weber/ZBL Potential
2.8. Comparative Analysis
3. MD Simulation Studies of Silicon Carbide Materials
3.1. MD Simulation Studies of Silicon Carbide Properties
3.1.1. MD Simulation Studies of SiC Thermal Properties
3.1.2. MD Simulation Studies of SiC Mechanical Properties
3.1.3. MD Simulation Studies of SiC Electrical Properties
3.2. MD Simulation Studies of Silicon Carbide Preparation
3.2.1. MD Simulation Studies of SiC Ion Implantation
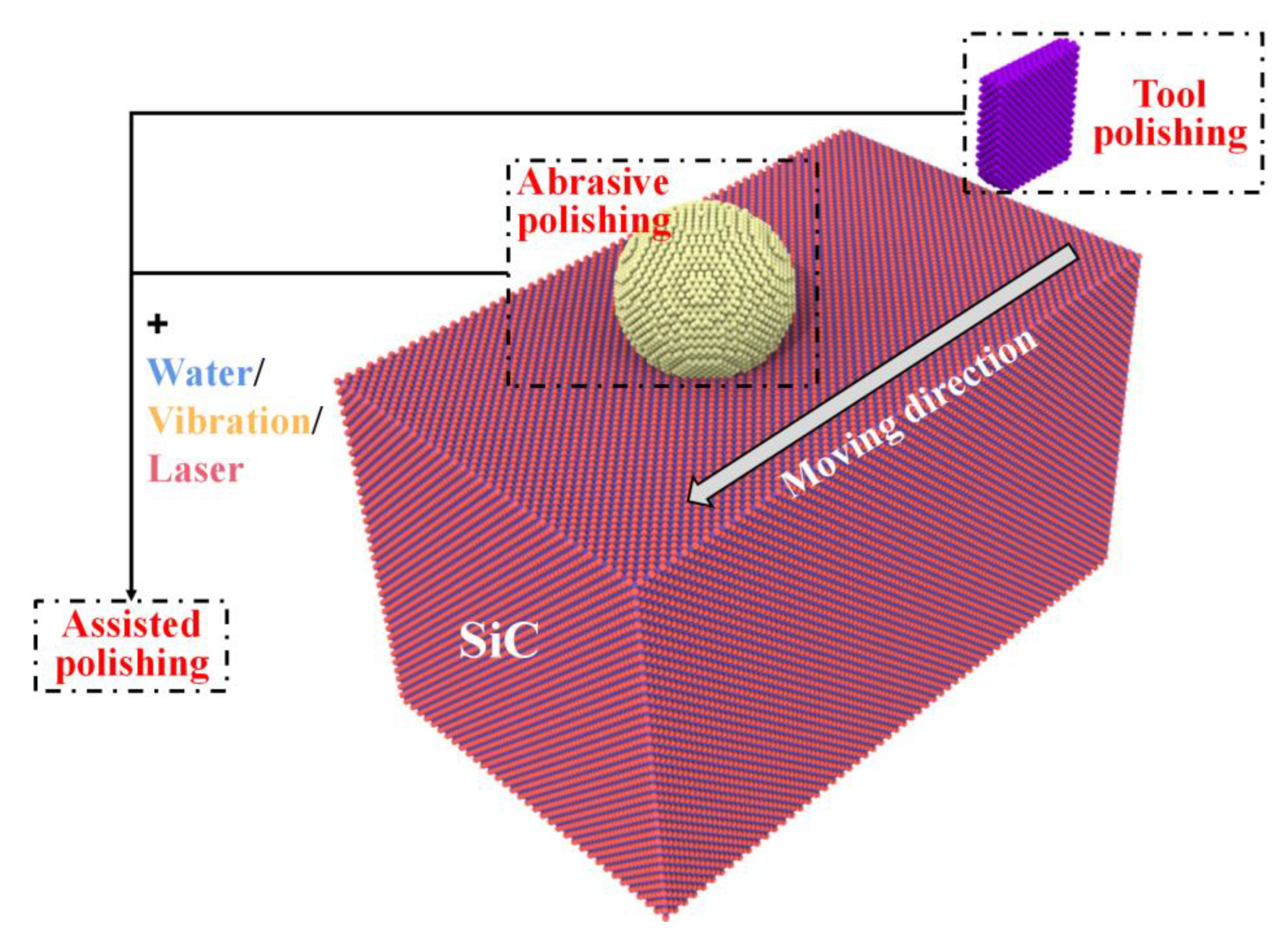
3.2.2. MD Simulation Studies of SiC Polishing
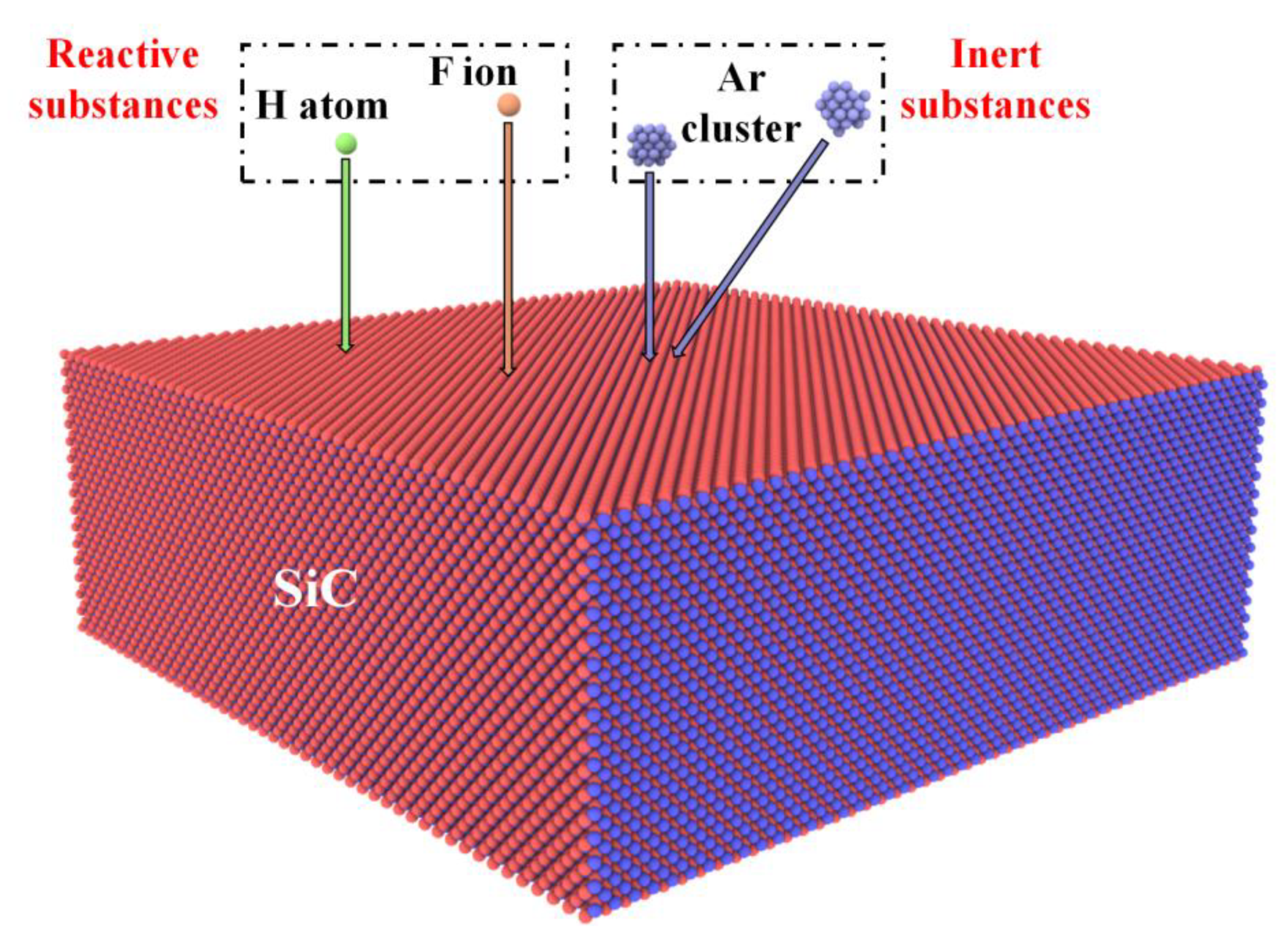
3.2.3. MD Simulation Studies of SiC Sputtering
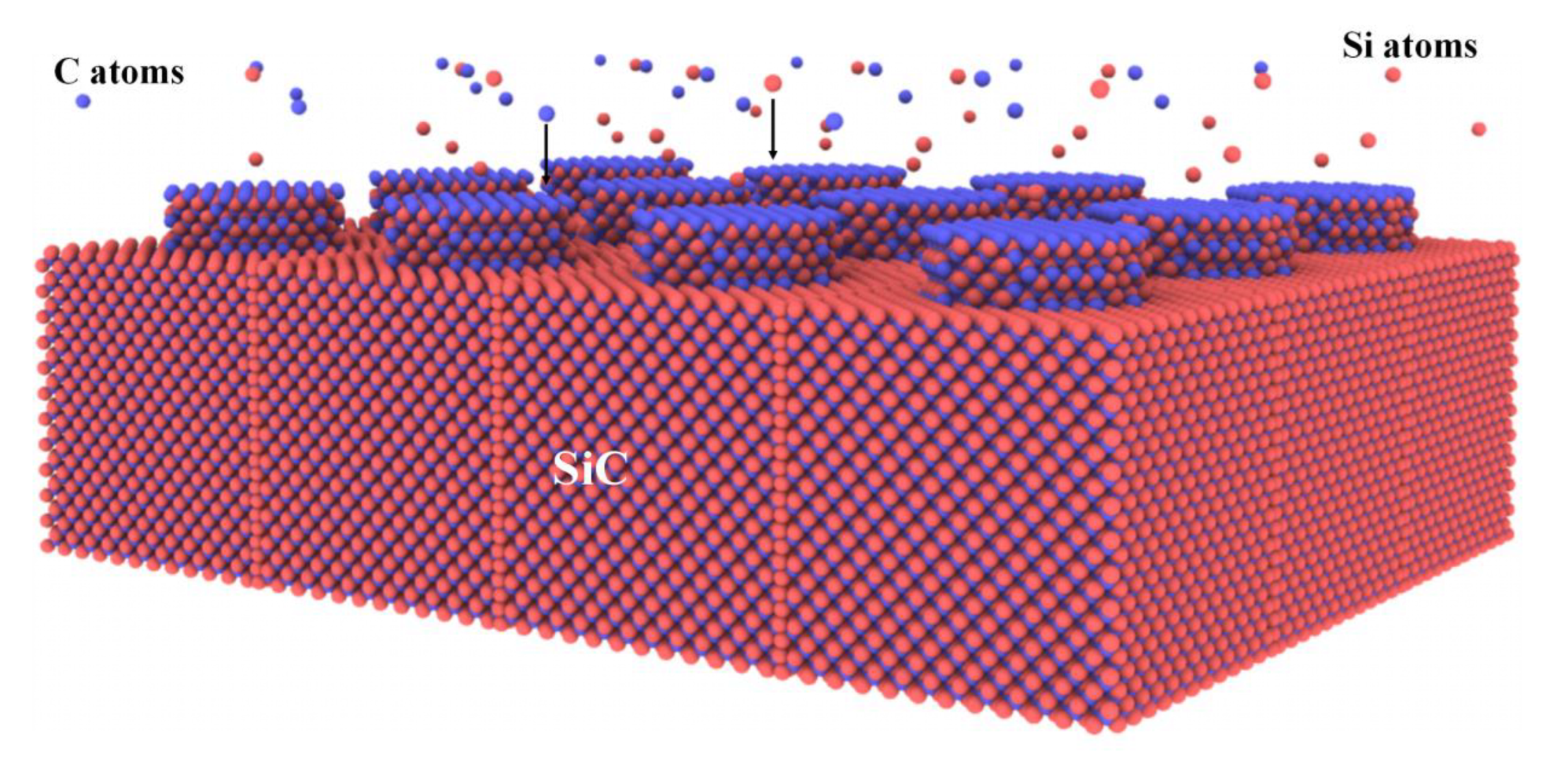
3.2.4. MD Simulation Studies of SiC Deposition
3.2.5. MD Simulation Studies of SiC Crystal Growth
3.2.6. MD Simulation Studies of SiC Amorphization
3.2.7. MD Simulation Studies of SiC Sintering
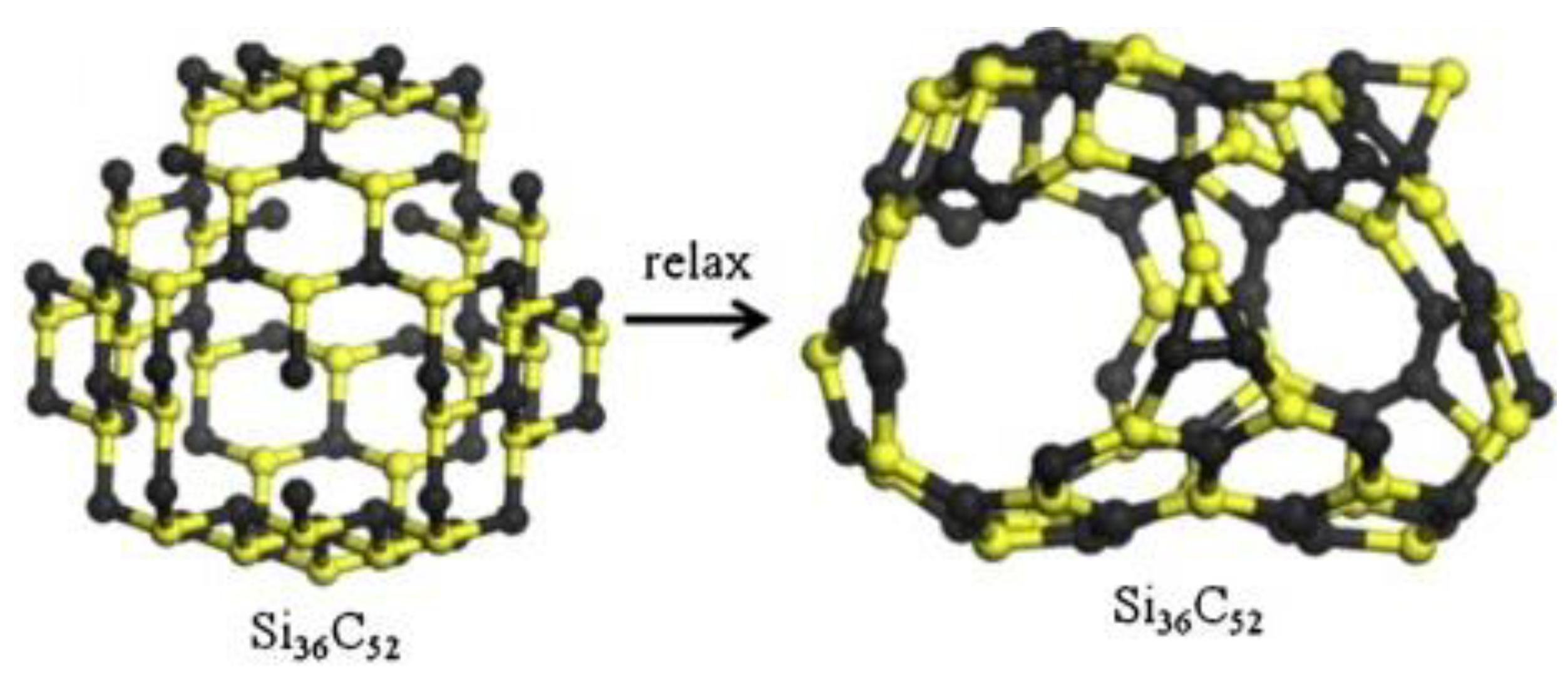
3.2.8. MD Simulation Studies of New-Type SiC Materials Preparation
3.3. MD Simulation Studies of Silicon Carbide Performance
