

Molecular Dynamics Method for Supercritical CO2 Heat Transfer: A Review

 ,
,  ,
,
Abstract
:1. Introduction
2. Key Developments in Force Field
2.1. MD Potentials
2.2. Comparison Criteria for Force Fields
2.3. Future Problems in Force Fields Development
3. Molecular Dynamics Method in Supercritical Heat Transfer
3.1. Thermal Relaxation Process
3.2. Principals on Thermophysical Properties
3.2.1. T/P Relationship
3.2.2. Thermal Conductivity
- (i)
- The first approach follows the generation of a heat flux by setting temperature gradients. Then, the simulated system can be set as constant temperature to maintain a constant thermal gradient. Consequently, the NEMD based on this approach is considered a conventional model [95].
- (ii)
- The second approach provide a thermal gradient following a constant heat flux for non-equilibrium process. Thus, the approach is identified as a reverse NEMD model (i.e., RNEMD) [96].
- (i)
- The thermal conductivity based on NEMD predictions should be extended to supercritical region more than that in the sub-critical;
- (ii)
- The uniform condition to control the number of atoms in MD predictions far from the empirical database so as to remove the non-adequality of equilibrium that may result, and the waste of computational resource while practicing.
3.3. Thermal Convection
3.4. Supercritical Mixtures
3.5. Supercritical Models and Chemical Application
4. Conclusions
- (i)
- Major force fields development has been summarized and compared. Based on recovery of vapor-liquid coexistence curves, the applicability of the model has been justified, while flexible-bond models provided larger uncertainties near the critical point;
- (ii)
- The predictions near the thermostats should be avoided since the curvature of the thermal gradient can be affected by thermostat gradient. Likewise, the thermal conductivity obtained from NEMD simulations can approve the numerical accuracy of the Green–Kubo equilibrium. This may support the local equilibrium hypothesis for transfer characteristics of supercritical CO2. Meanwhile, the amplitude of temperature fluctuation in EMD simulation was significantly dependent on the size of simulated MD cell; thus, the size effect should be treated with care near the critical point;
- (iii)
- On the other hand, the thermal behavior of combined molecules is still under debate. Supercritical mixtures may only provide similar heat transfer behavior with solo supercritical fluids above the critical line.
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Nomenclature
| CO2 | Carbon Dioxide |
| Cv | Specific heat at a constant volume (J/mol K) |
| Cp | Specific heat at a constant pressure (J/kg K) |
| cs | Speed of sound (m/s) |
| D | Diffusion coefficient (m2/s) |
| DOF | Degree of freedom for MD atoms |
| Jq | Instantaneous level of heat flux |
| kb | Constant of Boltzmann (J/K) |
| KE | Total kinetic energy of group of atoms (J/mole) |
| KT | Isothermal compressibility (m2/N) |
| L | Length of MD cell used for simulation (m) |
| N | Number of atoms |
| ∆n | Density difference at the two ends of simulated MD cell (kg/m3) |
| P | Pressure cell used during molecular dynamic simulations (MPa) |
| qi | Partial charges in center of each atom (Å) |
| rij | Distance between two atoms (Å) |
| T | Operational time (s) |
| T | Absolute temperature used in simulations (K) |
| uij | Interaction potential energy between molecules |
| V | Volume area for MD simulations (m3) |
| λ | Thermal conductivity (W/m K) |
| Γ | Sound attenuation coefficient (1/m) |
| η | Kinematic viscosity (m2/s) |
References
- Chen, L. Handbook of Research on Advancements in Supercritical Fluids Applications for Sustainable Energy System; IGI Global: Hershey, PA, USA, 2021. [Google Scholar]
- Chen, L. Microchannel Flow Dynamics and Heat Transfer of Near-Critical Fluid; Springer: Berlin/Heidelberg, Germany, 2017. [Google Scholar]
- Yan, H.; Zhang, J.; Zhou, N.; Li, M. Staged Numerical Simulations of Supercritical CO2 Fracturing of Coal Seams Based on the Extended Finite Element Method. J. Nat. Gas Sci. Eng. 2019, 65, 275–283. [Google Scholar] [CrossRef]
- She, X.; Cong, L.; Nie, B.; Leng, G.; Peng, H.; Chen, Y.; Zhang, X.; Wen, T.; Yang, H.; Luo, Y. Energy-Efficient and -Economic Technologies for Air Conditioning with Vapor Compression Refrigeration: A Comprehensive Review. Appl. Energy 2018, 232, 157–186. [Google Scholar] [CrossRef]
- Landelle, A.; Tauveron, N.; Haberschill, P.; Revellin, R.; Colasson, S. Organic Rankine Cycle Design and Performance Comparison Based on Experimental Database. Appl. Energy 2017, 204, 1172–1187. [Google Scholar] [CrossRef]
