Abstract
As the world aims to address the UN Sustainable Development Goals (SDGs), it is becoming more urgent for heavy transportation sectors, such as shipping and aviation, to decarbonise in an economically feasible way. This review paper investigates the potential fuels of the future and their capability to mitigate the carbon footprint when other technologies fail to do so. This review looks at the technologies available today, including, primarily, transesterification, hydrocracking, and selective deoxygenation. It also investigates the potential of fish waste from the salmon industry as a fuel blend stock. From this, various kinetic models are investigated to find a suitable base for simulating the production and economics of biodiesel (i.e., fatty acid alkyl esters) and renewable diesel production from fish waste. Whilst most waste-oil-derived biofuels are traditionally produced using transesterification, hydrotreating looks to be a promising method to produce drop-in biofuels, which can be blended with conventional petroleum fuels without any volume percentage limitation. Using hydrotreatment, it is possible to produce renewable diesel in a few steps, and the final liquid product mixture includes paraffins, i.e., linear, branched, and cyclo-alkanes, with fuel properties in compliance with international fuel standards. There is a wide range of theoretical models based on the hydrodeoxygenation of fatty acids as well as a clear economic analysis that a model could be based on.
1. Introduction
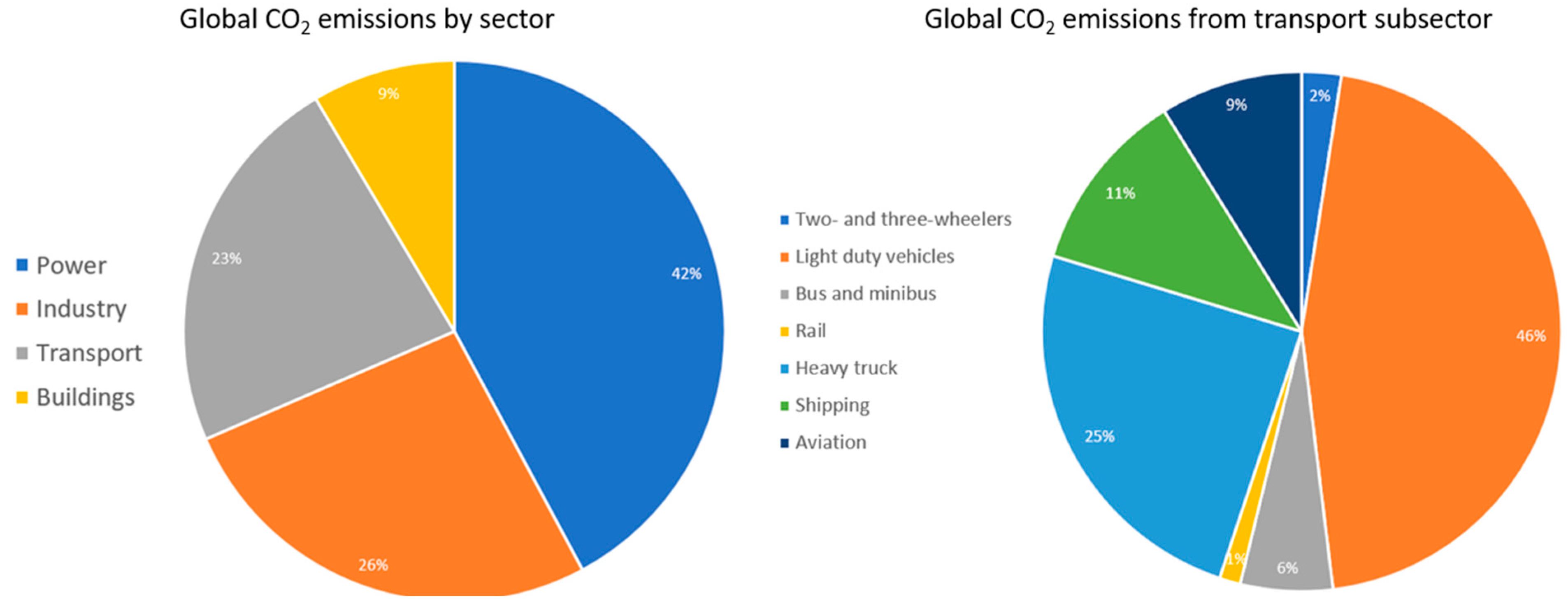
Transport fuels today make up 23% of all contributions to climate change [1], and the biggest contributors are light-duty vehicles, heavy trucks, and shipping. The global CO2 emissions by sector and transportation subsector is summarised in Figure 1.
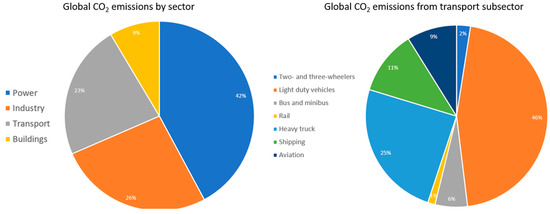
Figure 1.
Global CO2 emissions by sector and transportation subsector [1].
Biofuels have the potential to decarbonise these sectors, which is particularly important as they are much harder to decarbonise than other sectors, such as power [2]. Heavy trucks and marine vehicles currently rely on paraffinic liquid fuels from the oil and gas industry, such as diesel and heavy fuel oil, to power them due to their high energy density per unit volume. Smaller vehicles use fuels derived from crude oil, such as petrol and diesel, but they are much easier to decarbonise due to the shorter distances of travel. In electric vehicles, this allows for frequent recharging, and the weight of a battery is less impactful and simpler to implement [3]. The weight of a battery becomes a more significant factor in heavy trucks; however, for medium trips, it is still feasible to recharge the battery of these vehicles, but this method may require infrastructure upgrades to implement fast charging and to reach the high-power loads required, particularly in rural areas. For long-journey trucks and aviation and marine vessels, the energy density of the fuel is particularly important, making it much less feasible to utilise batteries, with the fundamental issue being the impact of the weight of the battery and the frequency of charging that would be required.
As a result, there is an increased interest in biofuels. For example, the current energy density of lithium-ion batteries is 300 Wh/kg [4], but to even have an impact on regional small planes, the minimum energy density would be 500 Wh/kg [5], a 166% increase, to decarbonise these transport sectors. It is expected that by 2050, biofuels will make up 27% of all transport fuel demands [6]. The main biofuels that are used today are categorised according to the feedstock used in their production: biodiesel, bioethanol, biomethane, hydrotreated vegetable oils and fats, and lignocellulosic-based fuels [7]. Furthermore, 90% of all biofuels used are either biodiesel, also known as fatty acid alkyl mixtures or bioethanol, and the majority of the feed for those fuels comes from edible food sources. The issue of the feedstock being primarily edible is that the overall impact may be negative, as the price of food may increase, having major effects on the local communities it is sourced from and across the world [8].
As a result of the aforementioned issues, this paper aims to review the feasibility of using inedible biological waste to produce liquid biofuels, with a particular interest in the hard-to-decarbonise marine transport industry.
2. Biodiesel
As mentioned in the introduction, biofuels come from various feedstocks and can be many different biologically derived chemicals.
Biodiesels are a promising alternative for traditionally derived petro-fuels. Biodiesel could be defined as a blend of any vegetable oil and diesel, while it also could be defined as blends of alkyl esters of vegetable oils or animal fats and diesel. These alkyl esters are typically produced through the transesterification [9] of oils or fats with short-chain alcohols, such as methanol or ethanol, which also leads to the production of glycerine, which is also known as glycerol, as a byproduct. Transesterification is a reaction where an ester is transformed into another through the interchange of its R group, often with an alcohol’s R group, and catalysed with the use of an acid or base. There are various feedstocks for this process, but they are typically waste-derived vegetable oils [10], such as sunflower, peanut, palm, coconut, and castor oil, and the alcohol is typically methanol. Ethanol can be used, but it is less reactive and can lead to technological problems, it is less toxic than methanol and can sourced from the fermentation of sugar cane [11]. Edible feedstocks raise ethical questions, as the use of these feedstocks may increase the price of food for the local population from where it is sourced, impacting their food security; this is especially the case for less developed countries. The use of biodiesel in engines has been understood since Rudolf Diesel theorised that vegetable oils could feed diesel engines from their inception in 1893 [11].
It is important to consider the behaviour of unblended biodiesel, i.e., B100, in engines without any modifications. Issues identified with the use of unblended biodiesel might include a decrease in power output, carbon deposits, oil ring sticking and thickening or gelling of lubrication oils, as well as the higher viscosity and lower volatility. Chemical and physical modifications can be implemented to overcome these issues [12]. For example, pyrolysis of the oil-derived feedstocks can lead to a feedstock made up of shorter chains, utilisation of microemulsions can improve the spray characteristics, dilution with ethanol can reduce the impact of CO, NOx, and SOx emissions, and transesterification reduces the molecular weight and viscosity while increasing volatility [12].
The transesterification reaction is summarised in Figure 2.
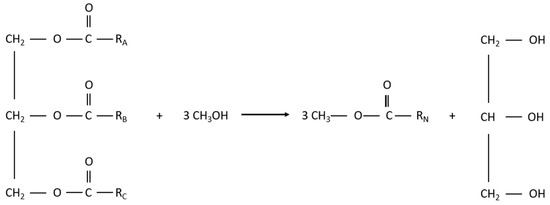
Figure 2.
Transesterification reaction.
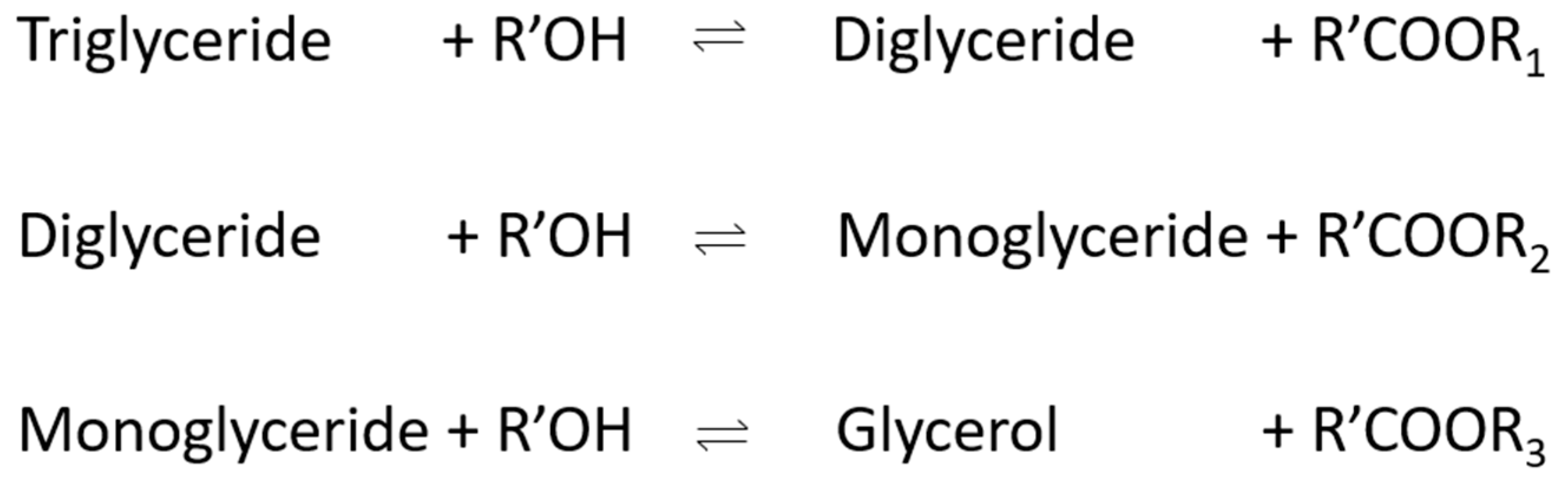
The catalyst can be either a base or an acid and homogenous or heterogeneous. Typically, sodium or potassium methylate due to its high activity and low cost. Additionally, vegetable oils are usually used in this process due to their advantages, including liquid portability, heat content (80% of diesel), availability, and renewability. The disadvantages are their high viscosity, lower volatility, and reactivity of unsaturated hydrocarbon chains [12]. In reality, the transesterification reaction is more complex, and it is made up of multiple reversible reactions as the triglyceride is converted to a diglyceride, monoglyceride, and, finally, glycerol, with 1 mole of ester being liberated at each step. The first step is characterised by an attack on the carbonyl carbon atom by the anion of alcohol (OH−), forming a tetrahedral intermediate. This intermediate reacts with alcohol to regenerate the anion, and, in the final step, the tetrahedral is rearranged with the formation of a fatty acid ester and a diglyceride [12].