3.3.1. MD Simulation Studies of SiC Irradiation Damage
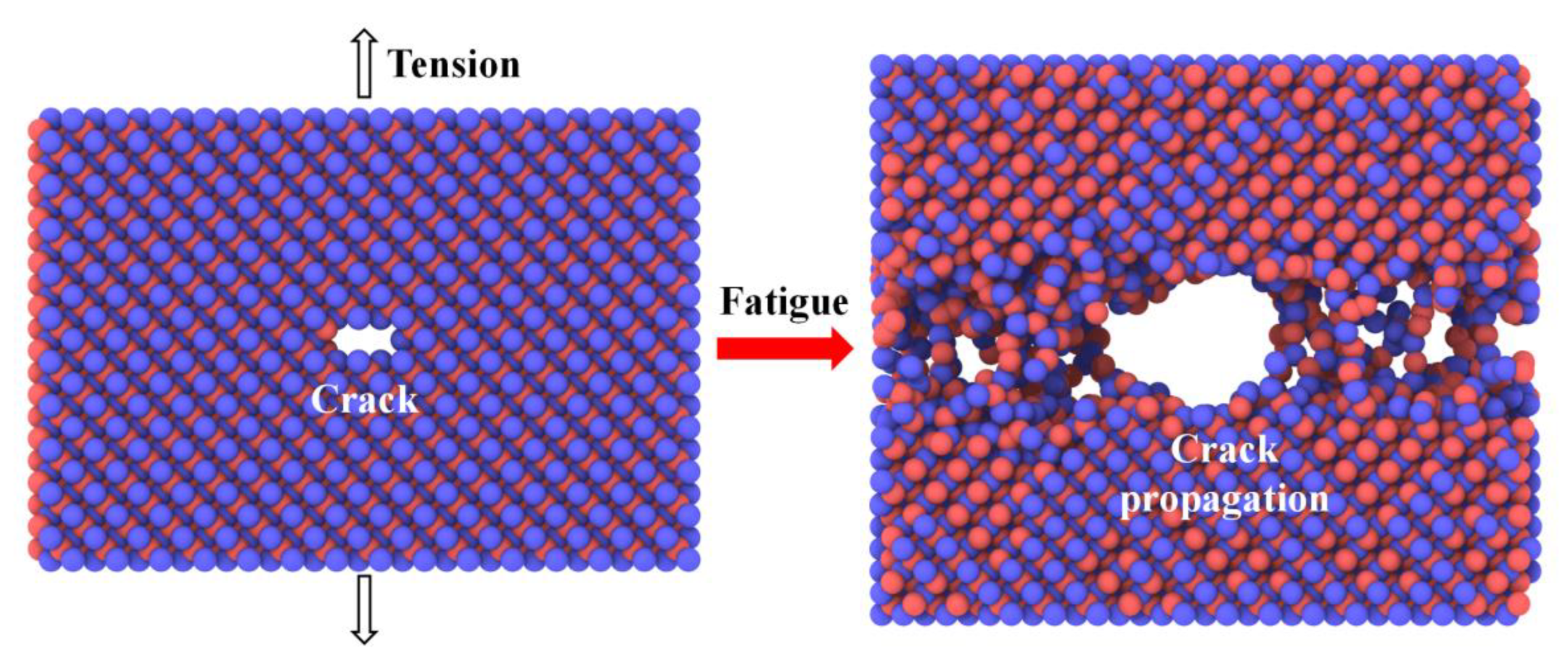
3.3.2. MD Simulation Studies of SiC Fatigue Damage
3.3.3. MD Simulation Studies of SiC Shock Damage
4. Conclusions
- (1)
- The Tersoff potential was the most popular interatomic potential function for SiC materials because it provided a satisfactory balance between computational accuracy and computational speed. Most of the MD simulation types in the properties, preparation, and properties of SiC materials were covered in the application scope of the Tersoff potential.
- (2)
- In the MD simulations of SiC materials’ properties, the thermal and mechanical properties, such as thermal conductivity, hardness, elastic modulus, etc., were studied mostly due to the limitation of the study scale. At present, mature simulation calculation methods were formed, and simulation results similar to the experimental measurements could be obtained.
- (3)
- In the MD simulations of SiC material’s preparation, ion implantation, polishing, sputtering, deposition, crystal growth, etc., were mainly studied. Most of these preparation methods were closely related to semiconductor fabrication processes and well-matched the nanoscale accuracy of semiconductor fabrication. The mechanism and quality in the preparation of SiC materials were mainly focused to obtain the guidance of preparation process optimization.
- (4)
- In the MD simulations of SiC materials performance, the irradiation damage was studied mostly. There are also some studies on the fatigue damage and impact damage. These studies are closely related to the performance of SiC materials in the field of nuclear energy. The effects of various damages (especially irradiation damage) on the properties of SiC materials during service, as well as the process and mechanism of damage, could be revealed by MD simulation from the atomic-level microscopic perspective. The theoretical guidance for the design optimization of SiC materials for nuclear energy and the improvement of nuclear reactor safety could be obtained.