- Chen, L.; Zhang, X.R. Experimental Analysis on a Novel Solar Collector System Achieved by Supercritical CO2 Natural Convection. Energy Conver. Manag. 2014, 77, 173–182. [Google Scholar] [CrossRef]
- Poppi, S.; Sommerfeldt, N.; Bales, C.; Madani, H.; Lundqvist, P. Techno-Economic Review of Solar Heat Pump Systems for Residential Heating Applications. Renew. Sustain. Energy Rev. 2018, 81, 22–32. [Google Scholar] [CrossRef]
- Ragui, K.; Chen, L. Identification of implicit dynamics of supercritical CO2 invasion in sub-regions of bench micromodels. J. Supercrit. Fluids 2023, 195, 105863. [Google Scholar] [CrossRef]
- Lin, Q.; Zhang, X.; Wang, T.; Zheng, C.; Gao, X. Technical Perspective of Carbon Capture, Utilization, and Storage. Engineering 2022, 14, 27–32. [Google Scholar] [CrossRef]
- Xue, J.P.; Nie, X.; Zhao, L.; Zhao, R.; Wang, J.; Yang, C.S.; Lin, A. Molecular Dynamics Investigation on Shear Viscosity of Mixed Working Fluid for Supercritical CO2 Brayton Cycle. J. Supercrit. Fluids 2022, 44, 2105–2116. [Google Scholar] [CrossRef]
- Ragui, K.; Bennacer, R.; Chen, L. Pore-Scale Modeling on Supercritical CO2 Invasion in 3D Micromodel with Randomly Arranged Spherical Cross-Sections. Energy Rep. 2021, 7, 33–42. [Google Scholar] [CrossRef]
- Chen, L.; Zhang, X.R. Experiments on Natural Convective Solar Thermal Achieved by Supercritical CO2/DME Mixture Fluid, Transactions of the ASME. J. Sol. Energy Eng. 2014, 136, 0130111. [Google Scholar] [CrossRef]
- Pipich, V.; Schwahn, D. Polymorphic Phase Transition in Liquid and Supercritical Carbon Dioxide. Sci. Rep. 2020, 10, 11861. [Google Scholar] [CrossRef]
- Chen, L.; Zhang, X.R.; Okajima, J.; Maruyama, S. Thermal Relaxation and Critical Instability of Near-Critical Fluid Microchannel Flow. Phys. Rev. E 2013, 87, 043016. [Google Scholar] [CrossRef] [PubMed]
- Marcus, Y. Supercritical Carbon Dioxide; Nova Science Publishers: New York, NY, USA, 2020. [Google Scholar]
- Cao, X.; Liang, X.; Shao, L.; Zhang, C. Performance Analysis of an Ejector-Assisted Two-Stage Evaporation Single-Stage Vapor-Compression Cycle. Appl. Therm. Eng. 2022, 205, 118005. [Google Scholar] [CrossRef]
- Shen, B.; Zhang, P. An Overview of Heat Transfer near the Liquid-Gas Critical Point under the Influence of the Piston Effect: Phenomena and Theory. Int. J. Therm. Sci. 2013, 71, 1–19. [Google Scholar] [CrossRef]
- Hasan, N.; Farouk, B. Thermoacoustic Transport in Supercritical Fluids at Near-Critical and near-Pseudo-Critical States. J. Supercrit. Fluids 2012, 68, 13–24. [Google Scholar] [CrossRef]
- Pettersen, J.; Hafner, A.; Skaugen, G.; Rekstad, H. Development of Compact Heat Exchangers for CO2 Air-Conditioning Systems. Int. J. Refrig. 1998, 21, 180–193. [Google Scholar] [CrossRef]
- Kim, M.-H.; Bullard, C.W. Development of a Microchannel Evaporator Model for a CO2 Air-Conditioning System. Energy 2001, 26, 931–948. [Google Scholar] [CrossRef]
- Xue, J.; Nie, X.; Du, Z.; Li, H.-R.; Zhao, L.; Zhu, Y.; Wang, J. Molecular Dynamics Investigation on Isobaric Heat Capacity of Working Fluid in Supercritical CO2 Brayton Cycle: Effect of Trace Gas. J. CO2 Util. 2022, 55, 101790. [Google Scholar] [CrossRef]
- Schuster, A.; Karellas, S.; Aumann, R. Efficiency Optimization Potential in Supercritical Organic Rankine Cycles. Energy 2010, 35, 1033–1039. [Google Scholar] [CrossRef]
- Milani, D.; Luu, M.T.; McNaughton, R.; Abbas, A. Optimizing an Advanced Hybrid of Solar-Assisted Supercritical CO2 Brayton Cycle: A Vital Transition for Low-Carbon Power Generation Industry. Energy Conver. Manag. 2017, 148, 1317–1331. [Google Scholar] [CrossRef]
- Ahn, Y.; Bae, S.J.; Kim, M.; Cho, S.K.; Baik, S.; Lee, J.I.; Cha, J.E. Review of Supercritical CO2 Power Cycle Technology and Current Status of Research and Development. Nucl. Eng. Technol. 2015, 47, 647–661. [Google Scholar] [CrossRef] [Green Version]
- Gharagheizi, F.; Ilani-Kashkouli, P.; Sattari, M.; Mohammadi, A.H.; Ramjugernath, D. A Group Contribution Method for Determination of Thermal Conductivity of Liquid Chemicals at Atmospheric Pressure. J. Mol. Liq. 2014, 190, 223–230. [Google Scholar] [CrossRef]
- Su, W.; Zhao, L.; Deng, S. Group Contribution Methods in Thermodynamic Cycles: Physical Properties Estimation of Pure Working Fluids. Renew. Sustain. Energy Rev. 2017, 79, 984–1001. [Google Scholar] [CrossRef]
- Nie, X.; Du, Z.; Zhao, L.; Deng, S.; Zhang, Y. Molecular Dynamics Study on Transport Properties of Supercritical Working Fluids: Literature Review and Case Study. Appl. Energy 2019, 250, 63–80. [Google Scholar] [CrossRef]