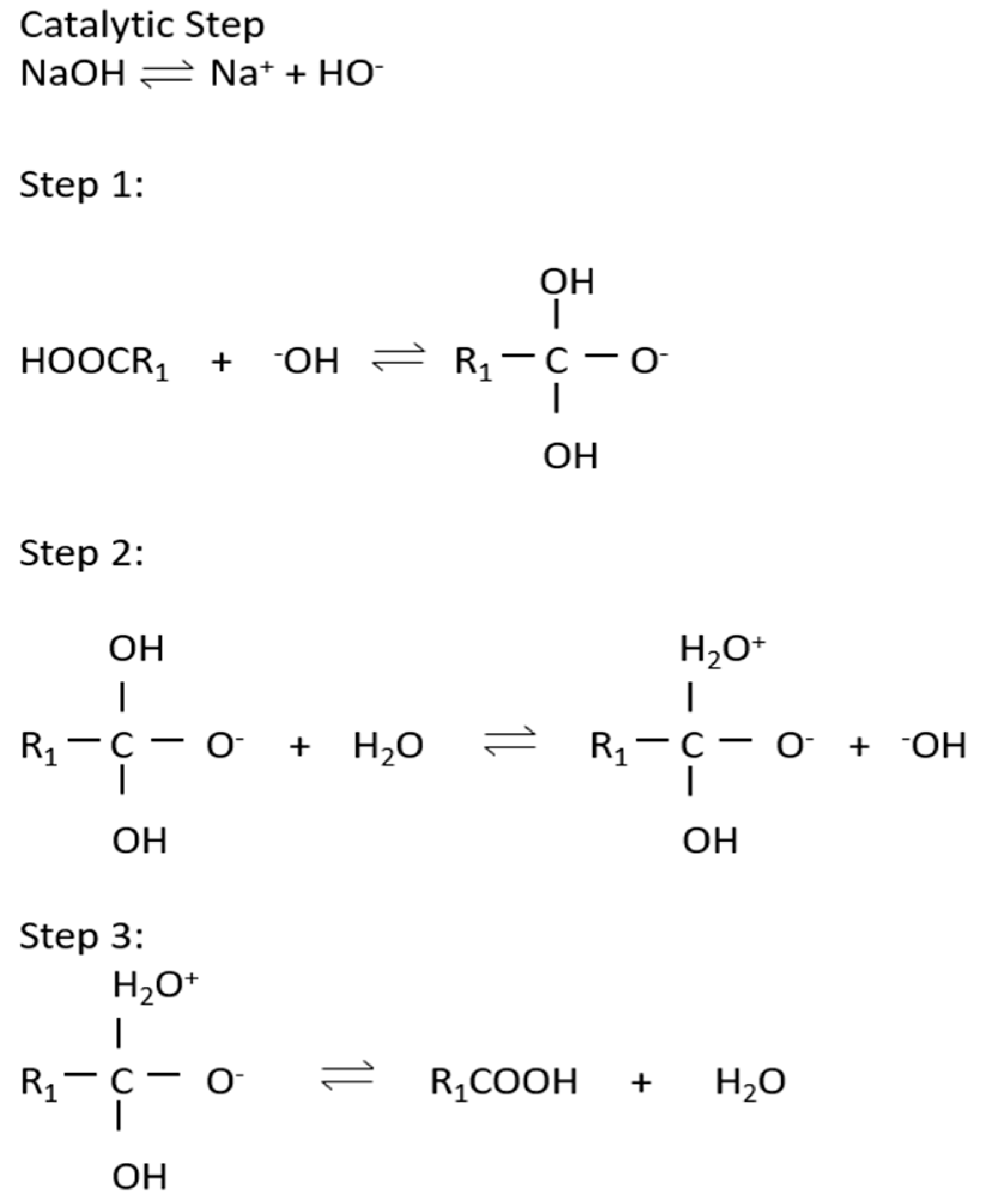
The cyclic regeneration of hydroxide ion and triglyceride to diglyceride mechanism is shown in Figure 3 and reversible reactions in the production of biodiesel are presented in Figure 4.
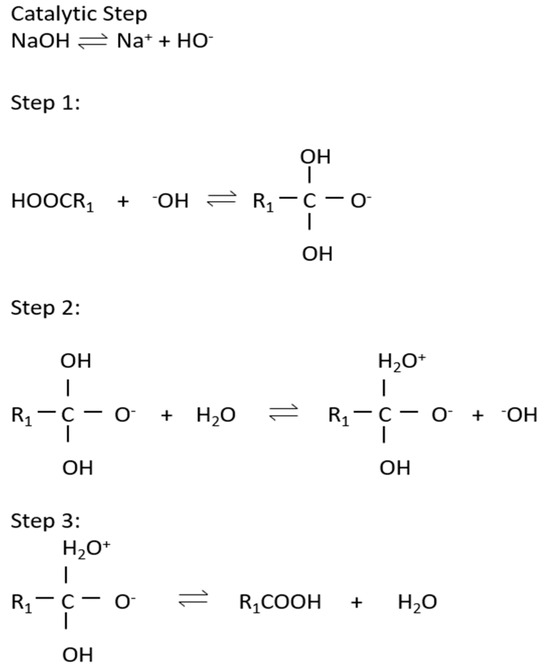
Figure 3.
Cyclic regeneration of hydroxide ion and triglyceride to diglyceride mechanism.

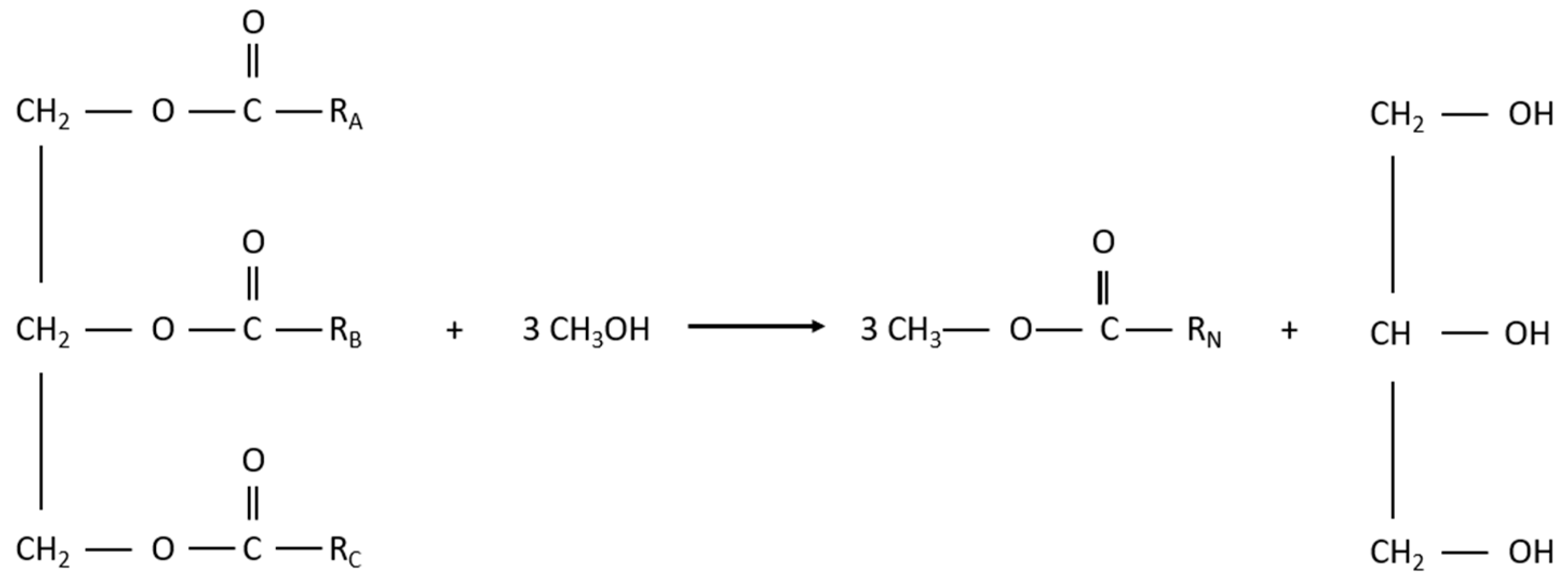
Figure 4.
Reversible reactions in the production of biodiesel.
3. Green Diesel
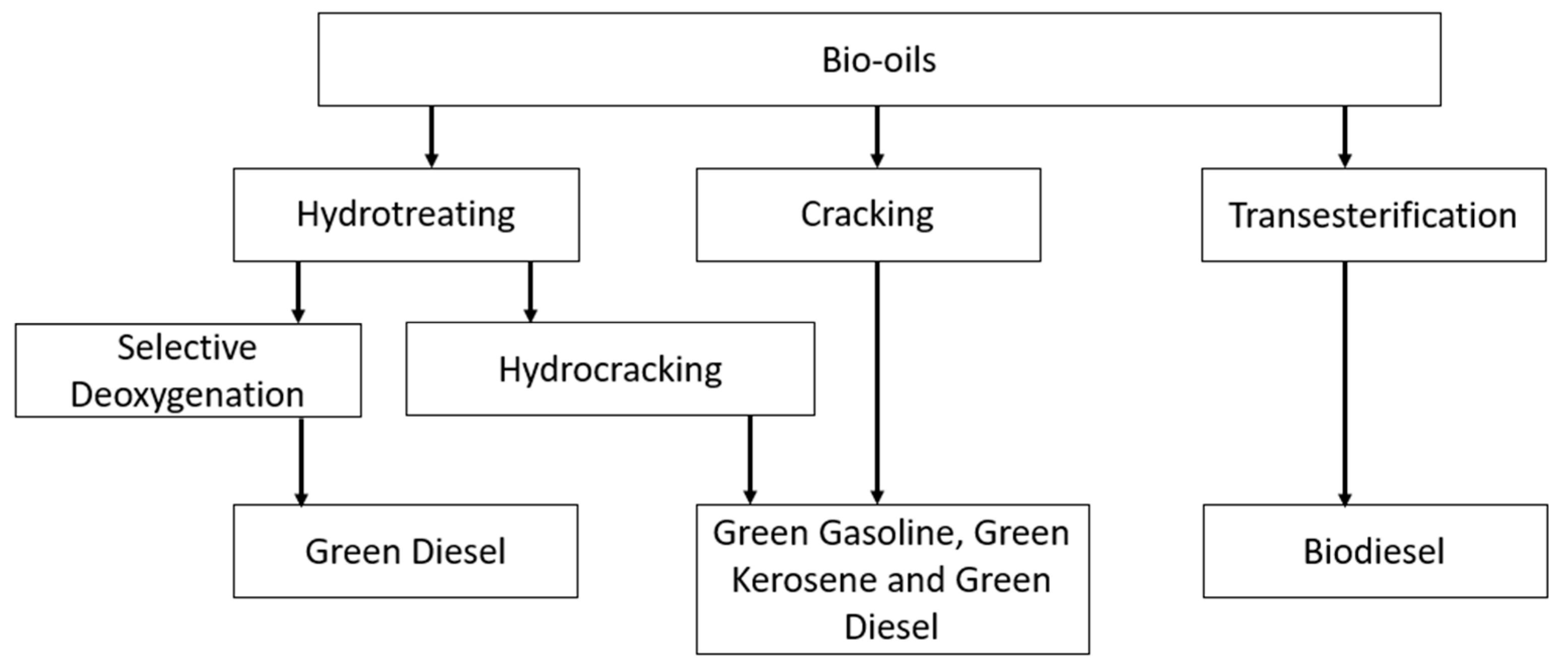
There are three main routes to produce biofuels from triglycerides [13]: (i) transesterification with alcohol to produce fatty acid alkyl esters (e.g., FAMEs); (ii) hydrocracking, where triglycerides are cracked in the presence of hydrogen over an acid catalyst, producing a liquid organic product consisting of a blend of paraffinic fuels, i.e., bionaphtha, kerosene, and green diesel; and (iii) selective deoxygenation (SDO), where the triglycerides are hydrotreated in the presence of a catalyst and hydrogen to remove oxygen (i.e., hydrodeoxygenation reaction).
