
Electron Paramagnetic Resonance in Lignocellulosic Biomass Pyrolysis Mechanism: Advancements, Applications, and Prospects



Abstract
1. Introduction
2. Electron Paramagnetic Resonance Spectroscopy
2.1. Principle of EPR Detection
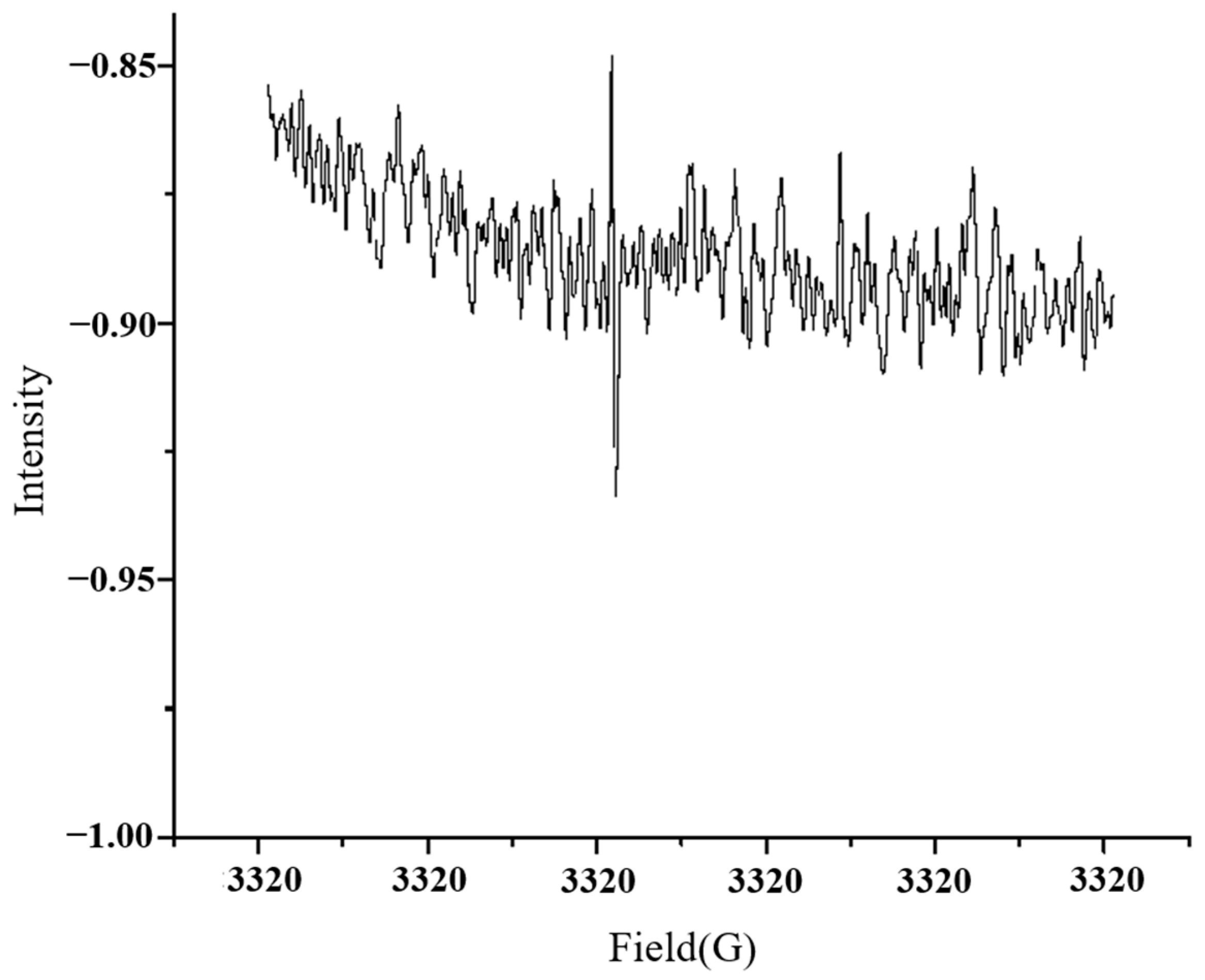
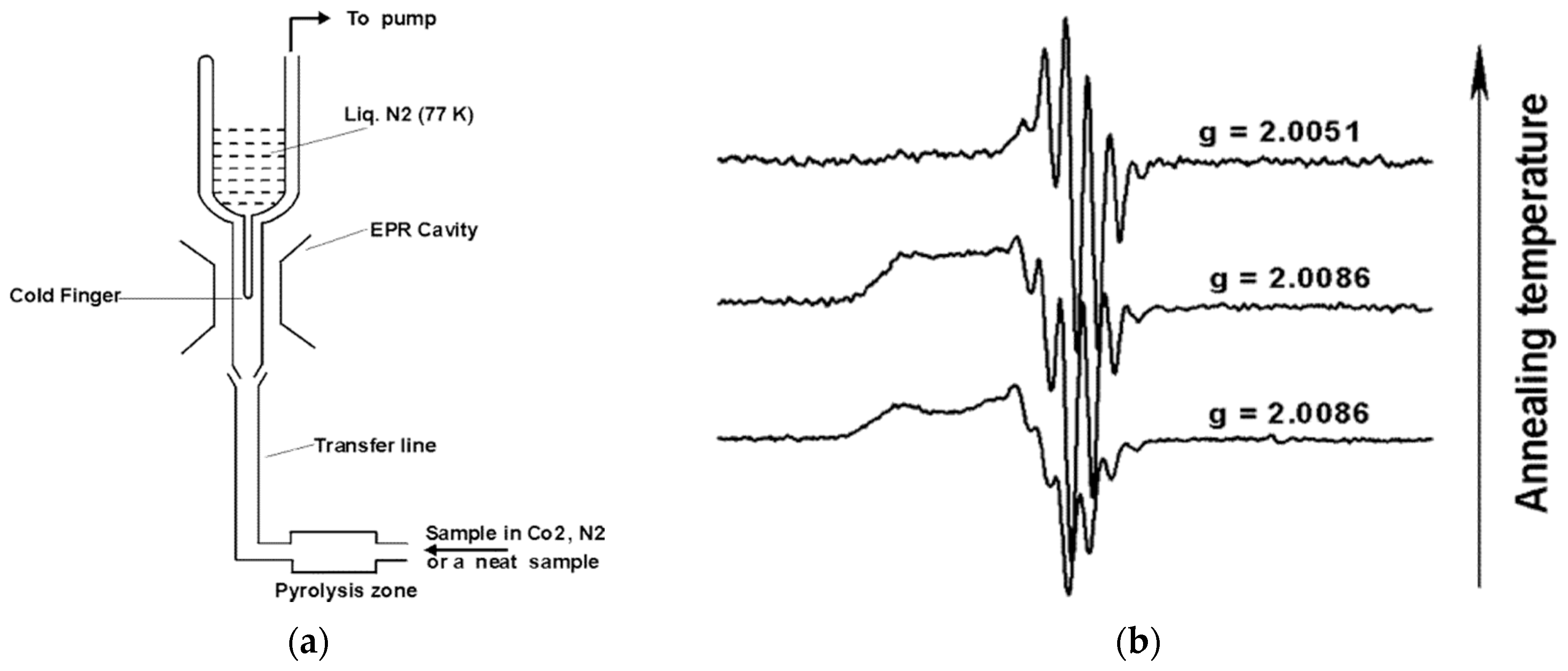
2.2. Radical Detection Methods