5. Prospect of Future Study
- (1)
- In the preparation of SiC materials, including the chemical vapor deposition, sintering, and embedding solid phase reaction preparation of SiC are common methods of nuclear fuel preparation. For example, in the fourth-generation nuclear energy system, SiC can be applied as the cladding material of tri-structural iso-tropic (TRISO) fuel or the matrix material of FCM-ATF. The former is prepared by fluidized bed chemical vapor deposition (FB-CVD) technology, and the latter is prepared by powder sintering. Therefore, it is necessary to strengthen the study on CVD and nanoparticle sintering in the MD simulation of SiC materials preparation. The appropriate microscopic process parameters to characterize the preparation process are necessary to find. They should be linked with the macroscopic parameters to provide reference and guidance for the preparation process optimization of nuclear fuel and SiC ceramics.
- (2)
- In the MD simulation of SiC properties, the accuracy of the simulation of thermal and mechanical parameters should be focused on. The development of the MD potential function and the accuracy of potential function parameters is involved. In the MD simulation study of SiC materials, the potential function is the basis and key aspect. The parameter optimization and verification, based on the existing potential functions of SiC, are necessary to strengthen to ensure the accuracy of the simulation results of properties, preparation, and properties. In addition, most of the current MD simulations of SiC materials stay at the level of physical phenomena but rarely involve chemical reactions. Therefore, the reactive force-field (ReaxFF) MD study and the development of ReaxFF potential for SiC materials is necessary to strengthen. It is particularly helpful for the preparation and performance simulation closer to reality. More importantly, more suitable potential functions for specific fields, environments, and purposes are needed to be developed. The microscopic and macroscopic connection parameters are necessary to develop, in particular. It means parameters that are more helpful for macroscopic experimental verification, such as melting point, elastic modulus, and Poisson’s ratio, etc., are obtained from the MD microscopic simulation results.
- (3)
- In the MD simulation of the SiC performance, there are many more scenarios that should be considered. The nuclear energy field (multiple ray collaborative irradiation), the semiconductor field (precision manufacturing, threshold characteristics), the bulletproof material field (mechanical shock), the chemical environmental protection machinery field (corrosion resistance, high-temperature resistance), etc., are included. It should be noted that the MD simulation of irradiation damage has been well studied and developed as a typical application scenario in the field of nuclear energy. However, more influencing factors should be considered to make the study closer to the real reactor environment and strengthen the verification of experimental results.
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liu, R.Z.; Liu, B.; Zhang, K.H.; Liu, M.L.; Shao, Y.L.; Tang, C.H. High temperature oxidation behavior of SiC coating in TRISO coated particles. J. Nucl. Mater. 2014, 453, 107–114. [Google Scholar] [CrossRef]
- Shi, L.; Zhao, J.Q.; Liu, B.; Li, X.W.; Luo, X.W.; Zhang, Z.M.; Zhang, P.; Sun, L.B.; Wu, X.X. Development strategy of key materials technology for the high temperature gas-cooled reactor. J. Tsinghua Univ. 2021, 61, 270–278. [Google Scholar] [CrossRef]
- Terrani, K.A.; Kiggans, J.O.; Katoh, Y.; Shimoda, K.; Montgomery, F.C.; Armstrong, B.L.; Parish, C.M.; Hinoki, T.; Hunn, J.D.; Snead, L.L. Fabrication and characterization of fully ceramic microencapsulated fuels. J. Nucl. Mater. 2012, 426, 268–276. [Google Scholar] [CrossRef]
- She, X.; Huang, A.Q.; Lucia, O.; Ozpineci, B. Review of Silicon Carbide Power Devices and Their Applications. IEEE Trans. Ind. Electron. 2017, 64, 8193–8205. [Google Scholar] [CrossRef]
- Li, C.R.; Xie, Z.P.; Kang, G.X.; An, D.; Wei, H.K.; Zhao, L. Research and Application Progress of SiC Ceramics: A Review. Bull. Chin. Ceram. Soc. 2020, 39, 1353–1370. [Google Scholar] [CrossRef]
- Zhai, F.R.; Shan, K.; Xie, Z.P.; Sun, J.L.; Yi, Z.Z. Preparation and Mechanical Properties of SiC/BN Multiphase Ceramics via Spark Plasma Sintering. J. Chin. Ceram. Soc. 2016, 44, 866–871. [Google Scholar] [CrossRef]
- Li, C.R.; Xie, Z.P.; Zhao, L. Research and Application of Sintering Technologies for SiC Ceramic Materials: A review. J. Ceram. 2020, 41, 137–149. [Google Scholar] [CrossRef]
- Thompson, A.P.; Aktulga, H.M.; Berger, R.; Bolintineanu, D.S.; Brown, W.M.; Crozier, P.S.; Veld, P.J.I.; Kohlmeyer, A.; Moore, S.G.; Nguyen, T.D.; et al. LAMMPS—A flexible simulation tool for particle-based materials modeling at the atomic, meso, and continuum scales. Comput. Phys. Commun. 2022, 271, 108171. [Google Scholar] [CrossRef]
- Tersoff, J. New Empirical-Approach for the Structure and Energy of Covalent Systems. Phys. Rev. B 1988, 37, 6991–7000. [Google Scholar] [CrossRef]
- Tersoff, J. Modeling Solid-State Chemistry: Interatomic Potentials for Multicomponent Systems. Phys. Rev. B 1989, 39, 5566–5568. [Google Scholar] [CrossRef]
- Devanathan, R.; de la Rubia, T.D.; Weber, W.J. Displacement threshold energies in β-SiC. J. Nucl. Mater. 1998, 253, 47–52. [Google Scholar] [CrossRef]
- Ziegler, J.F.; Biersack, J.P. The Stopping and Range of Ions in Matter. In Treatise on Heavy-Ion Science: Volume 6: Astrophysics, Chemistry, and Condensed Matter; Bromley, D.A., Ed.; Springer: Boston, MA, USA, 1985; pp. 93–129. [Google Scholar]
- Vashishta, P.; Kalia, R.K.; Rino, J.P.; Ebbsjo, I. Interaction Potential for SiO2—A Molecular-Dynamics Study of Structural Correlations. Phys. Rev. B 1990, 41, 12197–12209. [Google Scholar] [CrossRef] [PubMed]