Each of the processes and their products are summarised in Figure 5.
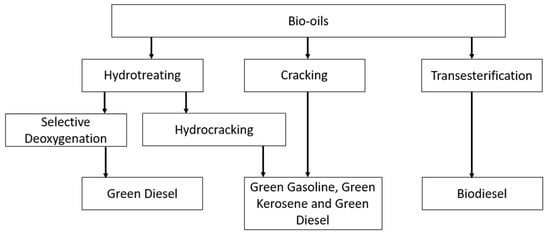
Figure 5.
Pathways for producing sustainable liquid fuels from animal fats and vegetable oils.
3.1. Transesterification
Biofuels from transesterification require a biologically derived oil, a catalyst, and alcohol as their feedstock. One patent claimed the use of 1.6 times the theoretical amount of alcohol in this reaction, and this is done to drive the equilibrium of the reaction forward, with 0.1 to 0.5% sodium or potassium methoxide to an oil or fat. When performed at 80 °C, this would lead to a 98% conversion to alkyl esters and high-quality glycerol. The study also found the following [14]:
- By conducting the reaction in steps by partially adding the alcohol and catalyst and removing the glycerol at each step, the amount of alcohol used can be reduced.
- Other alcohols can be used, such as ethanol, propanol, isopropanol, butanol, and pentanol. However, methanol is generally used due to its lower boiling point, low cost and availability.
- Water and free fatty acids inhibit the reaction, where higher alcohols are particularly sensitive to contamination with water.
Acid catalysts are much slower than alkali catalysts with respect to reaction kinetics.
Even with the 1.6 ratio of methanol, there will still be a significant amount of mono- and diglycerides, with a ratio of 6:1 required to reach a viable single-step reaction [14].
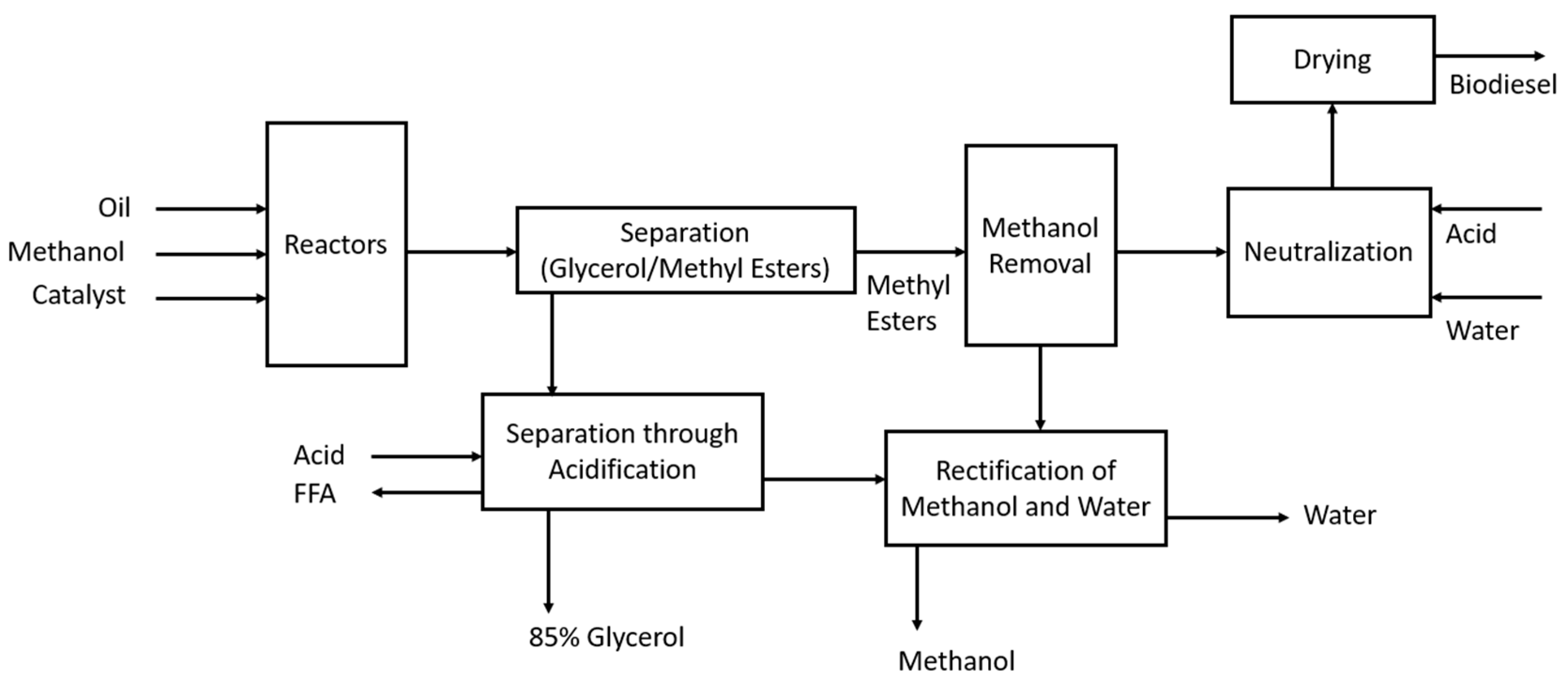
A typical process for biodiesel production through transesterification is represented in Figure 6.
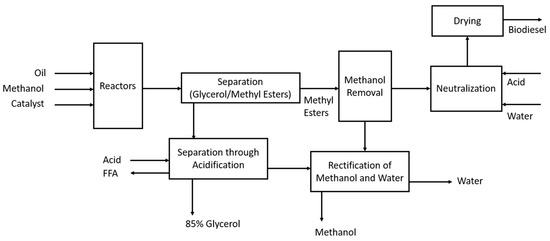
Figure 6.
Production of biodiesel (FAMEs) through transesterification.
This schematic [14] shows that the alcohol, catalyst, and oil enter a reactor where they are stirred for 1 h at 60 °C. Larger plants use a CSTR, and smaller plants use batch reactors. The reaction is typically performed in two steps, where 80% of the alcohol and catalyst is added in the first-stage CSTR, the glycerol is removed, and the remaining 20% is added in the second-stage reactor. Upon the completion of the reaction, glycerol is separated from the methyl esters, and this is easily completed due to glycerol’s low solubility in the esters through a centrifuge or settling tank. Any excess methanol can help the glycerol remain in the solution, thus slowing the separation process, but it is not typically removed due to the potential of reversing the transesterification reaction. Following the separation process, the mixture is neutralised and then stripped, typically with vacuum flashing or an evaporation process. Acid is then added to neutralise the residual catalyst and help with the removal of any soap and salts that may have formed; water washing helps with the removal of these components.
The crude glycerol stream leaving the separator is ~50% glycerol [14], with the remaining components being methanol, catalyst, and soap. In this makeup, glycerol has little commercial use, and the methanol content makes it hazardous waste. Acidification of the mixture will split the soaps in the fatty acids and salts, and the free fatty acids will be insoluble due to the high glycerol content. The methanol is then removed through a vacuum flash process or evaporation, making the glycerol 85% of the mixture, and it can then be sold for glycerol refining.
A key issue to be aware of is the free fatty acid (FFA) content of the feedstock, as a high amount of FFAs will result in saponification reactions, particularly if FFAs are above 5%. If this is the case, it is necessary to first use an acid catalyst to esterify the FFAs into methyl esters [14].
3.2. Hydrocracking
Cracking is the pyrolysis of an organic feedstock in the presence of a catalyst [15], and it leads to the production of lower-molecular-weight aliphatic and aromatic compounds with a reduced hydrogen content. Hydrocracking differs by conducting the cracking process in the presence of hydrogen, and these technologies have matured within the petrochemical industry but could still be applied to the production of biofuels. This process is generally indifferent to organic feedstocks, except for free fatty acids (FFAs), and they can be cost-effective, as demonstrated by the NExBTL commercial plant by NesteOil [15].
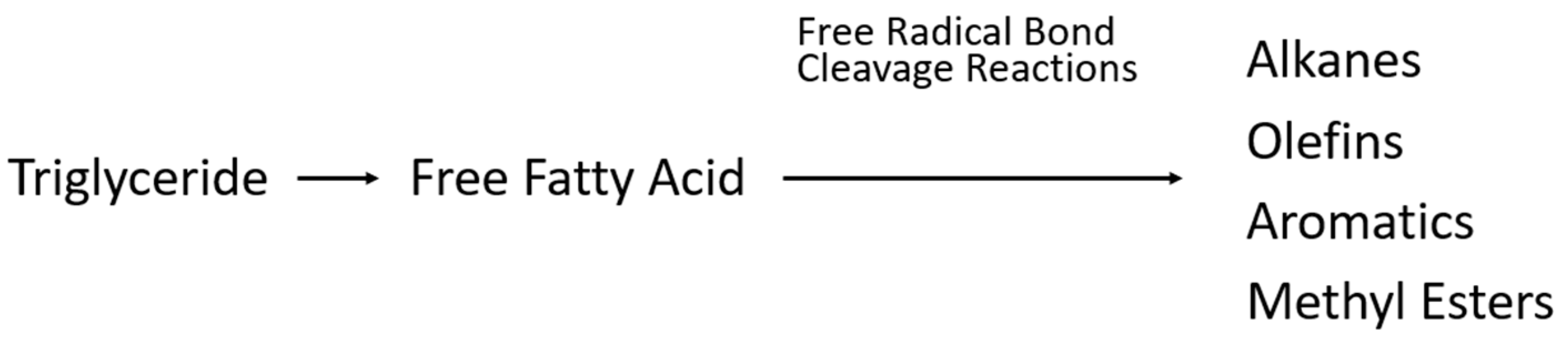
The general pathway of action [16] for the hydrocracking process of triglycerides is given in Figure 7.
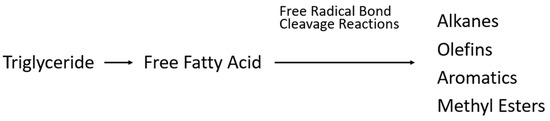
Figure 7.
Pathway of action for hydrocracking of triglycerides.
The first step is the formation of FFAs, followed by radical bond cleavage reactions, which form a series of products, including alkanes, olefins, and aromatics, to unsaturated and saturated methyl esters [15]. The resultant product has improved properties over the use of vegetable oils, but they are still inferior to diesel. The typical process conditions for thermal cracking are 350–500 °C and at pressures of atmospheric to several bars, with a yield of around 70%. As the temperature increases, the selectivity towards lighter hydrocarbons also increases.