- Zappoli, B.; Beysens, D.; Garrabos, Y. Heat Transfers and Related Effects in Supercritical Fluids; Springer: Dordrecht, The Netherlands, 2015; p. 108. [Google Scholar]
- Huang, D.; Li, W. A Brief Review on the Buoyancy Criteria for Supercritical Fluids. Appl. Ther. Eng. 2018, 131, 977–987. [Google Scholar] [CrossRef]
- Liao, G.; Du, Y.; Zhang, F.; Jiaqiang, E. Comprehensive review on physical properties of supercritical carbon dioxide calculated by molecular simulation. Korean J. Chem. Eng. 2023, 40, 11–36. [Google Scholar] [CrossRef]
- Zhu, H.; Li, Y.; Ren, H.; Zhou, D.; Yin, J. Molecular Dynamics Simulation of Supercritical CO2 Microemulsion with Ionic Liquid Domains: Structures and Properties. Chin. J. Chem. Eng. 2019, 27, 2653–2658. [Google Scholar] [CrossRef]
- Liao, G.; Du, Y.; Zhang, F.; Jiaqiang, E.; Chen, J.; Leng, E. Widom Line of Supercritical CO2 Calculated by Equations of State and Molecular Dynamics Simulation. J. CO2 Util. 2022, 62, 102075. [Google Scholar] [CrossRef]
- Nie, X.; Zhao, L.; Deng, S.; Su, W.; Zhang, Y. A Review of Molecular Simulation Applied in Vapor-Liquid Equilibria (VLE) Estimation of Thermodynamic Cycles Group Contribution. J. Mol. Liq. 2018, 264, 652–674. [Google Scholar] [CrossRef]
- Ungerer, P.; Nieto-Draghi, C.; Rousseau, B.; Ahunbay, G.; Lachet, V. Molecular Simulation of the Thermophysical Properties of Fluids: From Understanding toward Quantitative Predictions. J. Mol. Liq. 2007, 134, 71–89. [Google Scholar] [CrossRef]
- Kubo, R. Statistical-Mechanical Theory of Irreversible Processes. I. General Theory and Simple Applications to Magnetic and Conduction Problems. J. Phys. Soc. Japan. 1957, 12, 570–586. [Google Scholar]
- Zhong, H.; Lai, S.; Wang, J.; Qiu, W.; Lüdemann, H.D.; Chen, L. Molecular Dynamics Simulation of Transport and Structural Properties of CO2 Using Different Molecular Models. J. Chem. Eng. Data 2015, 60, 2188–2196. [Google Scholar] [CrossRef]
- Ciccotti, G.; Ferrario, M. Dynamical Non-Equilibrium Molecular Dynamics. Entropy 2013, 16, 233–257. [Google Scholar] [CrossRef] [Green Version]
- Maruyama, S. Molecular Dynamics Methods in Microscale Heat Transfer. In Advances in Numerical Heat Transfer; Minkowycz, W.J., Sparrow, E.M., Eds.; Taylor and Francis: New York, NY, USA, 2001; pp. 189–226. [Google Scholar]
- Seyyedattar, M.; Zendehboudi, S.; Butt, S. Invited review—Molecular dynamics simulations in reservoir analysis of offshore petroleum reserves: A systematic review of theory and applications. Earth Sci. Rev. 2019, 192, 194–213. [Google Scholar] [CrossRef]
- Zhu, S.-B.; Robinson, G.W. Molecular Dynamics Study of Liquid Carbon Monoxide. Comput. Phys. Commun. 1989, 52, 317–321. [Google Scholar] [CrossRef]
- Geiger, L.C.; Ladanyi, B.M.; Chapin, M.E. A Comparison of Models for Depolarized Light Scattering in Supercritical CO2. J. Chem. Phys. 1990, 93, 4533–4542. [Google Scholar] [CrossRef]
- Palmer, B.J.; Garrett, B.C. Constant Pressure–Constant Temperature Simulations of Liquid Water and Carbon Dioxide. J. Chem. Phys. 1993, 98, 4047–4058. [Google Scholar] [CrossRef]
- Harris, J.G.; Yung, K.H. Carbon Dioxide’s Liquid-Vapor Coexistence Curve and Critical Properties as Predicted by a Simple Molecular Model. J. Phys. Chem. 1995, 99, 12021–12024. [Google Scholar] [CrossRef]
- Destrigneville, C.M.; Brodholt, J.P.; Wood, B.J. Monte Carlo Simulation of H2O/CO2 Mixtures to 1073.15 K and 30 Kbar. Chem. Geol. 1996, 133, 53–65. [Google Scholar] [CrossRef]
- Errington, J.R. The Development of Novel Simulation Methodologies and Intermolecular Potential Models for Real Fluids. Ph.D. Thesis, Cornell University, Ithaca, NY, USA, 1999. [Google Scholar]
- Potoff, J.J.; Siepmann, J.I. Vapor-Liquid Equilibria of Mixtures Containing Alkanes, Carbon Dioxide, and Nitrogen. AIChE 2001, 47, 1676–1682. [Google Scholar] [CrossRef]
- Zhang, Z.; Duan, Z. An Optimized Molecular Potential for Carbon Dioxide. J. Chem. Phys. 2005, 122, 214507. [Google Scholar] [CrossRef]
- Merker, T.; Engin, C.; Vrabec, J.; Hasse, H. Molecular Model for Carbon Dioxide Optimized to Vapor-Liquid Equilibria. J. Chem. Phys. 2010, 132, 234512. [Google Scholar] [CrossRef]
- Perez-Blanco, M.E.; Maginn, E.J. Molecular Dynamics Simulations of CO2 at an Ionic Liquid Interface: Adsorption, Ordering, and Interfacial Crossing. J. Phys. Chem. 2010, 114, 11827–11837. [Google Scholar] [CrossRef] [PubMed]
- Tuckerman, M.E.; Langel, W. Multiple Time Scale Simulation of a Flexible Model of CO2. J. Chem. Phys. 1994, 100, 6368. [Google Scholar] [CrossRef]