- Vashishta, P.; Kalia, R.K.; Nakano, A.; Rino, J.P. Interaction potential for silicon carbide: A molecular dynamics study of elastic constants and vibrational density of states for crystalline and amorphous silicon carbide. J. Appl. Phys. 2007, 101, 103515. [Google Scholar] [CrossRef] [Green Version]
- Bazant, M.Z.; Kaxiras, E.; Justo, J.F. Environment-dependent interatomic potential for bulk silicon. Phys. Rev. B 1997, 56, 8542–8552. [Google Scholar] [CrossRef] [Green Version]
- Jiang, C.; Morgan, D.; Szlufarska, I. Carbon tri-interstitial defect: A model for the D-II center. Phys. Rev. B 2012, 86, 144118. [Google Scholar] [CrossRef] [Green Version]
- Baskes, M.I. Modified Embedded-Atom Potentials for Cubic Materials and Impurities. Phys. Rev. B 1992, 46, 2727–2742. [Google Scholar] [CrossRef]
- Huang, H.C.; Ghoniem, N.M.; Wong, J.K.; Baskes, M.I. Molecular-Dynamics Determination of Defect Energetics in β-SiC Using three Representative Empirical Potentials. Model. Simul. Mater. Sc. 1995, 3, 615–627. [Google Scholar] [CrossRef]
- Kang, K.H.; Eun, T.; Jun, M.C.; Lee, B.J. Governing factors for the formation of 4H or 6H-SiC polytype during SiC crystal growth: An atomistic computational approach. J. Cryst. Growth. 2014, 389, 120–133. [Google Scholar] [CrossRef]
- Gao, F.; Weber, W.J. Empirical potential approach for defect properties in 3C-SiC. Nucl. Instrum. Meth. B 2002, 191, 504–508. [Google Scholar] [CrossRef]
- Gao, F.; Devanathan, R.; Zhang, Y.; Weber, W.J. Annealing simulations of nano-sized amorphous structures in SiC. Nucl. Instrum. Meth. B 2005, 228, 282–287. [Google Scholar] [CrossRef]
- Tersoff, J. Carbon Defects and Defect Reactions in Silicon. Phys. Rev. Lett. 1990, 64, 1757–1760. [Google Scholar] [CrossRef] [PubMed]
- Tersoff, J. Chemical Order in Amorphous-Silicon Carbide. Phys. Rev. B 1994, 49, 16349–16352. [Google Scholar] [CrossRef] [PubMed]
- Erhart, P.; Albe, K. Analytical potential for atomistic simulations of silicon, carbon, and silicon carbide. Phys. Rev. B 2005, 71, 035211. [Google Scholar] [CrossRef] [Green Version]
- Stillinger, F.H.; Weber, T.A. Computer-Simulation of Local Order in Condensed Phases of Silicon. Phys. Rev. B 1985, 31, 5262–5271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, C.L.; Luo, X.Y.; Xie, Q.; Wu, Q.D. Molecular dynamics study of thermal conductivity of carbon nanotubes and silicon carbide nanotubes. Acta Phys. Sin. 2022, 71, 030202. [Google Scholar] [CrossRef]
- Mao, Y.C.; Li, Y.Y.; Xiong, Y.H.; Xiao, W. Point defect effects on the thermal conductivity of β-SiC by molecular dynamics simulations. Comput. Mater. Sci. 2018, 152, 300–307. [Google Scholar] [CrossRef]
- Wang, Q.; Gui, N.; Huang, X.L.; Yang, X.T.; Tu, J.Y.; Jiang, S.Y. The effect of temperature and cascade collision on thermal conductivity of 3C-SiC: A molecular dynamics study. Int. J. Heat Mass Transf. 2021, 180, 121822. [Google Scholar] [CrossRef]
- Dong, X.Y.; Shin, Y.C. Predictions of thermal conductivity and degradation of irradiated SiC/SiC composites by materials-genome-based multiscale modeling. J. Nucl. Mater. 2018, 512, 268–275. [Google Scholar] [CrossRef]
- Liu, Q.F.; Yu, W.S.; Luo, H.; Ren, X.; Shen, S.P. Tuning thermal resistance of SiC crystal/amorphous layered nanostructures via changing layer thickness. Comput. Mater. Sci. 2020, 184, 109868. [Google Scholar] [CrossRef]
- Samolyuk, G.D.; Golubov, S.I.; Osetsky, Y.N.; Stoller, R.E. Molecular dynamics study of influence of vacancy types defects on thermal conductivity of β-SiC. J. Nucl. Mater. 2011, 418, 174–181. [Google Scholar] [CrossRef]
- Wang, F.; Zhou, Y.; Gao, S.X.; Duan, Z.G.; Sun, Z.P.; Wang, J.; Zou, Y.; Fu, B.Q. Molecular dynamics study of effects of point defects on thermal conductivity in cubic silicon carbide. Acta Phys. Sin. 2022, 71, 036501. [Google Scholar] [CrossRef]
- Zhang, B.Y.; Qiu, H.P.; Tian, Y.; Chen, M.W.; Chen, S.H. Tension-Tension Fatigue of 2.5D-SiC/SiC Ceramic Matrix Composites at Elevated Temperatures. IOP Conf. Ser. Mater. Sci. Eng. 2019, 678, 012057. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.Z.; Yao, P.; Wang, J.; Huang, C.Z.; Zhu, H.T.; Liu, H.L.; Zou, B. Nanomechanical characterization of RB-SiC ceramics based on nanoindentation and modelling of the ground surface roughness. Ceram. Int. 2020, 46, 6243–6253. [Google Scholar] [CrossRef]
- Yang, B.; Deng, Q.B.; Su, Y.; Peng, X.H.; Huang, C.; Lee, A.; Hu, N. The effects of atomic arrangements on mechanical properties of 2H, 3C, 4H and 6H-SiC. Comput. Mater. Sci. 2022, 203, 111114. [Google Scholar] [CrossRef]
- Li, Y.Y.; Li, Y.; Xiao, W. Point defects and grain boundary effects on tensile strength of 3C-SiC studied by molecular dynamics simulations. Nucl. Eng. Technol. 2019, 51, 769–775. [Google Scholar] [CrossRef]
- Molaei, F.; Dehaghani, M.Z.; Salmankhani, A.; Fooladpanjeh, S.; Sajadi, S.M.; Safa, M.E.; Abida, O.; Habibzadeh, S.; Mashhadzadeh, A.H.; Saeb, M.R. Applying molecular dynamics simulation to take the fracture fingerprint of polycrystalline SiC nanosheets. Comput. Mater. Sci. 2021, 200, 110770. [Google Scholar] [CrossRef]
- Wu, W.L.; Hu, Y.; Meng, X.S.; Dai, J.B.; Dai, H.F. Molecular dynamics simulation of ion-implanted single-crystal 3C-SiC nano-indentation. J. Manuf. Process. 2022, 79, 356–368. [Google Scholar] [CrossRef]
- Kang, Q.; Fang, X.; Wu, C.; Verma, P.; Sun, H.; Tian, B.; Zhao, L.; Wang, S.; Zhu, N.; Maeda, R.; et al. Mechanical properties and indentation-induced phase transformation in 4H–SiC implanted by hydrogen ions. Ceram. Int. 2022, 48, 15334–15347. [Google Scholar] [CrossRef]
- Xue, L.H.; Feng, G.; Liu, S. Molecular dynamics study of temperature effect on deformation behavior of m-plane 4H-SiC film by nanoindentation. Vacuum 2022, 202, 111192. [Google Scholar] [CrossRef]