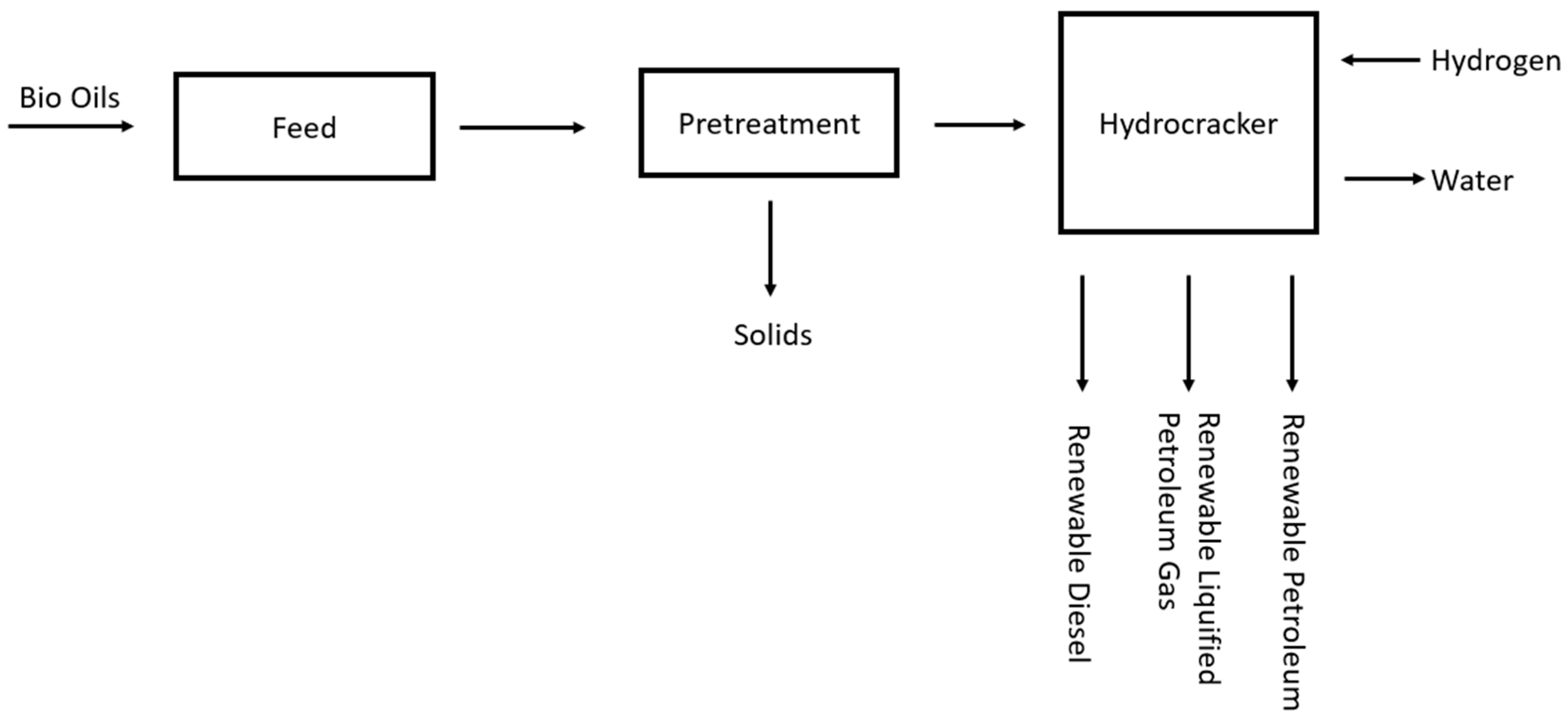
The process plant figure, adapted from Nylund et al. [17], for this production method is much simpler.
A schematic for the Neste Oil NExBTL plant is presented in Figure 8.
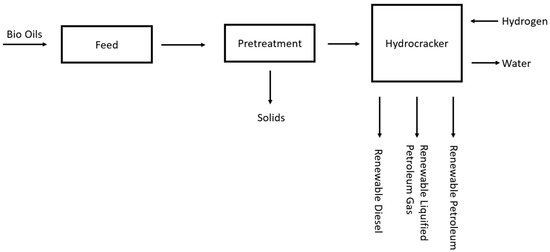
Figure 8.
Schematic for the Neste Oil NExBTL plant.
As the process plant is proprietary, it is unlikely that a specific plant process flow diagram will be made public; however, this plant schematic does give insight into the reduced number of units in the hydrocracking process. The reduced complexity of this process is also shown in pilot plants [18]. As shown in the figure, there is typically pretreatment of the feedstock, such as preheating and solid removal, followed by the hydrocracking reaction, which is essentially pyrolysis with hydrogen and a catalyst, ending with a small amount of downstream separation, thus removing any impurities.
3.3. Hydrodeoxygenation
Green diesel, also known as renewable diesel, is different to biodiesel as it consists of C15–C18 hydrocarbons; therefore, it is similar to fossil-derived diesel except for its narrower boiling point range [19]. It also has higher energy density and cetane values than biodiesel due to its paraffin content and a higher heating value.
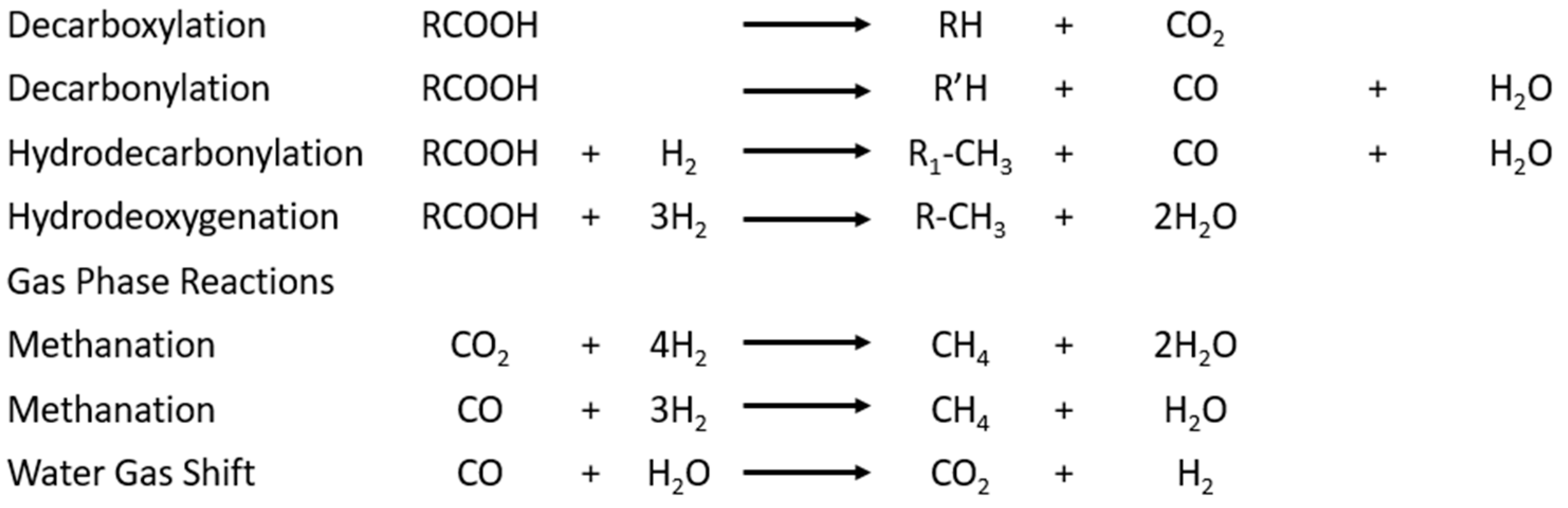
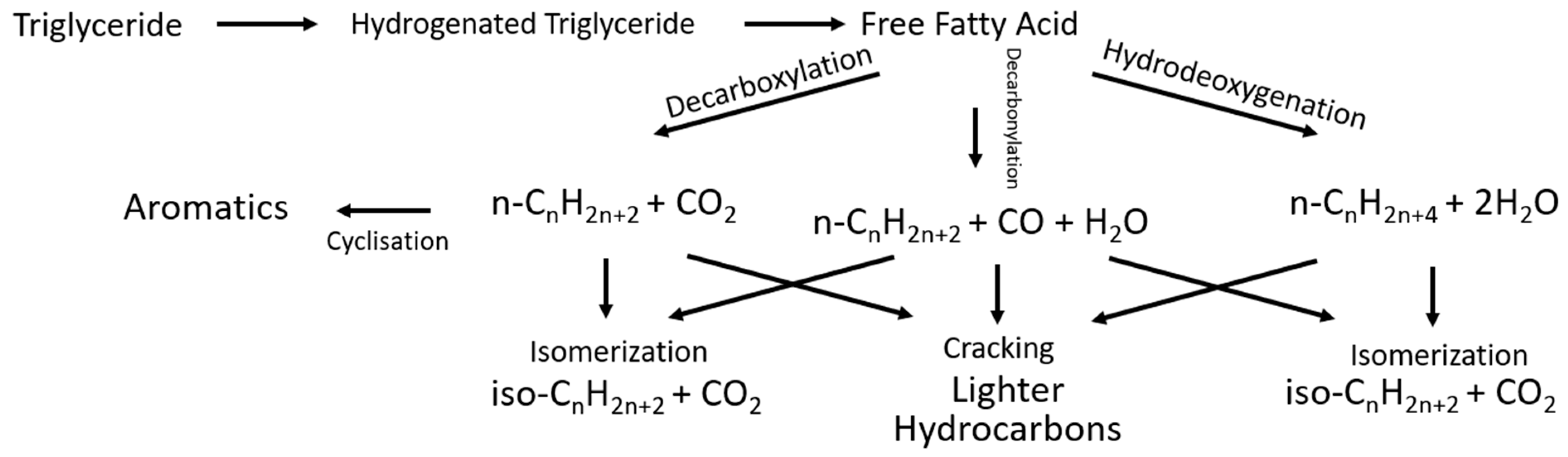
To deoxygenate the feedstock, temperatures in the range of 200 to 375 °C [20,21,22,23] are required in the presence of hydrogen, including instances when the temperature increase leads to an increase in conversion but also a higher operating cost. It has been found that low partial pressure of hydrogen rather than pure hydrogen improves the turnover frequency of the catalyst [24]. Additionally, deoxygenation leads to a higher selectivity than hydrotreating triglycerides [24]. There are many reactions involved in the deoxygenation of the triglycerides. The reactions involved in the deoxygenation of triglycerides are presented in Figure 9.
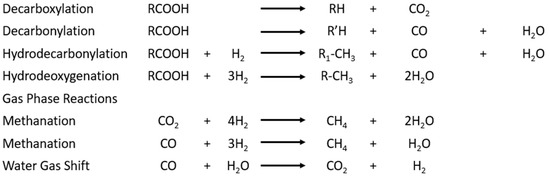
Figure 9.
Reactions involved in the deoxygenation of triglycerides.
The fatty acids can be directly decarboxylated, and activated carbon can provide catalytic activity for this reaction [24]. Furthermore, the decarboxylation and decarbonylation produce carbon dioxide and carbon monoxide, which are used in the water gas shift and methanation reactions. The gas phase reactions then produce hydrogen to enable hydrodecarbonylation (HDO) and hydrogenation of the fatty acids. If the feedstock contains unsaturated hydrocarbons, they are deoxygenated to form saturated diesel fuel hydrocarbons [24]. This is performed by the hydrogenation of the double bond followed by the deoxygenation of the now-saturated fatty acid.
For the type of reactor used in hydrodeoxygenation [24], it was found that tubular reactors suffered mass transfer limitations and therefore performed the worst. Semi-batch reactors outperformed batch reactors due to the utilisation of the purge gas. Catalysts can be deactivated during the deoxygenation reaction due to coking and poisoning by carbon monoxide and carbon dioxide.
4. Fish Waste as a Feedstock
One feedstock that could be used, as it is inedible and readily available, is fish waste. Every year, billions of tons [25] of fish wastes are sent to landfills, contributing to ground pollution and/or incinerated contributing negatively to air pollution. This waste has the potential to be utilised as biofuels, pharmaceuticals, fertilisers, and fodder. As fish waste is non-toxic, biodegradable, and inedible, it has potential, particularly in the production of biofuels. The waste from fish is typically the head, viscera, dorsal fins, tail, skin, and liver [25]. The high biological oxygen demand of this fish waste leads to the generation of hydrogen sulphide, scavengers, and noxious conditions from odours.
Fish waste oils that are rich in omega 3 are not typically available due to the utilisation of this compound in the pharmaceutical industry [25]. Besides the use of waste as a fertiliser [25], the only potentially profitable option for the waste is in the production of renewable fuels. Fish waste can be used to produce two types of fuel, biodiesel and biogas. Biodiesel is produced in the already discussed method of transesterification. The additional step is the extraction of the oils used in transesterification, which is performed by putting the wastes through a cooker at ~100 °C and then using an expeller to squeeze all of the fluids out and leave behind solid biomass. The liquid that leaves the expeller is estimated to be 54% water, 4% solids, and 42% oil [25]. As water is immiscible in oil, clarification is used to separate the mixture into three distinct layers. Alternatively, solvent extraction with the use of N-hexane is used, removing only the fish oil. This extracted fish oil can then be used for the transesterification process. The oil that is extracted has the composition summarised in Table 1 below.