- Cygan, R.T.; Romanov, V.N.; Myshakin, E.M. Molecular Simulation of Carbon Dioxide Capture by Montmorillonite Using an Accurate and Flexible Force Field. J. Phy. Chem. C 2012, 116, 13079–13091. [Google Scholar] [CrossRef]
- Cygan, R.T.; Liang, J.J.; Kalinichev, A.G. Molecular Models of Hydroxide, Oxyhydroxide, and Clay Phases and the Development of a General Force Field. J. Phys. Chem. B 2004, 108, 1255–1266. [Google Scholar] [CrossRef]
- Liu, Z.Y.; Chen, L.; Chen, H. Molecular Dynamics Study of CO2 Phase Change Transport in the Near-Critical Region: Model Parameter Optimization. Heat Transf. Eng. 2022, 1–10. [Google Scholar] [CrossRef]
- Liu, Z.Y.; Chen, L.; Chen, H. Characterization of Dynamic Fluctuations of CO2 fluid Parameters at Critical Regions near the Pseudo-Critical Line. Phys. Fluids 2022, 34, 062003. [Google Scholar] [CrossRef]
- Makimura, D.; Kunieda, M.; Liang, Y.; Matsuoka, T.; Takahashi, S.; Okabe, H. Application of Molecular Simulations to CO2-Enhanced Oil Recovery: Phase Equilibria and Interfacial Phenomena. SPE J. 2013, 18, 319–330. [Google Scholar] [CrossRef]
- Zendehboudi, S.; Rezaei, N.; Lohi, A. Applications of hybrid models in chemical, petroleum, and energy systems: A systematic review. Appl. Energy 2018, 228, 2539–2566. [Google Scholar] [CrossRef]
- Belonoshko, A.; Saxena, S.K. A Molecular Dynamics Study of the Pressure-Volume-Temperature Properties of Supercritical Fluids: II. CO2, CH4, CO, O2, and H2. Geochim. Cosmochim. Acta 1991, 55, 3191–3208. [Google Scholar] [CrossRef]
- Jiang, H.; Moultos, O.A.; Economou, I.G.; Panagiotopoulos, A.Z. Gaussian-Charge Polarizable and Nonpolarizable Models for CO2. J. Phys. Chem. B 2016, 120, 984–994. [Google Scholar] [CrossRef]
- Aimoli, C.G.; Maginn, E.J.; Abreu, C.R.A. Force Field Comparison and Thermodynamic Property Calculation of Supercritical CO2 and CH4 Using Molecular Dynamics Simulations. Fluid Phase Equilibria 2014, 368, 80–90. [Google Scholar] [CrossRef]
- Avendaño, C.; Lafitte, T.; Galindo, A.; Adjiman, C.S.; Jackson, G.; Müller, E.A. SAFT-γ Force Field for the Simulation of Molecular Fluids. 1. A Single-Site Coarse Grained Model of Carbon Dioxide. J. Phys. Chem. B 2011, 115, 11154–11169. [Google Scholar]
- Zhu, A.; Zhang, X.; Liu, Q.; Zhang, Q. A Fully Flexible Potential Model for Carbon Dioxide. Chin. J. Chem. Eng. 2009, 17, 268–272. [Google Scholar] [CrossRef]
- Zhenyu, D.; Deng, S.; Zhao, L.; Nie, X.; Li, S.; Zhang, Y.; Zhao, J.; Zheng, N. Molecular Dynamics Study on Viscosity Coefficient of Working Fluid in Supercritical CO2 Brayton Cycle: Effect of Trace Gas. J. CO2 Util. 2020, 38, 177–186. [Google Scholar] [CrossRef]
- Nieto-Draghi, C.; de Bruin, T.; Pérez-Pellitero, J.; Bonet Avalos, J.; Mackie, A.D. Thermodynamic and Transport Properties of Carbon Dioxide from Molecular Simulation. J. Chem. Phys. 2007, 126, 064509. [Google Scholar] [CrossRef]
- Tsuji, S.; Liang, Y.; Kunieda, M.; Takahashi, S.; Matsuoka, T. Molecular Dynamics Simulations of the CO2-Water-Silica Interfacial Systems. Energy Procedia 2013, 37, 5435–5442. [Google Scholar] [CrossRef] [Green Version]
- Fang, T.; Li, S.; Zhang, Y.; Su, Y.; Yan, Y.; Zhang, J. How the Oil Recovery in Deep Oil Reservoirs Is Affected by Injected Gas Types: A Molecular Dynamics Simulation Study. Chem. Eng. Sci. 2021, 231, 116286. [Google Scholar] [CrossRef]
- Adams, J.E.; Siavosh-Haghighi, A. Rotational Relaxation in Supercritical CO2. J. Phys. Chem. B 2002, 106, 7973–7980. [Google Scholar] [CrossRef]
- Hamanaka, T.; Yamamoto, R.; Onuki, A. Molecular Dynamics Simulation of Heat Conduction in Near-Critical Fluids. Phys. Rev. E 2005, 71, 011507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raabe, G. Molecular Simulation Studies on the Vapor-Liquid Phase Equilibria of Binary Mixtures of R-1234yf and R-1234ze(E) with R-32 and CO2. J. Chem. Eng. Data 2013, 58, 1867–1873. [Google Scholar] [CrossRef]
- Vaz, R.; Gomes, J.R.B.; Silva, C.M. Molecular Dynamics Simulation of Diffusion Coefficients and Structural Properties of Ketones in Supercritical CO2 at Infinite Dilution. J. Supercrit. Fluids 2016, 107, 630–638. [Google Scholar] [CrossRef]
- Hafskjold, B. Transient Heat Flow in a One-Component Lennard-Jones/Spline Fluid. A Non-Equilibrium Molecular Dynamics Study. Comput. Methods Sci. Technol. 2017, 23, 185–198. [Google Scholar]
- Mahdavi, E.; Khaledialidusti, R.; Barnoush, A. Rheological Properties of Super Critical CO2 with Al2O3: Material Type, Size and Temperature Effect. J. Mol. Liq. 2019, 289, 111037. [Google Scholar] [CrossRef]
- Oulebsir, F.; Vermorel, R.; Galli, G. Diffusion of Supercritical Fluids through Single-Layer Nanoporous Solids: Theory and Molecular Simulations. ACS J. Surf. Colloids 2018, 34, 561–571. [Google Scholar] [CrossRef]