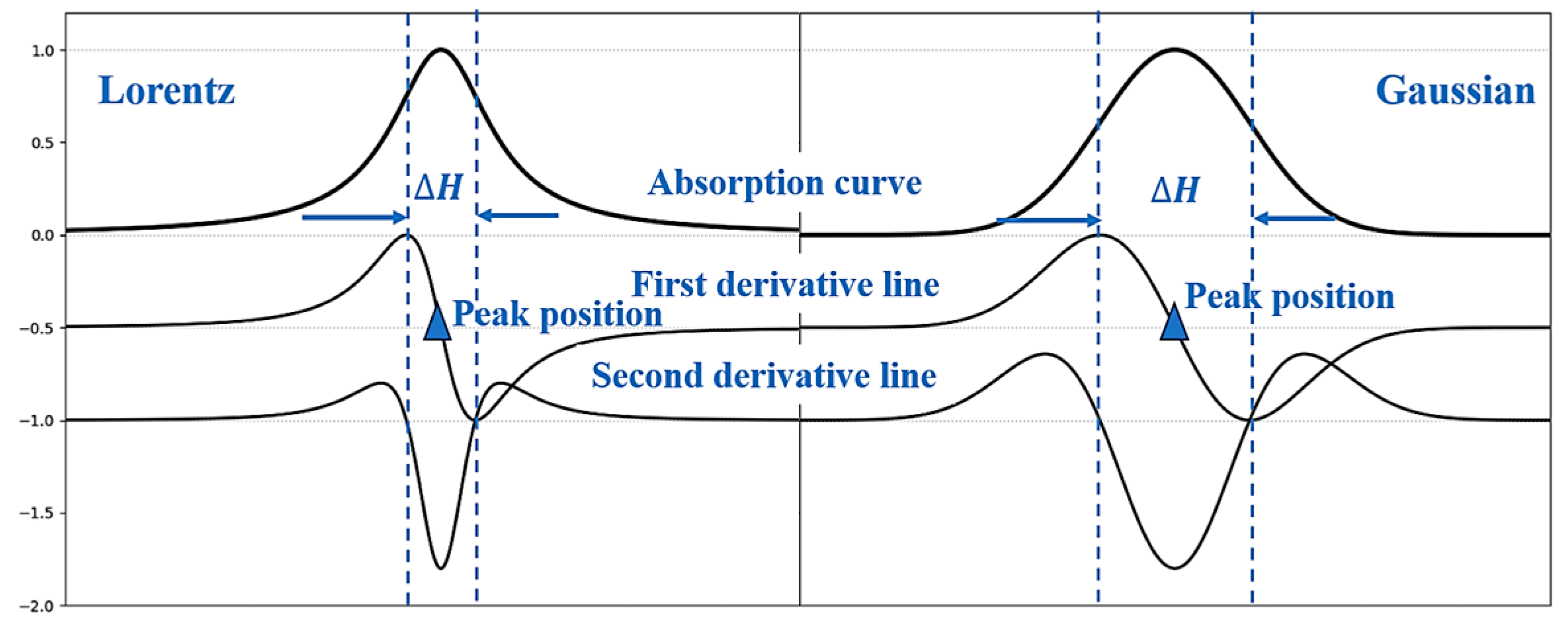
2.3. Spectral Analysis of EPR
3. EPR for Model Compounds’ Pyrolysis
3.1. Monomer Model Compound