Table 1.
Fatty acid composition of extracted oil extracted from various marine fish determined through gas chromatography [26].
Another study [27] looking at salmon oil specifically found the carbon length of the fatty acids extracted from salmon, also taking into account storage time and temperature.
The study found no significant difference in the composition of the oil over time or the temperature stored. Additionally, most of the oils were composed by C16:1, C18:1ω9cis, and C20:5ω3 EPA fatty acid chains. Given that omega 3 components are particularly valuable in the pharmaceutical industry, it may be important to consider separation of the hydrocarbons for sale in the pharmaceutical industry and for biodiesel production. The omega 3 components of the oil composition, comprising ~32% of the mixture, are C22:6ω3 DHA, C22:5ω3 DPA, C20:5ω3 EPA, C20:4ω3, C18:4ω3, and C18:3ω3. Wholesale diesel has a wholesale cost of GBP 1.0565 per L [28], which is equivalent to GBP 1.2884 per kg [29], assuming a low cost from recent prices. Whilst fish oil wholesale prices vary between GBP 1 and GBP 3 per kg [30], given the similarity in price, it is most likely not worth it to extract the omega components from the product stream.
Biofuels from fish waste offer the advantage of being a renewable non-edible feedstock suitable for use in most engines that already use diesel either as a drop in biofuel or independently as a biofuel. The fuel should follow the standards set out in EN ISO 14214. Parts that may need modifying are ones that biodiesel may interact with and therefore accelerate their damage, such as seals, rubber hoses, and gaskets [31], but this would be a low-cost modification.
5. Reaction Kinetics and Associated Models
The composition of fossil-derived diesel [29] is 75% C10–C15, with the remaining 25% being aromatic hydrocarbons, such as benzene and styrene, whilst also being 86% carbon and 14% hydrogen. Fossil diesel has a heating value of 42.6 MJ/kg, a density of 0.82–0.86 kg/L, and a viscosity of 2–5 mPa s. The flashpoint is greater than 55 °C. As discussed in Table 2, the composition of fish oil is C14–22, so this will require the breakdown of the fatty acids into the C10–C15 carbon chains. From Table 2, it can be calculated that the mass of carbon to hydrogen is 86.7% to 13.2%.
Table 2.
Fatty acid composition of oil extracted from salmon waste that is stored for four days under refrigerated conditions.
5.1. Hydrodeoxygenation
The feedstock for this process will be non-edible, meaning it is classified as a second-generation biofuel, and the two major reactions in the production of green diesel using this feedstock are hydrodeoxygenation and decarboxylation to form the correct chain-length alkanes from triglycerides [32]. The key aspects to review are the reaction mechanism, kinetic model, reaction conditions, and deactivation mechanism. Specifically, for hydrodeoxygenation (HDO), the overall reaction pathway is represented in Figure 10.
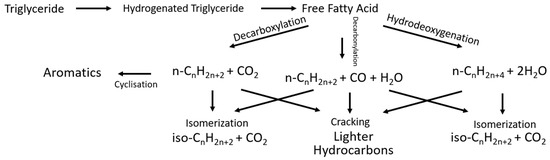
Figure 10.
Reaction pathway for HDO, adapted from [32].
This reaction pathway [32] includes the hydrogen saturation of the triglycerides before cracking through hydrodeoxygenation (HDO), decarboxylation (DCO2), and decarbonylation (DCO). For decarboxylation and decarbonylation, the alkanes formed have one less carbon atom, whilst the hydrodeoxygenation forms alkanes of the same chain length. However, the hydrodeoxygenation pathway has a higher demand for hydrogen, as all of the oxygen is removed from the alkane through hydroygenation, thus increasing costs. Additionally, the DCO and DCO2 pathways operate under milder conditions with a reduced byproduct of water, which are ideal conditions for catalysis. Essentially, the fatty acids are converted to alkanes through the three reaction pathways, and then they are cracked and isomerised into the correct type of alkanes to be cracked into the lighter hydrocarbons for suitable use as green diesel.
There are three general commercial processes [32] for the deoxygenation of fatty acids, including direct hydrodeoxygenation, hydrodeoxygenation and isomerisation, and the co-hydrotreatment process. For direct hydrodeoxygenation at 240–450 °C and 4–20 MPa [32], the feedstock is hydrogenated, typically with a sulfided Ni-Mo or Co-Mo catalyst, producing long-chain alkanes with a high cetane number, which is a good indicator of performance in engines. The resultant product can have poor cold flow properties, which will limit its use in practical applications. The hydrodeoxygenation and isomerisation process is what is used at the NExBTL plant [15]. This is similar to the previous process, just with the addition of isomerisation with a platinum catalyst, which results in good cold flow properties and a high cetane number. Co-hydrotreatment uses the existing hydrotreating unit in a refinery to blend green diesel products by mixing vegetable oils and animal fats with the diesel feed. The kinetics of decarbonylation were investigated for stearate (C18:0), and a Langmuir–Hinshelwood kinetic model was proposed [32]. The products’ adsorption products on the denominator were excluded.
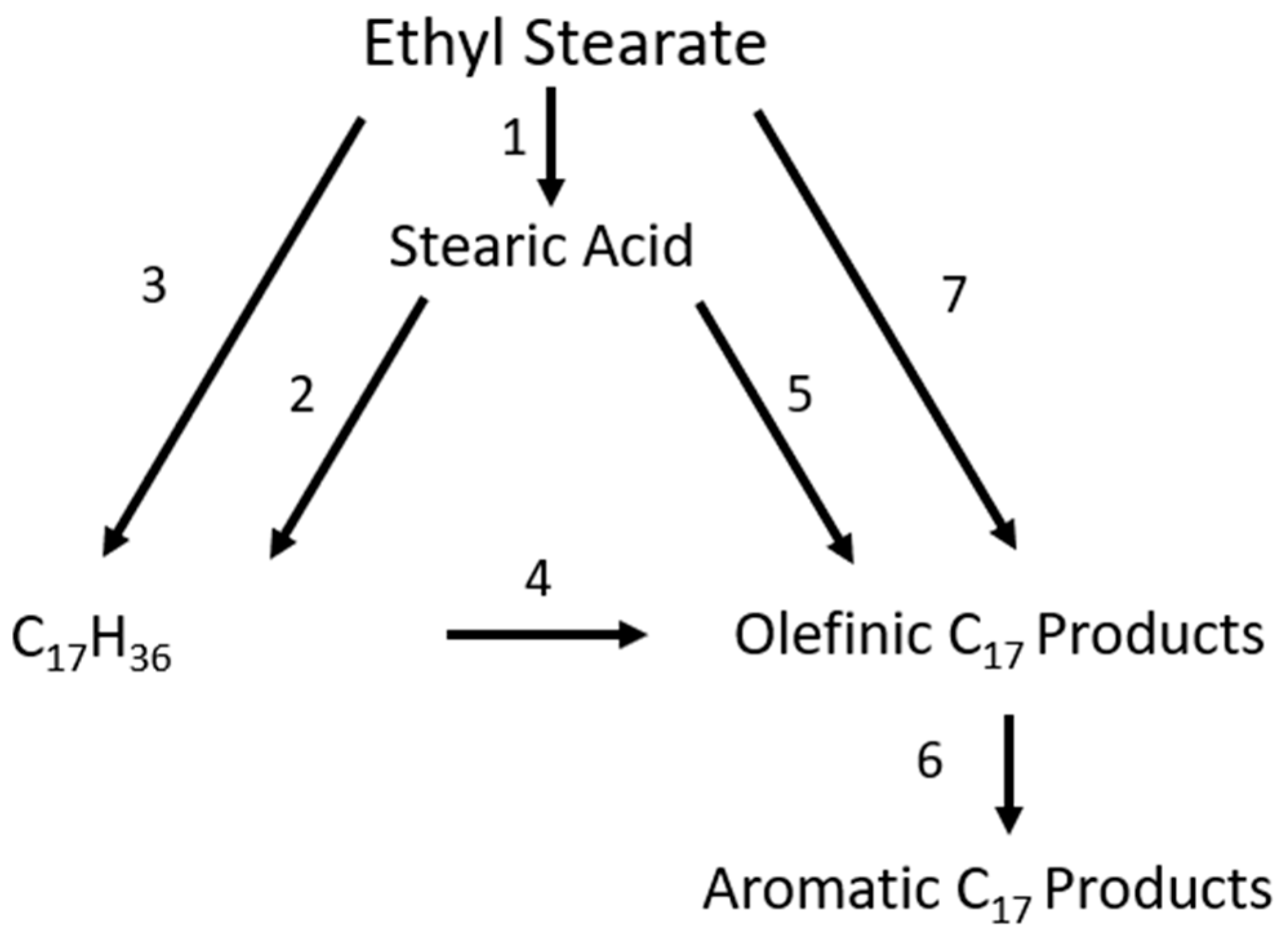
For the proposed reaction pathway for the conversion of ethyl stearate into alkane and aromatic products can be seen in Figure 11.
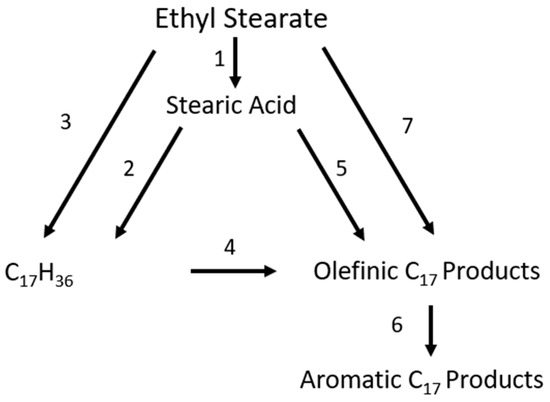
Figure 11.
Reaction pathway for decarbonylation of stearate, adapted from [32].
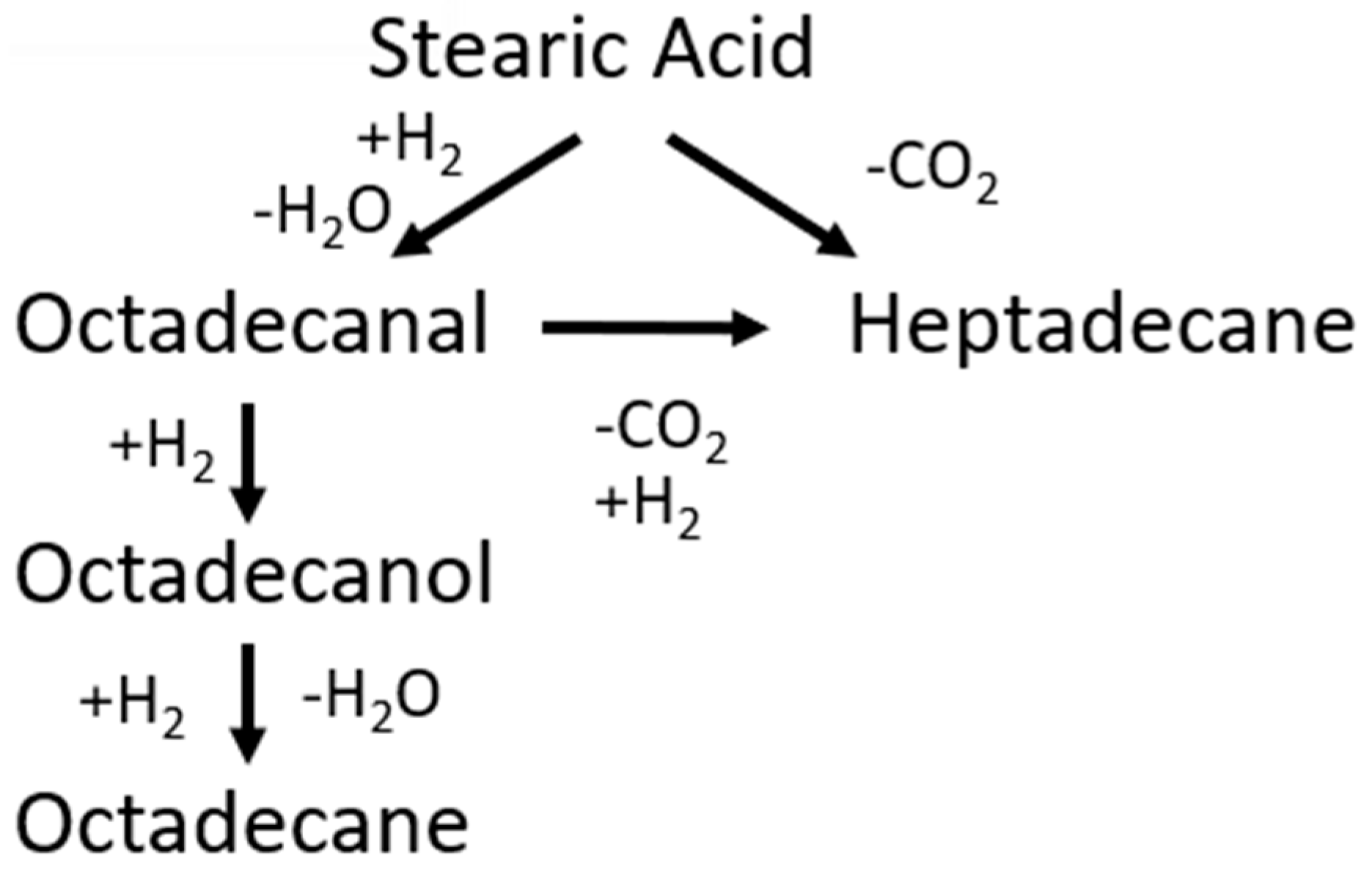
A catalytic mechanism was proposed by Kumar et al. [33] between 533 and 563 K in the presence of a nickel catalyst. The mechanism eliminated the external mass transfer but not the internal diffusion, leading to the model data and experimental data being well-aligned; however, they did not differentiate between DCO and DCO2 mechanisms [32]. Arora et al. [34] proposed the mechanism represented below (Figure 12) for stearic acid HDO, and rate expressions were found to be as follows (Table 3).