- Ortiz, F.J.G.; Kruse, A. The Use of Process Simulation in Supercritical Fluids Applications. React. Chem. Eng. 2020, 5, 424–451. [Google Scholar] [CrossRef]
- Allen, M.P.; Tildesley, D.J. Computer Simulation of Liquids; Oxford University Press: New York, NY, USA, 1987. [Google Scholar]
- Scheiner, B.S.; Baalrud, S.D. Mean Force Kinetic Theory Applied to Self-Diffusion in Supercritical Fluids. J. Chem. Phys. 2020, 152, 174102. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Fomin, Y.D.; Ryzhov, V.N.; Tsiok, E.N.; Brazhkin, V. Thermodynamic Properties of Supercritical Carbon Dioxide: Widom and Frenkel Lines. Phys. Rev. E 2015, 91, 022111. [Google Scholar] [CrossRef]
- Velasco, I.; Rivas, C.; Martínez-López, J.F.; Blanco, S.T.; Otín, S.; Artal, M. Accurate Values of Some Thermodynamic Properties for Carbon Dioxide, Ethane, Propane, and Some Binary Mixtures. J. Phys. Chem. B 2011, 115, 8216–8230. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Bao, H.; Ruan, X. Perspective: Predicting and Optimizing Thermal Transport Properties with Machine Learning Methods. Energy AI 2022, 8, 100153. [Google Scholar] [CrossRef]
- Algaer, E.A.; Alaghemandi, M.; Bohm, M.C.; Meller-Plathe, F. Anisotropy of the Thermal Conductivity of Stretched Amorphous Polystyrene in Supercritical Carbon Dioxide Studied by Reverse Nonequilibrium Molecular Dynamics Simulations. J. Phys. Chem. B 2009, 113, 14596–14603. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, L.; Wu, Q.; Yang, D.; Kanda, Y.; Zang, J.; Komiya, A.; Huang, Y. Preliminary Measurements of Transient Boundary Heat Transfer Process under Supercritical Pressures Using Pixelated Phase-Shifting Interferometry. Int. Commun. Heat Mass Transf. 2022, 138, 106396. [Google Scholar] [CrossRef]
- Luo, H.; Ciccotti, G.; Mareschal, M.; Meyer, M.; Zappoli, B. Thermal Relaxation of Supercritical Fluids by Equilibrium Molecular Dynamics. Phys. Rev. E 1995, 51, 2013–2021. [Google Scholar] [CrossRef] [PubMed]
- Panagiotopoulos, A.Z. Direct Determination of Phase Coexistence Properties of Fluids by Monte Carlo Simulation in a New Ensemble. J. Mol. Phys. 1987, 61, 813–826. [Google Scholar] [CrossRef]
- Smit, B. Computer Simulation of Phase Coexistence: From Atoms to Surfactants; Utrecht University: Utrecht, The Netherlands, 1990. [Google Scholar]
- Rovere, M.; Heermann, D.W.; Binder, K. The Gas-Liquid Transition of the Two-Dimensional Lennard-Jones Fluid. J. Phys. Condens. Matter 1990, 2, 7009–7032. [Google Scholar] [CrossRef]
- Chen, L.J.; Robert, M.; Shukla, K.P. Molecular Dynamics Study of the Temperature Dependence of the Inter-facial Thickness in Two-dimensional Fluid Phases. J. Chem. Phys. 1990, 93, 8254–8259. [Google Scholar] [CrossRef]
- Lebowitz, J.L.; Percus, J.K.; Verlet, L. Ensemble Dependence of Fluctuations with Application to Machine Computations. Phys. Rev. 1967, 153, 250–254. [Google Scholar] [CrossRef]
- Pathria, R.; Beale, P. Statistical Mechanics, 3rd ed.; Elsevier Pte Ltd.: Singapore, 2011. [Google Scholar]
- Leach, A.R. Molecular Modelling: Principles and Applications, 2nd ed.; Pearson Education Limited: Harlow, UK, 2001. [Google Scholar]
- Onsager, L. Reciprocal Relations in Irreversible Processes. I. Phys. Rev. 1931, 37, 405–426. [Google Scholar] [CrossRef]
- Ciccotti, G.; Kapral, R.; Sergi, A. Non-Equilibrium Molecular Dynamics. In Handbook of Materials Modeling; Springer: Dordrecht, The Netherlands, 2005; pp. 745–761. [Google Scholar]
- Palla, P.L.; Pierleoni, C.; Ciccotti, G. Bulk Viscosity of the Lennard-Jones System at the Triple Point by Dynamical Nonequilibrium Molecular Dynamics. Phys. Rev. E 2008, 78, 021204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, R.; Wu, W.; Liu, X.; Chen, K.; Yin, X. Dof Transcription Factors Are Involved in High CO2 Induced Persimmon Fruit Deastringency. Horticulturae 2022, 8, 643. [Google Scholar] [CrossRef]
- Zeeshan, A.; Atul, B. Thermal interfacial resistance and nanolayer effect on the thermal conductivity of Al2O3-CO2 nanofluid: A molecular dynamics approach. Appl. Phys. 2020, 6, 12805. [Google Scholar]
- Zhou, W.; Cheng, Y.; Chen, K.; Xie, G.; Wang, T.; Zhang, G. Thermal Conductivity of Amorphous Materials. Adv. Funct. Mater. 2020, 30, 1903829. [Google Scholar] [CrossRef]
- Li, X.; Zhao, L.; Cheng, T.; Liu, L.; Sun, H. One Force Field for Predicting Multiple Thermodynamic Properties of Liquid and Vapor Ethylene Oxide. Fluid Phase Equilibria 2008, 274, 36–43. [Google Scholar] [CrossRef]