- Xue, L.H.; Feng, G.; Wu, G.; Dong, F.; Liang, K.; Li, R.; Wang, S.Z.; Liu, S. Study of deformation mechanism of structural anisotropy in 4H-SiC film by nanoindentation. Mater. Sci. Semicond. Process. 2022, 146, 106671. [Google Scholar] [CrossRef]
- Domingues, G.; Monthe, A.M.; Guevelou, S.; Rousseau, B. Study by molecular dynamics of the influence of temperature and pressure on the optical properties of undoped 3C-SiC structures. J. Quant. Spectrosc. Radiat. Transf. 2018, 205, 220–229. [Google Scholar] [CrossRef]
- Chen, W.; Li, L.S. The study of the optical phonon frequency of 3C-SiC by molecular dynamics simulations with deep neural network potential. J. Appl. Phys. 2021, 129, 244104. [Google Scholar] [CrossRef]
- Fan, Y.X.; Xu, Z.W.; Song, Y.; Dong, B.; Xue, Z.F.; Liu, B.; Liu, L.; Tian, D.Y. Nano material removal mechanism of 4H-SiC in ion implantation-assisted machining. Comput. Mater. Sci. 2021, 200, 110837. [Google Scholar] [CrossRef]
- Zhang, X.D.; Li, Q.; Wang, M.; Zhang, Z.T.; Akhmadaliev, S.; Zhou, S.Q.; Wu, Y.Y.; Guo, B. Defects in hydrogen implanted SiC. Nucl. Instrum. Meth. B 2018, 436, 107–111. [Google Scholar] [CrossRef]
- Dai, H.F.; Hu, Y.; Wu, W.L.; Yue, H.X.; Meng, X.S.; Li, P.; Duan, H.G. Molecular dynamics simulation of ultra-precision machining 3C-SiC assisted by ion implantation. J. Manuf. Process. 2021, 69, 398–411. [Google Scholar] [CrossRef]
- Liu, B.; Xu, Z.W.; Wang, Y.; Gao, X.; Kong, R.J. Effect of ion implantation on material removal mechanism of 6H-SiC in nano-cutting: A molecular dynamics study. Comput. Mater. Sci. 2020, 174, 109476. [Google Scholar] [CrossRef]
- Fan, Y.X.; Xu, Z.W.; Song, Y.; Sun, T.Z. Molecular dynamics simulation of silicon vacancy defects in silicon carbide by hydrogen ion implantation and subsequent annealing. Diam. Relat. Mater. 2021, 119, 108595. [Google Scholar] [CrossRef]
- Kang, Q.; Fang, X.D.; Wu, C.; Sun, H.; Tian, B.; Zhao, L.B.; Wang, S.L.; Jiang, Z.D.; Zhu, N.; Maeda, R.; et al. Modification mechanism of collaborative ions implanted into 4H-SiC by atomic simulation and experiment. Int. J. Mech. Sci. 2021, 212, 106832. [Google Scholar] [CrossRef]
- Ma, G.L.; Li, S.J.; Liu, F.L.; Zhang, C.; Jia, Z.; Yin, X.C. A Review on Precision Polishing Technology of Single-Crystal SiC. Crystals 2022, 12, 101. [Google Scholar] [CrossRef]
- Zhou, P.; Zhu, N.N.; Xu, C.Y.; Niu, F.L.; Li, J.; Zhu, Y.W. Mechanical removal of SiC by multi-abrasive particles in fixed abrasive polishing using molecular dynamics simulation. Comput. Mater. Sci. 2021, 191, 110311. [Google Scholar] [CrossRef]
- Wu, Z.H.; Zhang, L.C.; Yang, S.Y.; Wu, C.H. Effects of grain size and protrusion height on the surface integrity generation in the nanogrinding of 6H-SiC. Tribol. Int. 2022, 171, 107563. [Google Scholar] [CrossRef]
- Zhou, Y.Q.; Huang, Y.H.; Li, J.M.; Zhu, F.L. The effect of contact types on SiC polishing process. Mater. Sci. Semicond. Process. 2022, 147, 106709. [Google Scholar] [CrossRef]
- Meng, B.B.; Yuan, D.D.; Xu, S.L. Study on strain rate and heat effect on the removal mechanism of SiC during nano-scratching process by molecular dynamics simulation. Int. J. Mech. Sci. 2019, 151, 724–732. [Google Scholar] [CrossRef]
- Luo, Q.F.; Lu, J.; Tian, Z.; Jiang, F. Controllable material removal behavior of 6H-SiC wafer in nanoscale polishing. Appl. Surf. Sci. 2021, 562, 150219. [Google Scholar] [CrossRef]
- Zhang, S.; Cheng, X.; Chen, J.Y. Surface deformation, phase transition and dislocation mechanisms of single crystalline 6H-SiC in oblique nano-cutting. Appl. Surf. Sci. 2022, 588, 152944. [Google Scholar] [CrossRef]
- Meng, B.B.; Yuan, D.D.; Zheng, J.; Qiu, P.; Xu, S.L. Tip-based nanomanufacturing process of single crystal SiC: Ductile deformation mechanism and process optimization. Appl. Surf. Sci. 2020, 500, 144039. [Google Scholar] [CrossRef]
- Chavoshi, S.Z.; Luo, X.C. Molecular dynamics simulation study of deformation mechanisms in 3C-SiC during nanometric cutting at elevated temperatures. Mater. Sci. Eng. A-Struct. 2016, 654, 400–417. [Google Scholar] [CrossRef] [Green Version]
- Zhou, P.; Li, J.; Wang, Z.K.; Chen, J.P.; Li, X.; Zhu, Y.W. Molecular dynamics study of the removal mechanism of SiC in a fixed abrasive polishing in water lubrication. Ceram. Int. 2020, 46, 24961–24974. [Google Scholar] [CrossRef]
- Nguyen, V.T.; Fang, T.H. Material removal and interactions between an abrasive and a SiC substrate: A molecular dynamics simulation study. Ceram. Int. 2020, 46, 5623–5633. [Google Scholar] [CrossRef]
- Hu, Z.W.; Chen, Y.; Lai, Z.Y.; Yu, Y.Q.; Xu, X.P.; Peng, Q.; Zhang, L. Coupling of double grains enforces the grinding process in vibration-assisted scratch: Insights from molecular dynamics. J. Mater. Process. Technol. 2022, 304, 117551. [Google Scholar] [CrossRef]
- Zhao, L.; Zhang, J.J.; Zhang, J.G.; Hartmaier, A. Atomistic investigation of machinability of monocrystalline 3C-SiC in elliptical vibration-assisted diamond cutting. Ceram. Int. 2021, 47, 2358–2366. [Google Scholar] [CrossRef]
- Meng, B.B.; Yuan, D.D.; Zheng, J.; Xu, S.L. Molecular dynamics study on femtosecond laser aided machining of monocrystalline silicon carbide. Mater. Sci. Semicond. Process. 2019, 101, 1–9. [Google Scholar] [CrossRef]
- Satake, S.; Yamashina, S.; Inoue, N.; Kunugi, T.; Shibahara, M. Large-scale molecular dynamics simulation of sputtering process with glancing-angle Ar cluster impact on 4H-SiC. Nucl. Instrum. Meth. B 2009, 267, 3258–3262. [Google Scholar] [CrossRef]
- Triendl, F.; Pfusterschmied, G.; Schwarz, S.; Pobegen, G.; Konrath, J.P.; Schmid, U. Barrier height tuning by inverse sputter etching at poly-Si/4H-SiC heterojunction diodes. Semicond. Sci. Technol. 2021, 36, 055021. [Google Scholar] [CrossRef]
- Prskalo, A.P.; Schmauder, S.; Ziebert, C.; Ye, J.; Ulrich, S. Molecular dynamics simulations of the sputtering of SiC and Si3N4. Surf. Coat. Technol. 2010, 204, 2081–2084. [Google Scholar] [CrossRef]