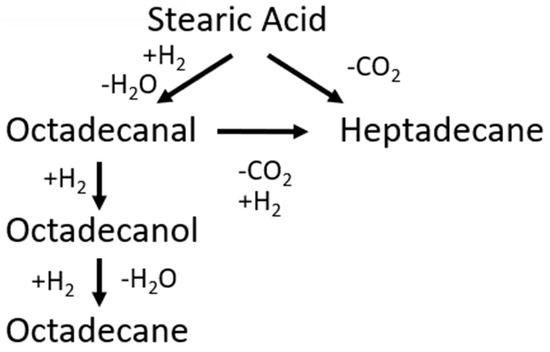
Figure 12.
Reaction scheme for HDO of stearic acid.
Table 3.
Reaction schemes and rate expressions.
This model [34] shows the HDO of stearic acid over a sulphide NiMo/Al2O3 catalyst and uses Langmuir–Hinshelwood kinetics. They also found that there was no oxygenate conversion in the absence of any catalyst; as a result, they did not consider non-catalytic reactions in the modelling. When modelling the reactor, they used a pseudo-homogeneous ideal batch reactor model with the following differential equations for each component [34]:
Nj is the number of moles of component j, is the stoichiometric coefficient for component j in reaction i, ri is the rate of reaction I, and W is the mass of the catalyst. The transport of gas to liquid and liquid to catalyst surface were considered negligible, and the reactor was under isothermal conditions. All rates were assumed to be dependent on the liquid-phase concentrations, in equilibrium with the gas phase. The vapour liquid equilibrium was calculated under the Predictive Soave–Relich–Kwong (PSRK) equations of state, as this is the most suitable estimation for describing mixtures of oxygenated oil feeds for HDO reactions. To improve the estimation of liquid density, the Peneloux volume translation was used to improve the liquid density. To calculate the amount of hydrogen at the inlet of the reactor, at the beginning of the experiment, it was iteratively calculated based on the total reactor volume, the total pressure for the experiment, and quantities of fatty acid and solvent. To calculate the equilibrium constants’ dependence on temperature, the equations were [34]
where A, B, C, etc. as published thermochemical properties were used, and, if they were not available, they were calculated using the Joback Method [35]. As it has been found that the conversion of octadecanal to octadecanol is equilibrium-limited, the rate of reaction was multiplied by a driving force factor [34]
where the activity is
is the fugacity of i in the mixture, is the liquid phase fugacity coefficient, and xi is the mole fraction in the liquid phase. They set the standard of fugacity to 1 bar as the standard state for the equilibrium constants for components in the gas phase.
To estimate the reaction rate constants for the differing temperatures, a modified Arrhenius equation was used [34]:
with a reference temperature of 300 °C, which was the mean experiment temperature. The parameters were optimised using non-linear regression, thus minimising the sum of the square residuals.
In the kinetic model, the reactions of hydrogenation of alkenes to alkanes for C17 and C18 were combined, as they were found in the experiment to be fast. Additionally, the isomers of C17 and C18 were also combined. When investigating the water gas shift and methanation reactions, it gave poor predictions of the gas phase reactions. This is believed to be because the prediction was sensitive to the equilibrium distribution, and the solubility of carbon dioxide was much greater than the solubility of carbon monoxide and methane, resulting in the gas phase being deficient in carbon dioxide but enriched in carbon monoxide and methane. However, as hydrogen was present in excess, the effects of the gas phase reactions were found to be negligible.
For the rate expressions, the stearic acid inhibition term is present on all of the reactions, and this is because all of the reactions are believed to occur on the same site, and stearic acid bonds strongly to the sites and inhibits further reactions for HDO.
To investigate mass transfer limitations [34], they began by changing the mixing rate at 500,900 and 1000 RPM; from this, it was found that there was little significant difference between 900 and 1000 RPM, suggesting that there was little external mass transfer limitation at this speed. To evaluate the gas–liquid mass transfer resistance, the Carberry number (Ca) was used [34]:
The total simulated rate of hydrogen () was found from kinetic model predictions r1, r3, and r5 in Table 3. The simulated rate of hydrogen was deemed appropriate for the calculation of the Carberry number as it was found to be similar to the experimental results. The interfacial area per liquid volume, a, and the gas–liquid mass transport coefficient, , were estimated from a lab-scale slurry reactor. was taken from phase equilibrium calculations, and m is the loading of the catalyst per liquid volume. The simulations found that the reaction rate peaks when enough stearic acid is converted into intermediates that consume hydrogen, and, around this time, the Ca number was also found to peak at 4.4 × 10−5. A Ca number greater than 0.1 indicates that the gas–liquid transport is limiting the reaction. To investigate the mass transfer resistance of hydrogen from the bulk liquid to the catalyst particle, the Weisz–Prater (WP) parameter was used [34]:
The density of the catalyst was estimated to be 1000 kg m−3, and the average catalyst particle diameter was 175 μm. The ratio of the void factor to tortuosity was set to 0.1. The diffusivity of H2 to the solvent ( was taken from the literature as 6.5 × 10−8 m2s−1 at 300 C. All-over values were taken from the simulation, as before. If WP is greater than 1, it could be rate-limiting, and it was found to peak at 3 and 1.8, indicating that pore mass transfer resistance is a factor in observed rate reactions, particularly with C18=O. The final mass transfer resistance, film mass transfer resistance, was evaluated using the Mears Parameter (MP) [34]:
All parameters besides km were estimated as before. km was found from the Sherwood number of 2, and the value it gives is a minimum of mass transfer as it assumes the particle is surrounded by a stagnant liquid. An MP value indicates that film mass transfer may be limiting, and, for the experiment, it was found that it peaked at 0.15 and 0.09 for the highest and lowest temperatures, respectively. From this, it was estimated that film mass transfer limitation was negligible.
For the reaction constants and activation energies that were estimated from the model, some have large confidence intervals. It is believed that this is due to high correlation or the fact that the experimental design could not resolve those values. For example, the rate constant for reaction 1 and the equilibrium constant for stearic acid were highly correlated, leading to large confidence intervals.
The model was then compared with the experimental data [34], and it does seem to predict the experiment well, as demonstrated by the faster consumption of stearic acid at higher temperatures and the increase in decarbonation, shown by the increase in C17 production at higher temperatures. The model follows the experimental data well.
However, this model did not consider all key catalytic steps, as it also indicated that the power law model was inconsistent with experimental results. Mechanistic models are derived from the surface catalytic mechanism for HDO reactions, where two active sites adsorb hydrogen and the dissociated oxygenated components. The model also had noncompetitive adsorption of heptanol and competitive adsorption of heptanoic acid. The mechanistic model had improved performance compared to power law models.
Overall, the mechanistic model seems more appropriate for giving more accurate results, and the Langmuir–Hinshelwood model was the most applied. Most of these models did not account for the deactivation of catalysts and competitive adsorption.
5.2. Isomerisation and Hydrocracking
Following the formation of alkanes from fatty acids through hydrodeoxygenation, the alkanes produced are long C15-C22 and form various isomers. To make the fuel suitable for use in diesel engines, it needs to be isomerised and cracked. This is performed under harsh conditions; in the literature, the values are 250–550 °C and 1–30 bar [36,37,38] in a plug flow reactor [39]. The mechanism of isomerisation and hydrocracking has been studied for crude oil feedstocks; it is, however, scarce for the mixtures of hydrocarbons that are biologically derived. The model proposed by Martinez-Hernandez et al. was based on the following hydrocracking reactions [39]:
CnH2n+2 + H2 → Cn−3H2n−4 + C3H8 for n = 13–18
This leads to the production of C10-C15 hydrocarbons. The production of C7-C9 is represented by the following reactions [39]:
CnH2n+2 + H2 → Cn−8H2n−14 + C8H18 for n = 17, 18
CnH2n+2 + H2 → Cn−7H2n−12 + C7H16 for n = 17, 18
CnH2n+2 + H2 → 2Cn/2Hn/2+2 for n = 16, 18
The first two reactions are the production of C8 and C7, with Equation (12) being the alkane split at the central bond. The final reaction is the isomerisation, as the alkane is converted into a linear form [39]:
CnH2n+2 → iso-CnH2n+2 for n = 6–18
This gives the typical symmetric cracking product distribution, with the major isomerisation products being C8+ hydrocarbons. To solve the reactions, a non-linear program problem was created to minimise the sum of square differences. The objective function was [39]
The calculated value, Ycalc,i, for the mass fraction of hydrocarbon i in the reactor effluent in relation to the conversion rates for each of the reactions is taken away from the experimental value, Yexp,I, of the mass fraction of hydrocarbon i in the isomerisation effluent. This objective function is subject to the following constraints [39]:
where mHT is the mass of hydrotreated steam fed into the reaction, mH2r is the stoichiometric mass of hydrogen for the reactions, and mproducts is the mass of products from the reactor. The model was specifically used for the production of bio-jet fuel, so the yield of the fuel was constrained to the following [39]:
The upper bound was 49.4%, and the lower bound was 12.8%.
The mass flows are then constrained so that the amount that is mass-produced is greater than or equal to the theoretical maximum produced during experimentation [39]:
The overall mass balance was [39]
- mi0 is the initial mass of component i fed into the reactor.
- vi,j is the stoichiometric coefficient of i in reaction j.
- mi is the mass of component i in the reaction mixture plus or minus any consumption.
- xj is the conversion, where it is assumed that the hydrocracking reactions occur in parallel, while isomerisation occurs after.
The conversion for hydrocracking reactions is
For the isomerisation reactions:
The range was set between 0 and 0.9 for both reactions; as the yields and compositions are not known, an additional constraint was used, the freezing point.
The standard freezing point for jet fuel is −47 °C, and, to find the freezing point, the sum of all of the freezing points of the components was multiplied by their mole fractions.
6. Catalysts
The two primary units in which catalysis is important are the hydrodeoxygenation unit, where the triglycerides are converted to alkanes, and the isomerisation/cracking unit, where the carbon chains are broken down and reorganised into linear alkanes C7–C15. They are heterogeneous catalysts, as these are used industrially.
A summary of various common catalysts and their conditions is given in Table 4 below.
Table 4.
Performance of catalysts in the hydrodeoxygenation of stearic acid and tristearin [40].