- Daivis, P.J.; Jou, D. Thermodynamic Considerations on Thermostats and Maxwell Relations in Steady Sheared Fluids. Contin. Mech. Thermodyn. 2012, 24, 37–48. [Google Scholar] [CrossRef]
- Jepps, O.G.; Ayton, G.; Evans, D.J. Microscopic Expressions for the Thermodynamic Temperature. Phys. Rev. E 2000, 62, 4757–4763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, N.; Rubi, J.M.; Bresme, F. Non-Equilibrium Molecular Dynamics Simulations of the Thermal Transport Properties of Lennard-Jones Fluids Using Configurational Temperatures. Mol. Simul. 2016, 42, 1214–1222. [Google Scholar] [CrossRef] [Green Version]
- Liang, Z.; Tsai, H.-L. The Vibrational Contribution to the Thermal Conductivity of a Polyatomic Fluid. Mol. Phys. 2010, 108, 1707–1714. [Google Scholar] [CrossRef]
- Liang, Z.; Tsai, H.-L. Calculation of Thermophysical Properties for CO2 Gas Using an Ab Initio Potential Model. Mol. Phys. 2010, 108, 1285–1295. [Google Scholar] [CrossRef]
- Trinh, T.T.; Vlugt, T.J.H.; Kjelstrup, S. Thermal Conductivity of Carbon Dioxide from Non-Equilibrium Molecular Dynamics: A Systematic Study of Several Common Force Fields. J. Chem. Phys. 2014, 141, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Span, R.; Wagner, W. A New Equation of State for Carbon Dioxide Covering the Fluid Region from the Triple-Point Temperature to 1100 K at Pressures up to 800 MPa. J. Phys. Chem. Ref. Data 1996, 25, 1509–1596. [Google Scholar] [CrossRef] [Green Version]
- Bresme, F.; Armstrong, J. Note: Local Thermal Conductivities from Boundary Driven Non-Equilibrium Molecular Dynamics Simulations. J. Chem. Phys. 2014, 140, 016102. [Google Scholar] [CrossRef] [PubMed]
- Onuki, A. Phase Transition Dynamics; Cambridge University Press: Cambridge, UK, 2002. [Google Scholar]
- Ohara, T. Contribution of Intermolecular Energy Transfer to Heat Conduction in a Simple Liquid. J. Chem. Phys. 1999, 111, 9667–9672. [Google Scholar] [CrossRef]
- Ohara, T. Intermolecular Energy Transfer in Liquid Water and Its Contribution to Heat Conduction: A Molecular Dynamics Study. J. Chem. Phys. 1999, 111, 6492–6500. [Google Scholar] [CrossRef]
- Tamba, J.; Ohara, T.; Aihara, T. Md Study on Interfacelike Phenomena in Supercritical Fluid. Microscale Thermophys. Eng. 1997, 1, 19–30. [Google Scholar]
- Tamba, J.; Takahashi, T.; Ohara, T.; Aihara, T. Transition from Boiling to Free Convection in Supercritical fluid. Exp. Therm. Fluid Sci. 1998, 17, 248–255. [Google Scholar] [CrossRef]
- Zhang, Y.; Yang, J.; Yu, Y.-X. Dielectric Constant and Density Dependence of the Structure of Supercritical Carbon Dioxide Using a New Modified Empirical Potential Model: A Monte Carlo Simulation Study. J. Phys. Chem. B 2005, 109, 13375–13382. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Wang, Y.; Ma, X. Phase Distribution Including a Bubblelike Region in Supercritical Fluid. Phys. Rev. E 2021, 104, 014142. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Ma, X.; Xu, J.; Li, M.; Wang, Y. The Three-Regime-Model for Pseudo-Boiling in Supercritical Pressure. Int. J. Heat Mass Transf. 2021, 181, 121875. [Google Scholar] [CrossRef]
- Guo, L.; Jin, H. Boiling Coal in Water: Hydrogen Production and Power Generation System with Zero Net CO2 Emission Based on Coal and Supercritical Water Gasification. Int. J. Hydrog. Energy 2013, 38, 12953–12967. [Google Scholar] [CrossRef]
- Guo, L.; Jin, H.; Ge, Z.; Lu, Y.; Cao, C. Industrialization Prospects for Hydrogen Production by Coal Gasification in Supercritical Water and Novel Thermodynamic Cycle Power Generation System with No Pollution Emission. Sci. China Technol. Sci. 2015, 58, 1989–2002. [Google Scholar] [CrossRef]
- Guo, L.; Jin, H.; Lu, Y. Supercritical Water Gasification Research and Development in China. J. Supercrit. Fluids 2015, 96, 144–150. [Google Scholar] [CrossRef]
- Yang, X.; Xu, J.; Wu, S.; Yu, M.; Hu, B.; Cao, B.; Li, J. A Molecular Dynamics Simulation Study of PVT Properties for H2O/H2/CO2 Mixtures in near-Critical and Supercritical Regions of Water. Int. J. Hydrog. Energy 2018, 43, 10980–10990. [Google Scholar] [CrossRef]
- Liu, Y.; Lafitte, T.; Panagiotopoulos, A.Z.; Debenedetti, P.G. Simulations of Vapor-Liquid Phase Equilibrium and Interfacial Tension in the CO2–H2O–NaCl System. AIChE 2013, 59, 3514–3522. [Google Scholar] [CrossRef]
- Chen, L.F.; Liu, D.; Zhang, H.L.; Li, Q. Theoretical Investigations on Heat Transfer to H2O/CO2 Mixtures in Supercritical Region. Sci. China Technol. Sci. 2020, 63, 1018–1024. [Google Scholar] [CrossRef]
- Saric, D.; Guevara-Carrion, G.; Vrabec, J. Thermodynamics of Supercritical Carbon Dioxide Mixtures Across the Widom Line. Phys. Chem. Chem. Phys. 2022, 24, 28257–28270. [Google Scholar] [CrossRef]