- Sun, W.; Liu, H.; Lin, L.; Zhao, C.; Lu, X.; He, P.; Gou, F. Molecular Dynamics Simulations of Atomic H Etching SiC Surface. Phys. Procedia 2012, 32, 539–544. [Google Scholar] [CrossRef] [Green Version]
- Gou, F.; Zen, L.T.; Meng, C.L. Molecular dynamics simulations of CF3 etching of SiC. Thin Solid Films 2008, 516, 1832–1837. [Google Scholar] [CrossRef]
- Lu, X.; Ning, J.; Qin, Y.; Qiu, Q.; Chuanwu, Z.; Ying, Y.; Ming, J.; Gou, F. Substrate temperature effect on F+ etching of SiC: Molecular dynamics simulation. Nucl. Instrum. Meth. B 2009, 267, 3235–3237. [Google Scholar] [CrossRef]
- Xue, L.H.; Feng, G.; Wu, G.; Wang, S.Z.; Li, R.; Han, X.; Sun, Y.M.; Liu, S. Study of the deposition of nanopillar-patterned 4H-SiC by molecular dynamics simulation. Appl. Surf. Sci. 2022, 579, 152209. [Google Scholar] [CrossRef]
- Teng, M.; He, X.D.; Sun, Y. Composition and nanohardness of SiC films deposited by electron beam physical vapor deposition. Int. J. Mod. Phys. B 2009, 23, 1910–1915. [Google Scholar] [CrossRef]
- Kim, J.Y.; Lee, B.W.; Nam, H.S.; Kwon, D. Effect of substrate temperature on structure and intrinsic stress in vapor-deposited amorphous silicon carbide film. Thin Solid Films 2004, 467, 294–299. [Google Scholar] [CrossRef]
- Kim, J.Y.; Lee, B.W.; Nam, H.S.; Kwon, D. Molecular dynamics analysis of structure and intrinsic stress in amorphous silicon carbide film with deposition process parameters. Mater. Sci. Forum 2004, 449–452, 97–100. [Google Scholar] [CrossRef]
- Gao, Y.; Yan, W.J.; Gao, T.H.; Chen, Q.; Yang, W.S.; Xie, Q.; Tian, Z.A.; Liang, Y.C.; Luo, J.; Li, L.X. Properties of the structural defects during SiC-crystal-induced crystallization on the solid-liquid interface. Mater. Sci. Semicond. Process. 2020, 116, 105155. [Google Scholar] [CrossRef]
- Narumi, T.; Shibuta, Y.; Yoshikawa, T. Molecular dynamics simulation of interfacial growth of SiC from Si-C solution on different growth planes. J. Cryst. Growth 2018, 494, 36–43. [Google Scholar] [CrossRef]
- Nguyen, H.T.T. Structural evolution of SiC sheet in a graphene-based in-plane hybrid system upon heating using molecular dynamics simulation. Thin Solid Films 2021, 739, 138992. [Google Scholar] [CrossRef]
- Tranh, D.T.N.; Hoang, V.V.; Hanh, T.T.T. Modeling glassy SiC nanoribbon by rapidly cooling from the liquid: An affirmation of appropriate potentials. Phys. B 2021, 608, 412746. [Google Scholar] [CrossRef]
- Hoang, V.V. Melting and pre-melting of two-dimensional crystalline SiC nanoribbons. Phys. E 2022, 137, 115012. [Google Scholar] [CrossRef]
- Hoang, V.V.; Giang, N.H.; Dong, T.Q.; Bubanja, V. Atomic structure and rippling of amorphous two-dimensional SiC nanoribbons—MD simulations. Comput. Mater. Sci. 2022, 203, 111123. [Google Scholar] [CrossRef]
- Liu, Y.T.; Liu, R.Z.; Liu, M.L.; Chang, J.X. Synthesis of SiC@Al2O3 core-shell nanoparticles for dense SiC sintering. Particuology 2019, 44, 80–89. [Google Scholar] [CrossRef]
- Xin, Z.H.; Zhang, C.Y.; Yu, M.; Jayanthi, C.S.; Wu, S.Y. Shedding light on the self-assembly of stable SiC based cage nanostructures: A comprehensive molecular dynamics study. Comput. Mater. Sci. 2014, 84, 49–62. [Google Scholar] [CrossRef]
- Wu, J.T.; Xu, Z.W.; Zhao, J.L.; Rommel, M.; Nordlund, K.; Ren, F.; Fang, F.Z. MD simulation of two-temperature model in ion irradiation of 3C-SiC: Effects of electronic and nuclear stopping coupling, ion energy and crystal orientation. J. Nucl. Mater. 2021, 557, 153313. [Google Scholar] [CrossRef]
- Nguyen, B.N.; Gao, F.; Henager, C.H.; Kurtz, R.J. Prediction of thermal conductivity for irradiated SiC/SiC composites by informing continuum models with molecular dynamics data. J. Nucl. Mater. 2014, 448, 364–372. [Google Scholar] [CrossRef]
- Zarkadoula, E.; Samolyuk, G.; Zhang, Y.W.; Weber, W.J. Electronic stopping in molecular dynamics simulations of cascades in 3C-SiC. J. Nucl. Mater. 2020, 540, 152371. [Google Scholar] [CrossRef]
- Samolyuk, G.D.; Osetsky, Y.N.; Stoller, R.E. Molecular dynamics modeling of atomic displacement cascades in 3C-SiC: Comparison of interatomic potentials. J. Nucl. Mater. 2015, 465, 83–88. [Google Scholar] [CrossRef] [Green Version]
- He, B.; Xue, J.M.; Zou, X.Q.; Wang, Y.G. Molecular Dynamics Study on the Irradiation-Induced Damage in SiC. Acta Sci. Nat. Univ. Pekin. 2009, 45, 385–389. [Google Scholar] [CrossRef]
- Li, B.S.; Zhang, C.; Liu, H.P.; Xu, L.J.; Wang, X.; Yang, Z.; Ge, F.F.; Gao, W.; Shen, T.L. Microstructural and elemental evolution of polycrystalline α-SiC irradiated with ultra-high-fluence helium ions before and after annealing. Fusion. Eng. Des. 2020, 154, 111511. [Google Scholar] [CrossRef]
- Ran, Q.; Zhou, Y.; Zou, Y.; Wang, J.; Duan, Z.A.; Sun, Z.P.; Fu, B.Q.; Gao, S.X. Molecular dynamics simulation of displacement cascades in cubic silicon carbide. Nucl. Mater. Energy 2021, 27, 100957. [Google Scholar] [CrossRef]
- Backman, M.; Toulemonde, M.; Pakarinen, O.H.; Juslin, N.; Djurabekova, F.; Nordlund, K.; Debelle, A.; Weber, W.J. Molecular dynamics simulations of swift heavy ion induced defect recovery in SiC. Comput. Mater. Sci. 2013, 67, 261–265. [Google Scholar] [CrossRef]
- Peterson, R.; Senesky, D. Modeling of radiation-induced defect recovery in 3C-SiC under high field bias conditions. Comput. Mater. Sci. 2019, 161, 10–15. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.Y.; Xiao, W.; Li, H.L. Molecular dynamics simulation of C/Si ratio effect on the irradiation swelling of β-SiC. J. Nucl. Mater. 2016, 480, 75–79. [Google Scholar] [CrossRef]
- Tian, J.T.; Feng, Q.J.; Zheng, J.; Zhou, W.; Li, X.; Liang, X.B.; Liu, D.F. Molecular Dynamics Simulations of Radiation-induced Swelling and Amorphization in a Single Crystal of 3C-SiC. Mater. Rep. 2022, 36, 20100248-5. [Google Scholar] [CrossRef]