To ensure a significant yield is obtained without a catalyst, the temperature typically must be greater than 400 °C. It was found that high rates were occurring with carbonaceous supports due to the ability to promote the reaction through amphoteric properties and surface properties and its ability to minimise the effect of coke-induced deactivation through the large surface area. It was also found that the most active metals, in order of greatest to least, were palladium, platinum, nickel, rhodium, iridium, ruthenium, and osmium [41].
The literature on the effect of loading is less clear, with some sources stating that a difference between 0.5 wt% and 5 wt% has little effect on selectivity and conversion. Other sources state that there is a significant change when they increase the catalyst from 1 wt% to 5 wt% [40]. For the dispersion of the catalyst [47], 18, 47, 65, and 72% were investigated, and 65% was found to be the most effective, with the others providing insufficient surface area or strong interactions that change the activity of the catalyst. An increase in the alkalinity of the catalyst was found to increase the number of desirable products, such as n-heptadecane, but also the number of undesirable products, such as aromatics.
Although palladium and platinum are effective catalysts, they present a considerable cost compared to other types of catalysts. Nickel looks like it could be a promising alternative, as, in Table 4, 20 wt% Ni/C outperformed 5 wt% Pd/C and 1 wt% Pt/C. Even though platinum and palladium are more active than nickel, nickel is much lower in cost. Although the reactions in Table 4 were in batch reactors, which led to significant deactivation of the catalysts, nickel is 1000–2500 times cheaper than palladium and platinum [40]. Another potential advantage is that utilising a nickel catalyst can induce some cracking within the hydrocarbons, thus increasing the yield of hydrocarbons within the green diesel range.
Another compound that has shown potential is the CoMo [48], cobalt–molybdenum, catalyst. With this catalyst, activity is promoted by cobalt; however, it produces products that do not facilitate deoxygenation reactions. Hence, they suggest the addition of sulphur for a CoMoS, the cobalt-molybdenum-sulphur catalyst, but this has a flaw of deactivation as oxygen reacts with the sulphur molecule. Additionally, tungsten has also shown promise in the HDO reaction [49], especially when mixed with nickel, as it can lead to an increased surface area as they interact with each other. However, the study focused on the HDO of aromatics, so the use of tungsten and nickel on carbon may not be feasible for triglycerides from oils. The price of tungsten is not substantially higher than that of nickel, so the blend could be investigated to see if the improvement in activity is worth the cost. It is also important to note that the investigations into cobalt, molybdenum, and tungsten have only been conducted at a laboratory scale.
Catalyst Deactivation
Catalyst deactivation is important as it is a key factor in the determination of the lifetime of the catalyst. Deactivation could be due to poisoning, specifically from the interactions with metals, metalloids, and chemicals, leaching of active phases, coking, and sintering. Due to the oxygenates content [32] and conditions, coking is the most likely to happen, and the higher the number of unsaturated triglycerides, the more coking occurs. Pre-hydrogenation was beneficial for producing high yields of diesel-range alkanes and preventing the coking of catalysts. Another way to reduce the coking effect is by increasing the space velocity, which reduces the secondary reaction occurring. Lowering the temperature, increasing the hydrogen partial pressure, and supporting larger pores, which are mildly acidic, can prevent coking.
Another mechanism to poison the catalyst [32], particularly the CoMoS catalyst, occurs when the oxygen within the triglyceride interacts with sulphur vacancies on the catalyst and releases hydrogen sulphide molecules. The oxygen replaces the sulphur in the catalyst. Water could also cause this interaction, and, in fact, may enhance the process. This was also found in NiMo/Al2O3 and CoMo/Al2O3, where an increase in water by 1.9% reduced conversion by 15%. As this reaction is reversible, the addition of H2S in the feed could prevent this interaction from occurring, but this introduces separation and corrosion problems.
Leaching and sintering can also impact deactivation, but it largely depends on the catalyst and the support selected.
7. Economic Analysis
For any product to be sustainable, it must be economically feasible. To conduct a techno-economic analysis, one performed by Martinez-Hernandez et al. used the following parameters [39]: annual operating time, dollar exchange rate, interest rate, lifetime of the plant, feedstock cost, hydrogen cost, electricity cost, cooling water cost, steam cost, wastewater cost, labour rate, sell price in comparison to propane, naphtha, bio-jet fuel, and green diesel. In this economic analysis, the key parameters were the sensitivity to oil feedstock price, the minimum selling price, and an uncertainty analysis using Monte Carlo simulations. From the simulation of the production using an HDO reactor and a cracking and isomerisation reactor all based in Mexico, they obtained a table of products, including, primarily, carbon chain length C8–C15. The software used to calculate the sensitivity to oil price and the minimum selling price was the “SuperPro Designer Version 11”, and they created their own Monto Carlo simulations for the uncertainty analysis.
As the oil feed is the largest cost component of fuel production, a study [39] looked at how low the price of the product should be to be profitable.
It shows that for a 10% Initial Rate of Return (IRR) of USD 0.263 per litre of oil a production capacity of 95,000 barrels per year would be required. As the price increases, the capacity of production must increase to achieve a viable IRR. Green diesel seems much less sensitive to the price, as all but USD 0.5 per litre of oil can achieve a 10% IRR with production above 60,000 barrels per year. Given that the cost of salmon [50] is GBP 7.3 per kg, it is feasible to achieve a low cost of production, although it does depend on other costs, such as utilities and labour.
Another aspect investigated was the minimum selling price of the fuel depending on the production capacity of the plant. The study [39] also investigated the minimum selling price.
It shows that to produce bio-jet fuel below 150,000 barrels per year, the minimum selling price rapidly increases, but, after that, the decrease is slower and more linear, looking more likely to plateau. For green diesel, the minimum selling price rapidly decreases below 100,000 barrels per year, and, after that, it plateaus. This means that for production above 100,000 barrels per year of green diesel, the increase in capacity may not lead to an increase in Return on Investment, or ROI. Green diesel is also attractive, as this minimum selling price is competitive with fossil fuels, which have a minimum selling price of USD 0.95 per litre [39].
In the final aspect of their techno-economic analysis [39], they investigated the uncertainty of the results using a Monte Carlo simulation on the 63,000 barrels per year produced by the green diesel plant and the 75,000 barrels per year produced by the bio-jet fuel plant. The parameters analysed were the oil price, hydrogen price, direct fixed capital costs, and the jet and diesel selling prices. For green diesel, there was a 50% chance of failure to reach the 10% IRR and a 24% chance that there would be a negative IRR [39]. It was assumed that the high distributions are likely due to the high sensitivity of the IRR to variations in capital costs and oil and hydrogen costs. To reduce the risk and increase the likelihood of profitability, the selling price for the products was required, and to identify a suitable price, they carried out another Monte Carlo simulation to identify what price was sufficient for a 95% probability of a positive IRR. For green diesel, it was found that a sell price of USD 1 per litre would provide this, and this price is close to the bulk cost of fossil fuels [39].
8. Conclusions
The need for biofuels has been demonstrated by the fact that heavy transport makes up a significant portion of transport emissions, and, for this reason, biofuels are predicted to make up 27% of fuels by 2050. This will ensure that the hard-to-decarbonise sectors, such as the marine sector, aviation, and heavy transport, can utilise sustainable fuels. This is essential, as current sustainable technologies, such as batteries, are inadequate for these sectors due to their power-to-weight ratios. Furthermore, batteries use limited resources, such as lithium, compounded by the fact the extraction of lithium is harmful to local environments and communities.
The production of sustainable liquid biofuels with similar properties to fossil-derived fuels faces several challenges, especially around the renewable feedstock used. If the renewable feedstock is a vegetable or animal oil from edible biomass, it can impact the local communities, as it can increase the local price of the food. For this reason, this study looks at inedible biomass, including, specifically, the inedible waste produced from salmon. This waste typically ends up in landfills, contributing to climate change, noxious fumes, and the accumulation of scavengers, whilst they still contain valuable oils that could be used for biofuel production or pharmaceuticals. It has been identified that the cost of production of these fuels means that the separation of the pharmaceutically active ingredients is not feasible, as the bulk sell prices of these products are similar.
There are three viable processes to produce biofuels from fish waste: transesterification, hydrocracking, and hydrodeoxygenation. Transesterification is the traditional method for biofuel production, and it is a chemical reaction with an alcohol and catalyst, resulting in biodiesel. The advantages of this method are that it is well-established, that commercial viability has already been proven, as well as the fact that the method of production is well-understood. Hydrocracking also exists commercially and in the Neste Oil plant where hydrogen is added under cracking conditions to create kerosene and green diesel hydrocarbons. However, the process investigated in this paper is hydrodeoxygenation. It is promising, as it has a good yield of green diesel, and, in the economic analysis, green diesel is competitive in price with traditional fossil fuels. As a result, green diesel production through hydrodeoxygenation provides a good opportunity to disrupt the traditional fossil-fuel-based fuel economy.
The kinetics of hydrodeoxygenation have been investigated and found to primarily be due to hydrodeoxygenation, decarboxylation, and decarbonylation. In hydrodeoxygenation, hydrogen attacks the oxygen molecule on the triglyceride to produce water and an alkane. In decarboxylation, the conditions force carbon dioxide to be removed from the triglyceride, producing the alkane and carbon dioxide. Decarbonylation is similar, but carbon monoxide is produced. These three pathways produce various isomers of alkanes in the range of C15–C20 from the oil feedstock. As green diesel needs to be linear, alkanes with some aromatics, cracking, and isomerisation reactions are required. In this process, high temperature and pressure conditions crack and isomerise the alkanes so that they have suitable characteristics for use in diesel engines.
Various catalysts have been investigated for this process, and the work from this has typically found that metals in carbon show the most success in yield and conversion. Specifically, palladium, platinum, and nickel show promising properties in the process. Palladium and platinum are very active; however, they are extremely expensive, as they are 1000–2500 times more expensive than nickel. Furthermore, nickel loading on carbon of 20 wt% has shown similar activity to palladium and platinum loading of 5 wt%. This means that nickel is a promising economic catalyst to produce biodiesel, and its use will likely be present in the work. Other catalysts, such as CoMoS, have also been investigated, but their activities do not justify their use in the process due to their use primarily being proven at only the lab scale or their proprietary nature making them prohibitively expensive. CoMoS has shown promising properties in activity, but this has only been proven at a lab scale, and it has a unique poisoning mechanism where the sulphur reacts with oxygen to poison the catalyst, which means it is unlikely to be suitable for the process.
Typically, catalyst deactivation methods are primarily centred around poisoning and coking due to the carbonaceous nature of the reactions; however, it has been found that loading on carbon reduces the impact of these methods significantly.
Overall, there are various methods to produce green fuels. However, selective deoxygenation shows the most potential due to its high yield and high-value product.
Author Contributions
N.O.: methodology, visualisation, formal analysis, writing—original draft preparation. D.K.: methodology, visualisation, formal analysis, writing—review and editing. S.L.: conceptualisation, visualisation, formal analysis, writing—review and editing. B.S.: conceptualisation, visualisation, formal analysis, writing—review and editing, supervision, and project administration. All authors have read and agreed to the published version of the manuscript.
Funding
The authors would like to acknowledge the MarRI-UK initiative supported by the UK Department for Transport for developing the ideas for this review paper (original project title: SALMO—Sustainable Aquaculture Leading to Marine Opportunities).