- Fan, M.; Lu, Y. Insights into carbon monoxide oxidation in supercritical H2O/CO2 mixtures using reactive molecular dynamics simulations, J. Supercrit. Fluids 2022, 15, 33–38. [Google Scholar] [CrossRef]
- Wang, X.; Peng, F.; Song, K.; Feng, G.; Guo, Z. Molecular Dynamics Study of Interfacial Properties in CO2 Enhanced Oil Recovery. Fluid Phase Equilibria 2018, 467, 25–32. [Google Scholar] [CrossRef]
- Gharagheizi, F.; Eslamimanesh, A.; Mohammadi, A.H.; Richon, D. Artificial Neural Network Modeling of Solubilities of 21 Commonly Used Industrial Solid Compounds in Supercritical Carbon Dioxide. Int. Eng. Chem. Res. 2011, 50, 221–226. [Google Scholar] [CrossRef]
- Rad, H.B.; Sabet, J.K.; Varaminian, F. Determination of Valsartan Solubility in Supercritical Carbon Dioxide: Experimental Measurement and Molecular Dynamics Simulation. J. Mol. Liq. 2020, 42, 851–865. [Google Scholar]
- Ismadji, S.; Bhatia, S.K. Solubility of Selected Esters in Supercritical Carbon Dioxide. J. Supercrit. Fluids 2003, 27, 1–11. [Google Scholar] [CrossRef]
- Yamini, Y.; Arab, J.; Asghari-khiavi, M. Solubilities of Phenazopyridine, Propranolol, and Methimazole in Supercritical Carbon Dioxide. J. Pharm. Biomed. Anal. 2003, 32, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Housaindokht, M.R.; Haghighi, B.; Bozorgmehr, M.R. A Comparison among Three Equations of State in Predicting the Solubility of Some Solids in Supercritical Carbon Dioxide. Korean J. Chem. Eng. 2007, 24, 102–105. [Google Scholar] [CrossRef]
- Guennec, Y.; Privat, R.; Lasala, S.; Jaubert, J.-N. On the Imperative Need to Use a Consistent α-Function for the Prediction of Pure-Co mpound Supercritical Properties with a Cubic Equation of State. Fluid. Phase. Equilibria 2017, 445, 45–53. [Google Scholar] [CrossRef]
- Bonyadi, M.; Esmaeilzadeh, F. A Modification of the Alpha Function (α), and the Critical Compressibility Factor (Ζc) in ER (Esmaeilzadeh–Roshanfekr) Equation of State. Fluid Phase Equilibria 2008, 273, 31–37. [Google Scholar] [CrossRef]
- Kurnik, R.T.; Reid, R.C. Solubility of Solid Mixtures in Supercritical Fluids. Fluid Phase Equilibria 1982, 8, 93–105. [Google Scholar] [CrossRef]
- Kosal, E.; Holder, G.D. Solubility of Anthracene and Phenanthrene Mixtures in Supercritical Carbon Dioxide. J. Chem. Eng. Data 1987, 32, 148–150. [Google Scholar] [CrossRef]
- Cortesi, A.; Kikic, I.; Alessi, P.; Turtoi, G.; Garnier, S. Effect of Chemical Structure on the Solubility of Antioxidants in Supercritical Carbon Dioxide: Experimental Data and Correlation. J. Supercrit. Fluids 1999, 14, 139–144. [Google Scholar] [CrossRef]
- Goodarznia, I.; Esmaeilzadeh, F. Solubility of an Anthracene, Phenanthrene, and Carbazole Mixture in Supercritical Carbon Dioxide. J. Chem. Eng. Data 2002, 47, 333–338. [Google Scholar] [CrossRef]
- Ingrosso, F.; Ruiz-López, M.F. Modeling Solvation in Supercritical CO2. Chem. Phys. 2017, 18, 2560–2572. [Google Scholar]
- Ingrosso, F.; Altarsha, M.; Dumarçay, F.; Kevern, G.; Barth, D.; Marsura, A.; Ruiz-López, M.F. Driving Forces Controlling Host-Guest Recognition in Supercritical Carbon Dioxide Solvent. Chem. A Eur. J. 2016, 22, 2972–2979. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y. Two-Dimensional Direct Numerical Simulations of Fingering Double Diffusive Convection Turbulence. Kongqi Donglixue Xuebao. Acta Aerodynam. Sinica 2022, 40, 182–187. [Google Scholar]
- Fang, T.; Wang, M.; Gao, Y.; Zhang, Y.; Yan, Y.; Zhang, J. Enhanced Oil Recovery with CO2/N2 Slug in Low Permeability Reservoir: Molecular Dynamics Simulation. Chem. Eng. Sci. 2019, 197, 204–211. [Google Scholar] [CrossRef]
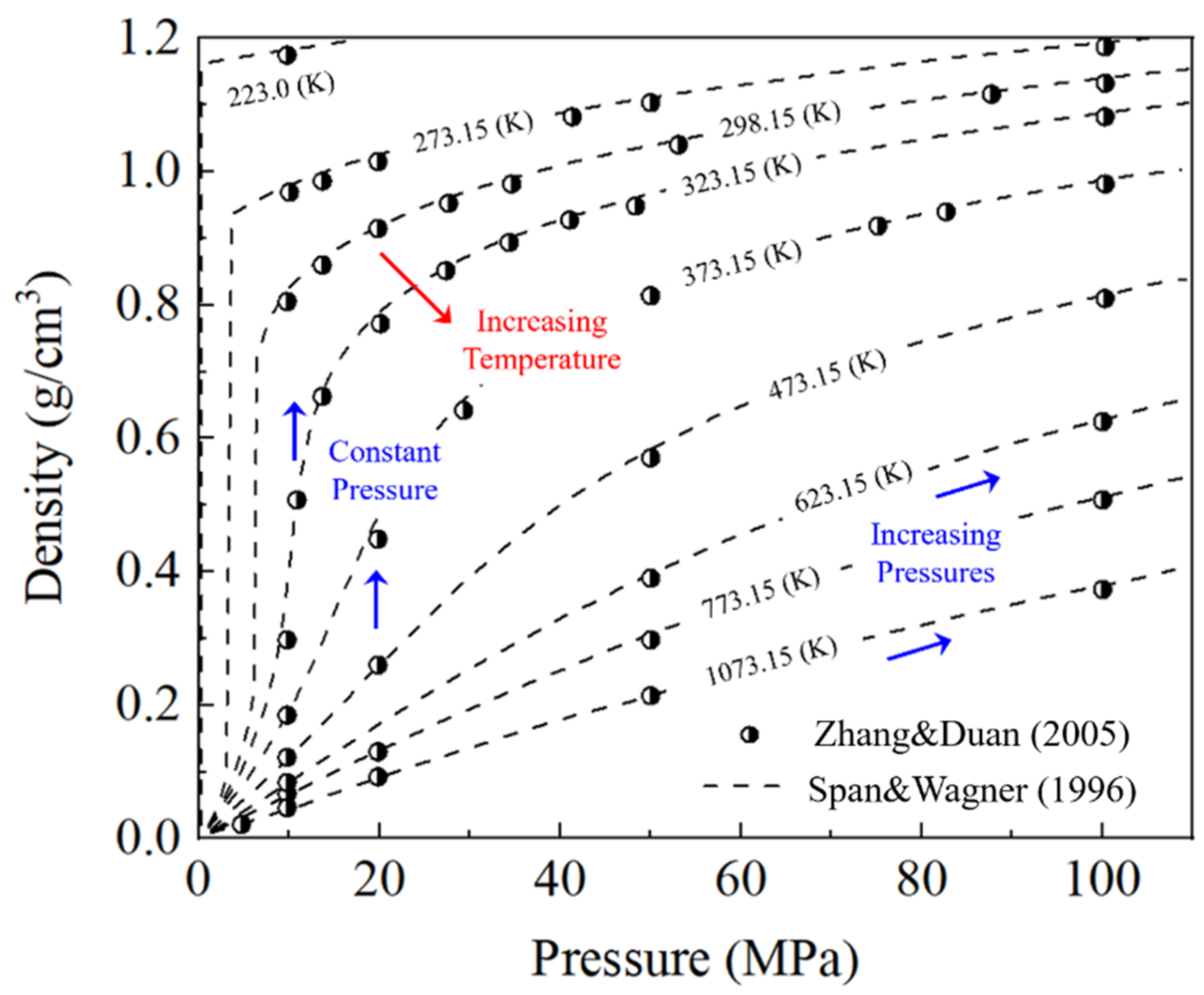




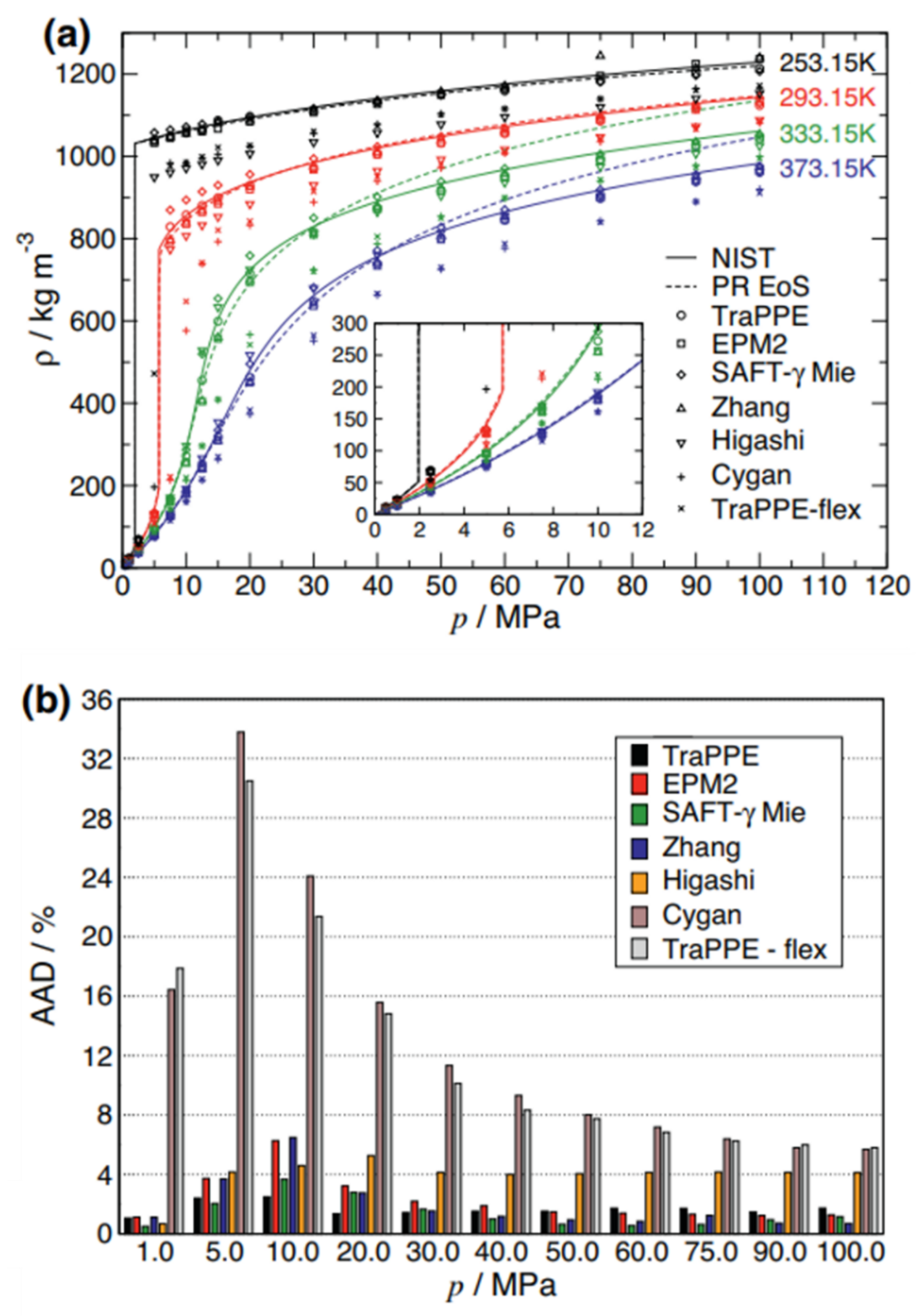
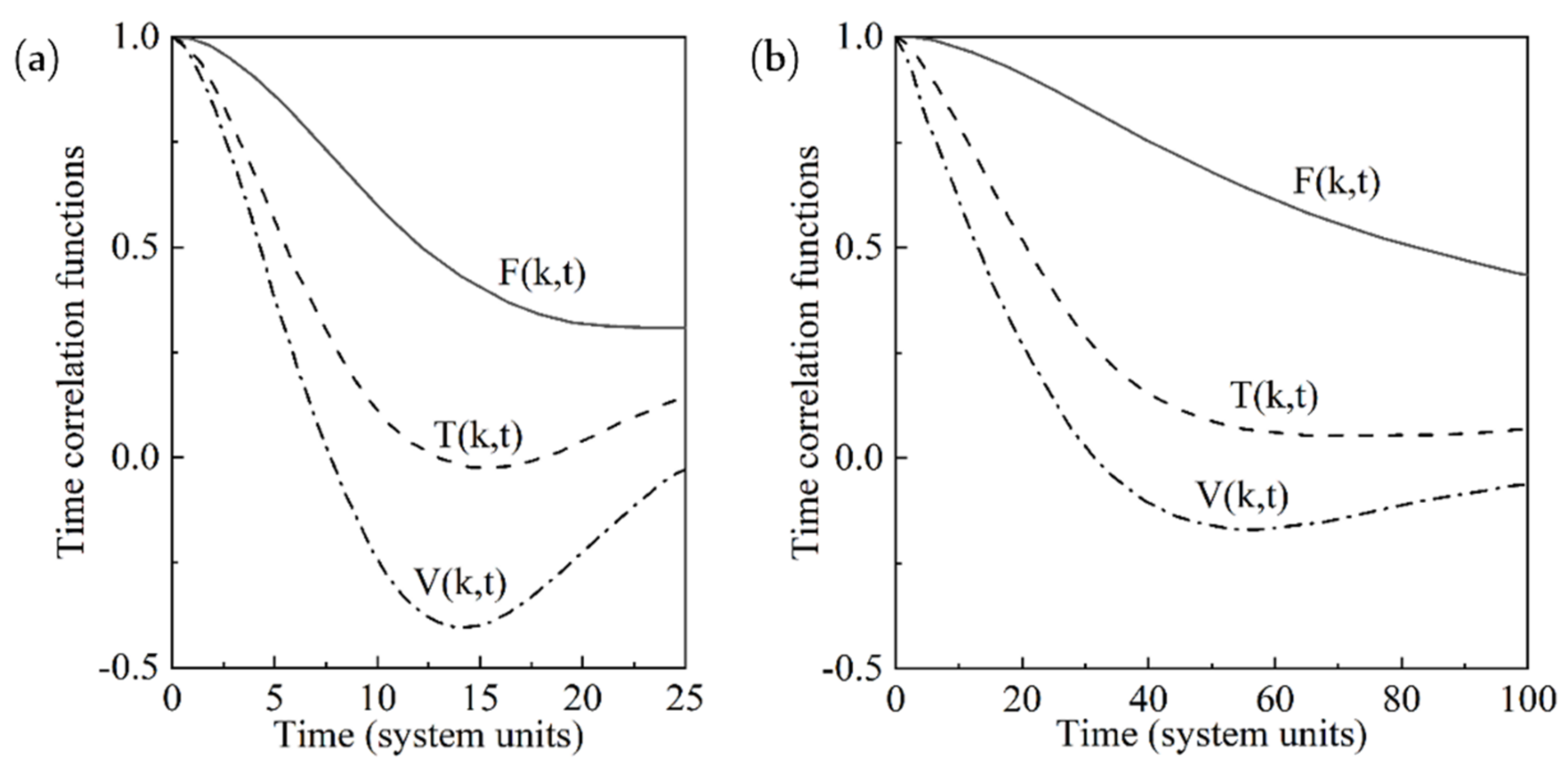







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reference | MD Technology | Key Findings and Observations | Knowledge Gap | ||
|---|---|---|---|---|---|
| MD Cell | Fluid | Potentials | |||
| Tsuji et al. [64] | 3D | CO2/H2O/ Silica | EPM2 (CO2)/ SPC/E (H2O)/ (silica) | Interfacial tension changes with wettability of CO2/brine and CO2/H2O/Silica systems. | Large-scale T/p effects on interfacial tension property with experimental validation. |
| Fang et al. [65] | 3D | CO2/Oil/ Silica | EPM2 (CO2)/ OPLS-AA/ CLAYFF | Detachment order of oil and gases from silica surface during injection process of CO2. | Microscopic interaction mechanism between oil and CO2 at reliable reservoirs conditions. |