- Zhu, S.; Mizuno, M.; Kagawa, Y.; Mutoh, Y. Monotonic tension, fatigue and creep behavior of SiC-fiber-reinforced SiC-matrix composites: A review. Compos. Sci. Technol. 1999, 59, 833–851. [Google Scholar] [CrossRef]
- Feng, L.X.; Li, W.H.; Hahn, E.N.; Branicio, P.S.; Zhang, X.Q.; Yao, X.H. Structural phase transition and amorphization in hexagonal SiC subjected to dynamic loading. Mech. Mater. 2022, 164, 104139. [Google Scholar] [CrossRef]
- Lee, W.H.; Yao, X.H.; Jian, W.R.; Han, Q. High-velocity shock compression of SiC via molecular dynamics simulation. Comput. Mater. Sci. 2015, 98, 297–303. [Google Scholar] [CrossRef]
- Zhang, J.Y.; Branicio, P.S. Molecular Dynamics Simulations of Plane Shock Loading in SiC. Procedia Eng. 2014, 75, 150–153. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Potential Function | Key Parameter | Complexity | Application |
|---|---|---|---|
| Tersoff [9,10] | Cutoff function, Two-body potential, Three-body potential | Simple | Thermal properties, Mechanical properties, Electrical properties, Ion implantation, Polishing, Sputtering, Crystal growth, Amorphization, Sintering, Irradiation damage, Fatigue damage, Shock damage |
| Tersoff/Ziegler-Biersack-Littmark (ZBL) [11,12] | Cutoff function, Two-body potential, Three-body potential, Connection function, ZBL potential | General | Ion implantation, Irradiation damage |
| Vashishta [13,14] | Two-body potential, Three-body potential | Simple | Mechanical properties, Electrical properties, Polishing, Deposition, Shock damage |
| Environment-Dependent Interatomic Potential (EDIP) [15,16] | Two-body potential, Three-body potential | General | Mechanical properties |
| Modified Embedded-Atom Method (MEAM) [17,18,19] | Pair potential interaction, Embedding energy | Complex | Thermal properties, Crystal growth, Irradiation damage |
| Gao-Weber [20] | Cutoff function, Two-body potential, Three-body potential | Simple | Irradiation damage |
| Gao-Weber/ZBL [21] | Cutoff function, Two-body potential, Three-body potential, Connection function, ZBL potential | General | Crystal growth, Irradiation damage |
| Simulation Field | Study Content | Type of SiC | Simulation Characteristic | |
|---|---|---|---|---|
| Properties | Thermology [26,27,28,29,30,31,32] | Thermal conductivity | 3C-SiC, SiC nanotubes, SiC/SiC composites, SiC crystal/amorphous layers | Calculation of thermal conductivity by the equilibrium molecular dynamics (EMD), the nonequilibrium molecular dynamics (NEMD), and the reverse nonequilibrium molecular dynamics (RNEMD). |
| Mechanics [33,34,35,36,37,38,39,40,41] | Hardness, Elastic modulus, Strength | 3C-SiC, 2H-SiC, 4H-SiC, 6H-SiC, SiC nanosheets | Calculation of hardness, elastic modulus, and strength by uniaxial tension/compression, nanoindentation. | |
| Electricity [42,43] | Dielectric constant | 3C-SiC | Calculation of dielectric constant of SiC by the linear response theory. | |
| Preparation | Ion implantation [38,39,44,45,46,47,48,49] | Si ion implantation | 3C-SiC, 4H-SiC, 6H-SiC | Effects of ion implantation on defect evolution, mechanical properties, and machinability. |
| H ion implantation | ||||
| Metal ion implantation | ||||
| Polishing [50,51,52,53,54,55,56,57,58,59,60,61,62,63] | Abrasive polishing | 3C-SiC, 4H-SiC, 6H-SiC | Removal and deformation mechanism of surface in polishing. | |
| Tool polishing | ||||
| Assisted polishing | ||||
| Sputtering [64,65,66,67,68,69] | Inert substance sputtering | 3C-SiC, 4H-SiC | Sputtering mechanism, sputtering yield, and sputtering products on surface. | |
| Reactive substance sputtering | ||||
| Deposition [70,71,72,73] | Physical deposition | 4H-SiC, Amorphous SiC | Effects of atomic incident energy, substrate morphology, and substrate temperature on the quality of deposited films. | |
| Crystal growth [19,21,74,75] | Crystal growth at solid/liquid interface | 3C-SiC, 4H-SiC, 6H-SiC | Crystallization behavior and mechanism. | |
| Amorphization [76,77,78,79] | Heating amorphization | SiC nanosheets, SiC nanoribbons, SiC nanobelts | Structural evolution and mechanism of amorphization. | |
| Cooling amorphization | ||||
| Sintering [80] | Nanoparticle sintering | SiC nanoparticles | Sintering process and mechanism of nanoparticles. | |
| New-type SiC materials [81] | Preparation of nanocage | SiC nanocages | Properties and preparation feasibility of nanocage structure. | |
| Performance | Irradiation damage [28,29,31,36,82,83,84,85,86,87,88,89,90,91,92] | Effect of irradiation on properties | 3C-SiC, 4H-SiC, 6H-SiC, SiC/SiC composites | Effects of defects induced by irradiation damage on thermal and mechanical properties. |
| Mechanism of irradiation damage | Formation process and mechanism of irradiation defects, recovery mechanism of irradiation defects, and formation mechanism of irradiation swelling. | |||
| Fatigue damage [37,93] | Crack | SiC nanosheets | Evolution of cracks under external force and its effect on mechanical properties. | |
| Shock damage [94,95,96] | Shock response | 3C-SiC, 4H-SiC, 6H-SiC | Effects of shock velocity on shock response. | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yan, Z.; Liu, R.; Liu, B.; Shao, Y.; Liu, M. Molecular Dynamics Simulation Studies of Properties, Preparation, and Performance of Silicon Carbide Materials: A Review. Energies 2023, 16, 1176. https://doi.org/10.3390/en16031176
Yan Z, Liu R, Liu B, Shao Y, Liu M. Molecular Dynamics Simulation Studies of Properties, Preparation, and Performance of Silicon Carbide Materials: A Review. Energies. 2023; 16(3):1176. https://doi.org/10.3390/en16031176
Chicago/Turabian StyleYan, Zefan, Rongzheng Liu, Bing Liu, Youlin Shao, and Malin Liu. 2023. "Molecular Dynamics Simulation Studies of Properties, Preparation, and Performance of Silicon Carbide Materials: A Review" Energies 16, no. 3: 1176. https://doi.org/10.3390/en16031176