Conflicts of Interest
Author Sergio Lima was employed by the company Green Fuels Research Ltd. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- International Energy Agency. CO2 Emissions in 2022; IEA: Paris, France, 2023. [Google Scholar]
- International Energy Agency. Biofuels; IEA: Paris, France, 2023. [Google Scholar]
- Gross, S. The Challenge of Decarbonizing Heavy Transport; Brookings: Washington, DC, USA, 2020. [Google Scholar]
- Clean Energy Institute. Lithium-Ion Battery; Clean Energy Institute: Washington, DC, USA, 2023. [Google Scholar]
- Bills, A.; Sripad, S.; Fredericks, W.L.; Singh, M.; Viswanathan, V. Performance Metrics Required of Next-Generation Batteries to Electrify Commercial Aircraft. ACS Energy Lett. 2020, 5, 663–668. [Google Scholar] [CrossRef]
- Hajilary, N.; Rezakazemi, M.; Shirazian, S. Biofuel types and membrane separation. Environ. Chem. Lett. 2019, 17, 1–18. [Google Scholar] [CrossRef]
- Oh, Y.; Hwang, K.; Kim, C.; Kim, J.R.; Lee, J. Recent developments and key barriers to advanced biofuels: A short review. Bioresour. Technol. 2018, 257, 320–333. [Google Scholar]
- Gui, M.M.; Lee, K.T.; Bhatia, S. Feasibility of edible oil vs. non-edible oil vs. waste edible oil as biodiesel feedstock. Energy 2008, 33, 1646–1653. [Google Scholar] [CrossRef]
- Chen, W.-H.; Lee, K.T.; Ong, H.C. Biofuel and Bioenergy Technology. Energies 2019, 12, 290. [Google Scholar] [CrossRef]
- Abd Rabu, R.; Janajreh, I.; Honnery, D. Transesterification of waste cooking oil: Process optimization and conversion rate evaluation. Energy Convers. Manag. 2013, 65, 764–769. [Google Scholar] [CrossRef]
- Pinto, A.C.; Guarieiro, L.L.N.; Rezende, M.J.C.; Ribeiro, N.M.; Torres, E.A.; Lopes, W.A.; Pereira, P.A.d.P.; Andrade, J.B.d. Biodiesel: An overview. J. Braz. Chem. Soc. 2005, 16, 1313–1330. [Google Scholar] [CrossRef]
- Ma, F.; Hanna, M.A. Biodiesel production: A review. Bioresour. Technol. 1999, 70, 1–15. [Google Scholar] [CrossRef]
- Plazas-González, M.; Guerrero-Fajardo, C.A.; Sodré, J.R. Modelling and simulation of hydrotreating of palm oil components to obtain green diesel. J. Clean. Prod. 2018, 184, 301–308. [Google Scholar] [CrossRef]
- Gerpen, J.V. Biodiesel processing and production. Fuel Process. Technol. 2005, 86, 1097–1107. [Google Scholar] [CrossRef]
- Sivasamy, A.; Cheah, K.; Fornasiero, P.; Kemausuor, F.; Zinoviev, S.; Miertus, S. Catalytic Applications in the Production of Biodiesel from Vegetable Oils. ChemSusChem 2009, 2, 278–300. [Google Scholar] [CrossRef]
- Schwab, A.W.; Dykstra, G.J.; Selke, E.; Sorenson, S.C.; Pryde, E.H. Diesel Fuel from Thermal Decomposition of Soybean Oil. J. Am. Oil Chem. Soc. 1988, 65, 1781–1786. [Google Scholar] [CrossRef]
- Nylund, N.; Erkkilä, K.; Ahtiainen, M.; Murtonen, T.; Saikkonen, P.; Amberla, A.; Aatola, H. Optimized usage of NExBTL renewable diesel fuel. OPTIBIO 2011, 2604, 1–172. [Google Scholar]
- Bezergianni, S.; Kalogianni, A. Hydrocracking of used cooking oil for biofuels production. Bioresour. Technol. 2009, 100, 3927–3932. [Google Scholar] [CrossRef]
- Tsiotsias, A.I.; Hafeez, S.; Charisiou, N.D.; Al-Salem, S.M.; Manos, G.; Constantinou, A.; AlKhoori, S.; Sebastian, V.; Hinder, S.J.; Baker, M.A.; et al. Selective catalytic deoxygenation of palm oil to produce green diesel over Ni catalysts supported on ZrO2 and CeO2–ZrO2: Experimental and process simulation modelling studies. Renew. Energy 2023, 206, 582–596. [Google Scholar] [CrossRef]
- Zhou, Y.; Liu, X.; Yu, P.; Hu, C. Temperature-tuned selectivity to alkanes or alcohol from ethyl palmitate deoxygenation over zirconia-supported cobalt catalyst. Fuel 2020, 278, 118–295. [Google Scholar] [CrossRef]
- Kim, S.K.; Lee, H.; Hong, M.H.; Lim, J.S.; Kim, J. Low-temperature, Selective Catalytic Deoxygenation of Vegetable Oil in Supercritical Fluid Media. ChemSusChem 2014, 7, 492–500. [Google Scholar] [CrossRef]
- Wang, S.; Zhou, P.; Jiang, L.; Zhang, Z.; Deng, K.; Zhang, Y.; Zhao, Y.; Li, J.; Bottle, S.; Zhu, H. Selective deoxygenation of carbonyl groups at room temperature and atmospheric hydrogen pressure over nitrogen-doped carbon supported Pd catalyst. J. Catal. 2018, 368, 207–216. [Google Scholar] [CrossRef]
- Rogers, K.A.; Zheng, Y. Selective Deoxygenation of Biomass-Derived Bio-oils within Hydrogen-Modest Environments: A Review and New Insights. ChemSusChem 2016, 9, 1750–1772. [Google Scholar] [CrossRef] [PubMed]
- Morales-Delarosa, S.; Campos-Martin, J.M. 6—Catalytic processes and catalyst development in biorefining. In Advances in Biorefineries; Waldron, K., Ed.; Woodhead Publishing: Sawston, UK, 2014; p. 152. [Google Scholar]
- Yuvaraj, D.; Bharathiraja, B.; Rithika, J.; Dhanasree, S.; Ezhilarasi, V.; Lavanya, A.; Praveenkumar, R. Production of biofuels from fish wastes: An overview. Biofuels 2019, 10, 301–307. [Google Scholar] [CrossRef]
- Tanwar, D.; Tanwar, A.; Sharma, D.; Mathur, Y.P.; Khatri, K.K.; Soni, S.L. Production and characterization of fish oil methyl ester. Int. J. Innov. Technol. Res. 2013, 1, 209–217. [Google Scholar]
- Wu, T.H.; Bechtel, P.J. Salmon by-product storage and oil extraction. Food Chem. 2008, 111, 868–871. [Google Scholar] [CrossRef]
- RAC. Petrol and Diesel Prices; RAC: London, UK, 2023. [Google Scholar]
- Huth, M.; Heilos, A. 14—Fuel flexibility in gas turbine systems: Impact on burner design and performance. In Modern Gas Turbine Systems; Jansohn, P., Ed.; Woodhead Publishing: Sawston, UK, 2013; p. 635. [Google Scholar]
- Selina Wamucii. United Kingdom Fish Oil Prices. 2023. Available online: https://www.selinawamucii.com/insights/prices/united-kingdom/fish-oil/#google_vignette (accessed on 7 November 2023).
- Tyrovola, T.; Dodos, G.S.; Kalligeros, S.; Zannikos, F.E. The Introduction of Biofuels in Marine Sector. J. Environ. Sci. Eng. 2017, 6, 415–421. [Google Scholar] [CrossRef]
- Lin, D.; Mao, Z.; Feng, X.; Zhou, X.; Yan, H.; Zhu, H.; Liu, Y.; Chen, X.; Tuo, Y.; Peng, C.; et al. Kinetic insights into deoxygenation of vegetable oils to produce second-generation biodiesel. Fuel 2023, 333, 126–416. [Google Scholar] [CrossRef]
- Kumar, P.; Yenumala, S.R.; Maity, S.K.; Shee, D. Kinetics of hydrodeoxygenation of stearic acid using supported nickel catalysts: Effects of supports. Appl. Catal. A Gen. 2014, 471, 28–38. [Google Scholar] [CrossRef]
- Arora, P.; Grennfelt, E.L.; Olsson, L.; Creaser, D. Kinetic study of hydrodeoxygenation of stearic acid as a model compound for renewable oils. Chem. Eng. J. 2019, 364, 376–389. [Google Scholar] [CrossRef]
- CheGuide. Property Estimation Joback Method. 2023. Available online: https://cheguide.com/ (accessed on 8 November 2023).
- Kissin, Y.V. Chemical mechanisms of catalytic cracking over solid acidic catalysts: Alkanes and alkenes. Catal. Rev. 2001, 43, 85–146. [Google Scholar] [CrossRef]
- Kotrel, S.; Knözinger, H.; Gates, B.C. The Haag–Dessau mechanism of protolytic cracking of alkanes. Microporous Mesoporous Mater. 2000, 35–36, 11–20. [Google Scholar] [CrossRef]
- Tang, X.; Huang, G.; Zhang, M.; Han, J. Compositional Characteristics and Geochemical Significance of N-alkanes in Process of Crude Oil Cracking. Earth Sci. Front. 2009, 16, 372–378. [Google Scholar] [CrossRef]
- Martinez-Hernandez, E.; Ramírez-Verduzco, L.F.; Amezcua-Allieri, M.A.; Aburto, J. Process simulation and techno-economic analysis of bio-jet fuel and green diesel production—Minimum selling prices. Chem. Eng. Res. Des. 2019, 146, 60–70. [Google Scholar] [CrossRef]
- Santillan-Jimenez, E.; Crocker, M. Catalytic deoxygenation of fatty acids and their derivatives to hydrocarbon fuels via decarboxylation/decarbonylation. J. Chem. Technol. Biotechnol. 2012, 87, 1041–1050. [Google Scholar] [CrossRef]
- Snåre, M.; Kubičková, I.; Mäki-Arvela, P.; Eränen, K.; Murzin, D.Y. Heterogeneous Catalytic Deoxygenation of Stearic Acid for Production of Biodiesel. Ind. Eng. Chem. Res. 2006, 45, 5708–5715. [Google Scholar] [CrossRef]
- Berenblyum, A.S.; Podoplelova, T.A.; Shamsiev, R.S.; Katsman, E.A.; Danyushevsky, V.Y. On the mechanism of catalytic conversion of fatty acids into hydrocarbons in the presence of palladium catalysts on alumina. Pet. Chem. 2011, 51, 336–341. [Google Scholar] [CrossRef]
- Berenblyum, A.S.; Danyushevsky, V.Y.; Katsman, E.A.; Podoplelova, T.A.; Flid, V.R. Production of engine fuels from inedible vegetable oils and fats. Pet. Chem. 2010, 50, 305–311. [Google Scholar] [CrossRef]
- Immer, J.G.; Kelly, M.J.; Lamb, H.H. Catalytic reaction pathways in liquid-phase deoxygenation of C18 free fatty acids. Appl. Catal. A Gen. 2010, 375, 134–139. [Google Scholar] [CrossRef]
- Lestari, S.; Mäki-Arvela, P.; Eränen, K.; Beltramini, J.; Max Lu, G.Q.; Murzin, D.Y. Diesel-like Hydrocarbons from Catalytic Deoxygenation of Stearic Acid over Supported Pd Nanoparticles on SBA-15 Catalysts. Catal. Lett. 2010, 134, 250–257. [Google Scholar] [CrossRef]
- Morgan, T.; Grubb, D.; Santillan-Jimenez, E.; Crocker, M. Conversion of Triglycerides to Hydrocarbons Over Supported Metal Catalysts. Top. Catal. 2010, 53, 820–829. [Google Scholar] [CrossRef]
- Mäki-Arvela, P.; Snåre, M.; Eränen, K.; Myllyoja, J.; Murzin, D.Y. Continuous decarboxylation of lauric acid over Pd/C catalyst. Fuel 2008, 87, 3543–3549. [Google Scholar] [CrossRef]
- Bui, V.N.; Laurenti, D.; Afanasiev, P.; Geantet, C. Hydrodeoxygenation of guaiacol with CoMo catalysts. Part I: Promoting effect of cobalt on HDO selectivity and activity. Appl. Catal. B Environ. 2011, 101, 239–245. [Google Scholar] [CrossRef]
- Echeandia, S.; Arias, P.L.; Barrio, V.L.; Pawelec, B.; Fierro, J.L.G. Synergy effect in the HDO of phenol over Ni–W catalysts supported on active carbon: Effect of tungsten precursors. Appl. Catal. B Environ. 2010, 101, 1–12. [Google Scholar] [CrossRef]
- MOWI. Salmon Farming Industry Handbook; Springer: Berlin/Heidelberg, Germany, 2019; pp. 1–56. [Google Scholar]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).