| Adams and Siavosh-Haghighi [66] | 3D | CO2 | EPM2 | Diverse response of density and related low-variation zones according to a large series of governing parameters. | Local density enhancement to be solute-induced in the near-critical regime with inhomogeneity degree. |
| Hamanaka [67] | 2D | L-J | - | Thermodynamic characters in near-critical fluids by predicting steady-state density and temperature profiles. | Gas–liquid interface reaction to applied heat flow where latent heat transport is critical at convection mode. |
| Raabe et al. [68] | 3D | Refrigerant/ CO2 | AMBER | Comparison of force field simulations with experimental basis at subcritical conditions. | Large-scale experimental data near critical point for validity of chosen force field. |
| Vaz et al. [69] | 3D | Ketone/ CO2 | OPLS-AA (ketone)/ EPM2 (CO2) | Mass transfer process of ketones in supercritical CO2 environment. | Mechanism of local interactions that can play a role in the dynamics of the system. |
| Hafskjold [70] | 3D | L-J | - | Supercritical temperature-pressure wave coupling transfer process. | Thermodynamic states far from critical point with no projection to sCO2 heat transfer. |
| Mahdavi et al. [71] | 3D | Al2O3/ CO2 | COMPASS/ COMB | Effect of aluminum oxide structure on rheology of supercritical CO2 with self-diffusion coefficient. | Rheology far from critical point conditions with no projection to sCO2 heat transfer characteristics near critical region. |
| Scheme | Bond | Angle | Bond-Bond | Bond-Angle | Bond-Torsion | Dihedral Angle | Coulomb Force | vdW | Electron Transfer |
|---|---|---|---|---|---|---|---|---|---|
| COMPASS | √ | √ | √ | √ | √ | √ | √ | √ | |
| COMB | √ | √ | √ | ||||||
| CHARMM | √ | √ | √ | √ | √ | √ | |||
| CLAYFF | √ | √ | √ | √ | |||||
| OPLS-AA | √ | √ | √ | √ | √ | √ |
| Models | εC (K) | σC (Å) | εO (K) | σO (Å) | qC (e) | qO (e) | dC-O (Å) |
|---|---|---|---|---|---|---|---|
| MSM | 29.00 | 2.79 | 83.10 | 3.01 | 0.60 | −0.30 | 1.16 |
| EPM2 | 28.13 | 2.76 | 80.51 | 3.03 | 0.65 | −0.33 | 1.15 |
| TraPPE | 27.00 | 2.80 | 79.00 | 3.05 | 0.70 | −0.35 | 1.16 |
| EPM2_Flex 1 | ks = 10,739 (kJ/mol·Å2), kb = 10,739 (kJ/mol·rad2) | ||||||
| EPM2_Flex 2 | km = 2015.75 (kJ/mol·Å2), α = 2.35, kb = 10,739 (kJ/mol·rad2) | ||||||
| Chemical Compounds | Tc (K) | Pc (bar) | Ω | vs (cm3/mol) | Refs. |
|---|---|---|---|---|---|
| Triphenylene | 1013.6 | 29.28 | 0.492 | 175 | [125] |
| Benzoin | 853.52 | 26.6 | 0.599 | 162 | [126] |
| Mandelic acid | 903.79 | 34.73 | 0.645 | 117 | [126] |
| Propyl 4-hydroxybenzoate | 815.92 | 31.3 | 0.722 | 131.6 | [126] |
| Hexamethylbenzene | 758 | 24.4 | 0.515 | 152.7 | [126] |
| Henanthrene | 882.65 | 31.72 | 0.437 | 182 | [126] |
| Anthracene | 869.15 | 30.8 | 0.353 | 142.6 | [126] |
| Carbazole | 899.1 | 32.65 | 0.496 | 151.5 | [126] |
| Fluorene | 826.4 | 29.5 | 0.406 | 139.3 | [126] |
| Fyrene | 936 | 25.7 | 0.509 | 158.5 | [126] |
| O-hydroxy benzoic acid | 739 | 51.8 | 0.832 | 95.7 | [126] |
| 1-0ctadecanol | 777 | 13.4 | 0.863 | 333 | [126] |
| Phenazopyridine | 1148.4 | 27.56 | 0.735 | 160.3 | [127] |
| Propranolol | 958.5 | 22 | 1.061 | 214.3 | [127] |
| Methimazole | 731.7 | 60.75 | 0.442 | 162.1 | [127] |
| Benzoic acid | 752 | 45.6 | 0.62 | 92.51 | [128] |
| Acenaphthene | 803.15 | 31 | 0.38 | 126.19 | [128] |
| Perylene | 863 | 8.68 | 0.915 | 201.85 | [128] |
| Methyl salicylate | 700 | 40.7 | 0.631 | 130 | [128] |
| Fluoranthene | 905 | 26.1 | 0.587 | 161.55 | [128] |
| Phenol | 692.2 | 60.5 | 0.45 | 89 | [128] |
| p-chloropheno | 724.75 | 53.61 | 0.456 | 101.4 | [128] |
| 2,4-dichlorophenol | 718.38 | 53.04 | 0.608 | 117.9 | [128] |
| 2.6-dichlorogheno | 718.38 | 53.04 | 0.608 | 117.9 | [128] |
| 2,4,6-trichlorophenol | 745.96 | 51.52 | 0.522 | 132.6 | [128] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, L.; Zhang, Y.; Ragui, K.; Hou, C.; Zang, J.; Huang, Y. Molecular Dynamics Method for Supercritical CO2 Heat Transfer: A Review. Energies 2023, 16, 2902. https://doi.org/10.3390/en16062902
Chen L, Zhang Y, Ragui K, Hou C, Zang J, Huang Y. Molecular Dynamics Method for Supercritical CO2 Heat Transfer: A Review. Energies. 2023; 16(6):2902. https://doi.org/10.3390/en16062902
Chicago/Turabian StyleChen, Lin, Yizhi Zhang, Karim Ragui, Chaofeng Hou, Jinguang Zang, and Yanping Huang. 2023. "Molecular Dynamics Method for Supercritical CO2 Heat Transfer: A Review" Energies 16, no. 6: 2902. https://doi.org/10.3390/en16062902
APA StyleChen, L., Zhang, Y., Ragui, K., Hou, C., Zang, J., & Huang, Y. (2023). Molecular Dynamics Method for Supercritical CO2 Heat Transfer: A Review. Energies, 16(6), 2902. https://doi.org/10.3390/en16062902