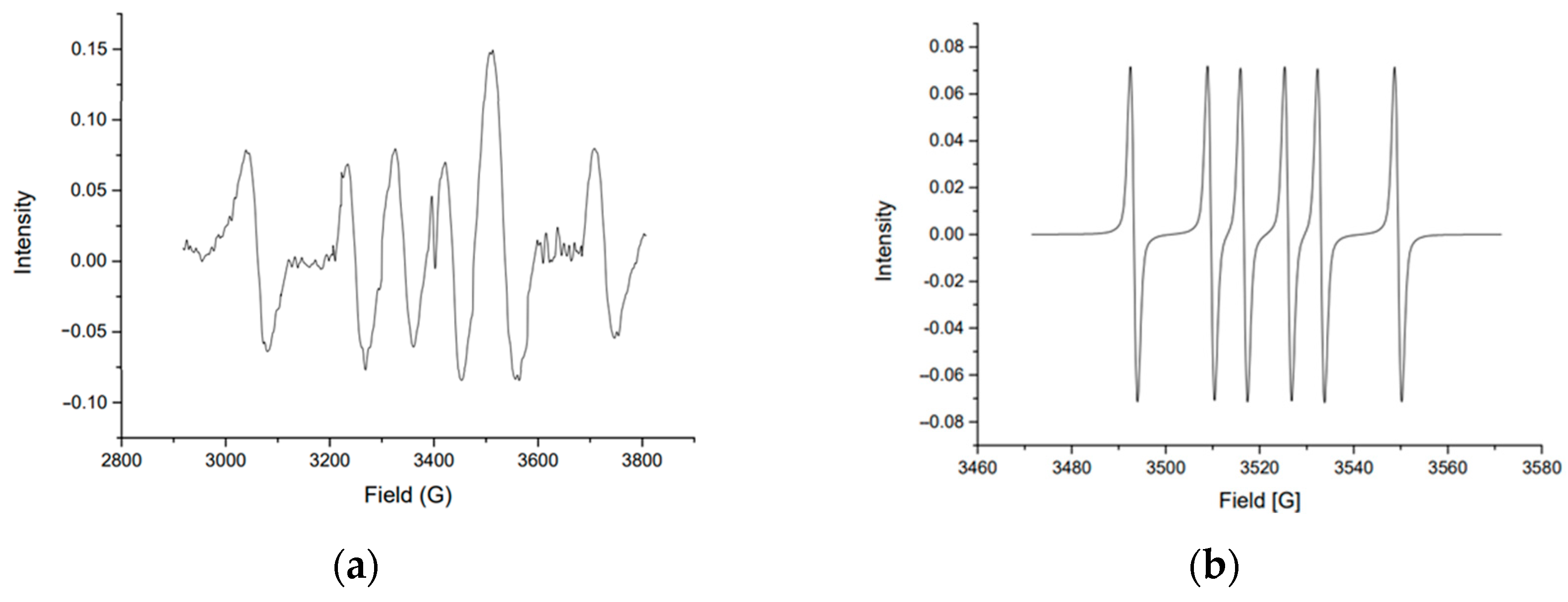
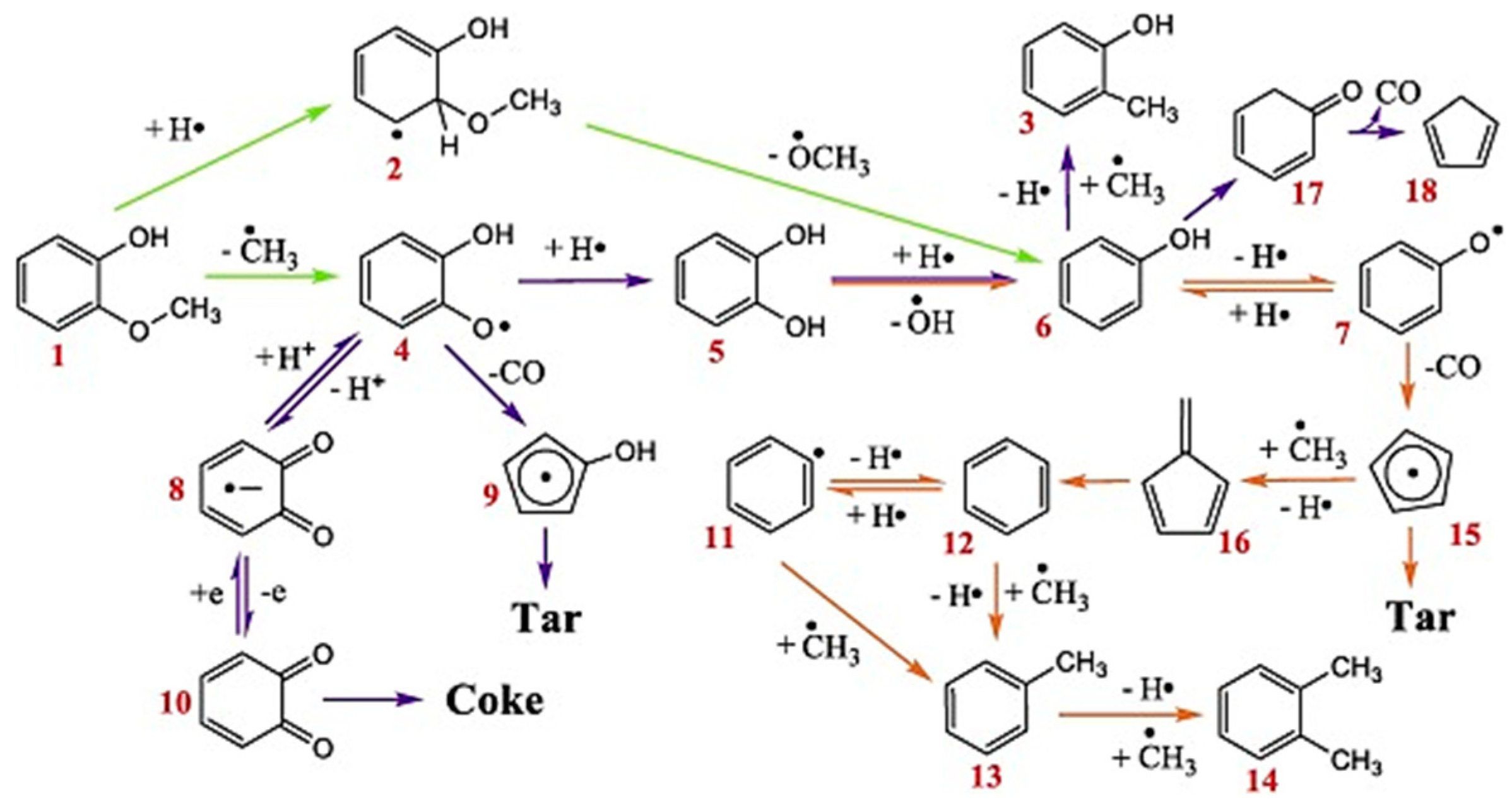
3.1.1. Guaiacol
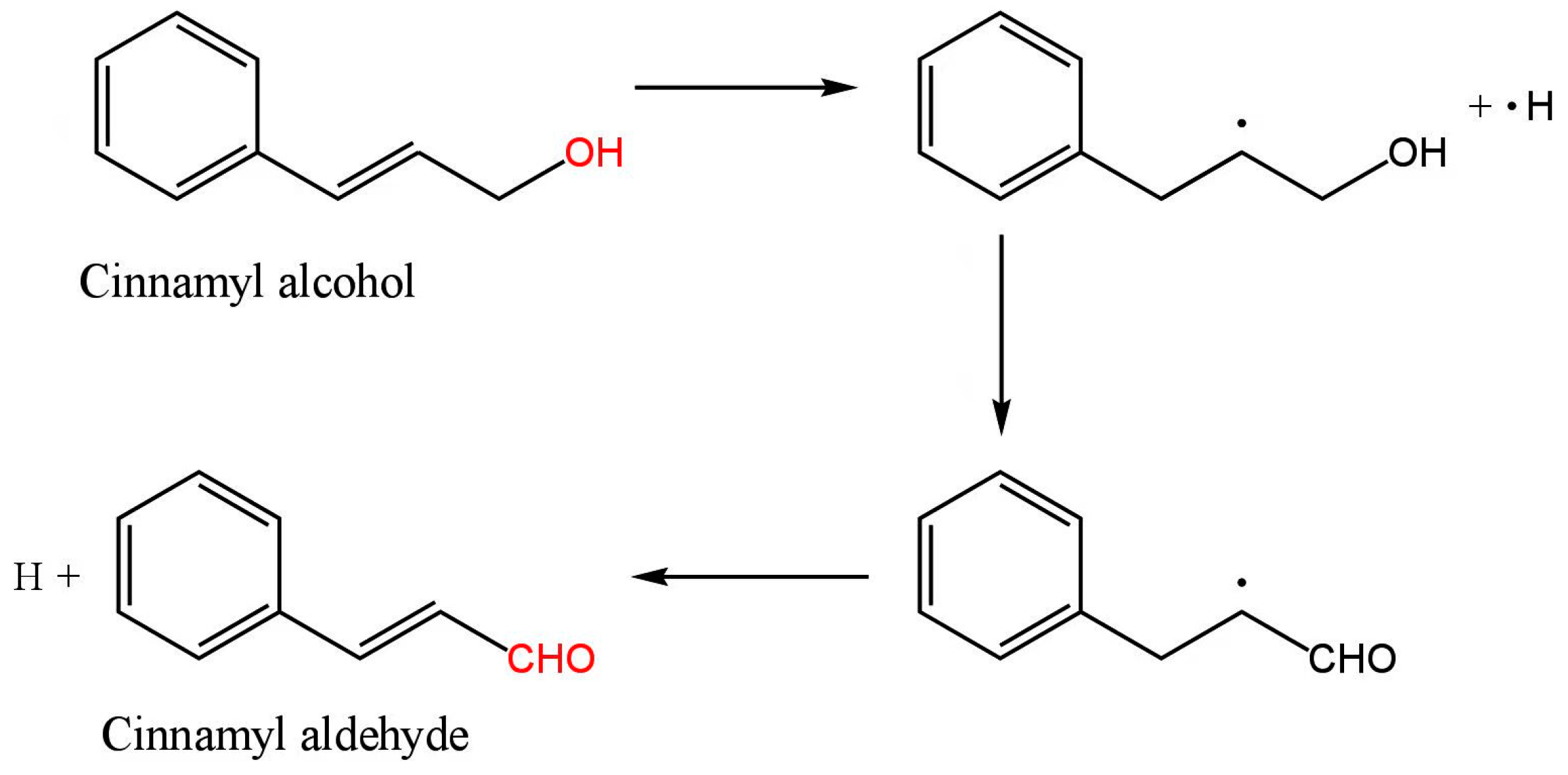
3.1.2. Cinnamyl Alcohol
3.1.3. p-Coumaryl Alcohol
3.1.4. Coniferyl Alcohol
3.2. Dimeric Model Compound
3.2.1. Dimeric Model Compound Containing α-O-4 Bond
3.2.2. Dimeric Model Compound Containing β-O-4 Bond
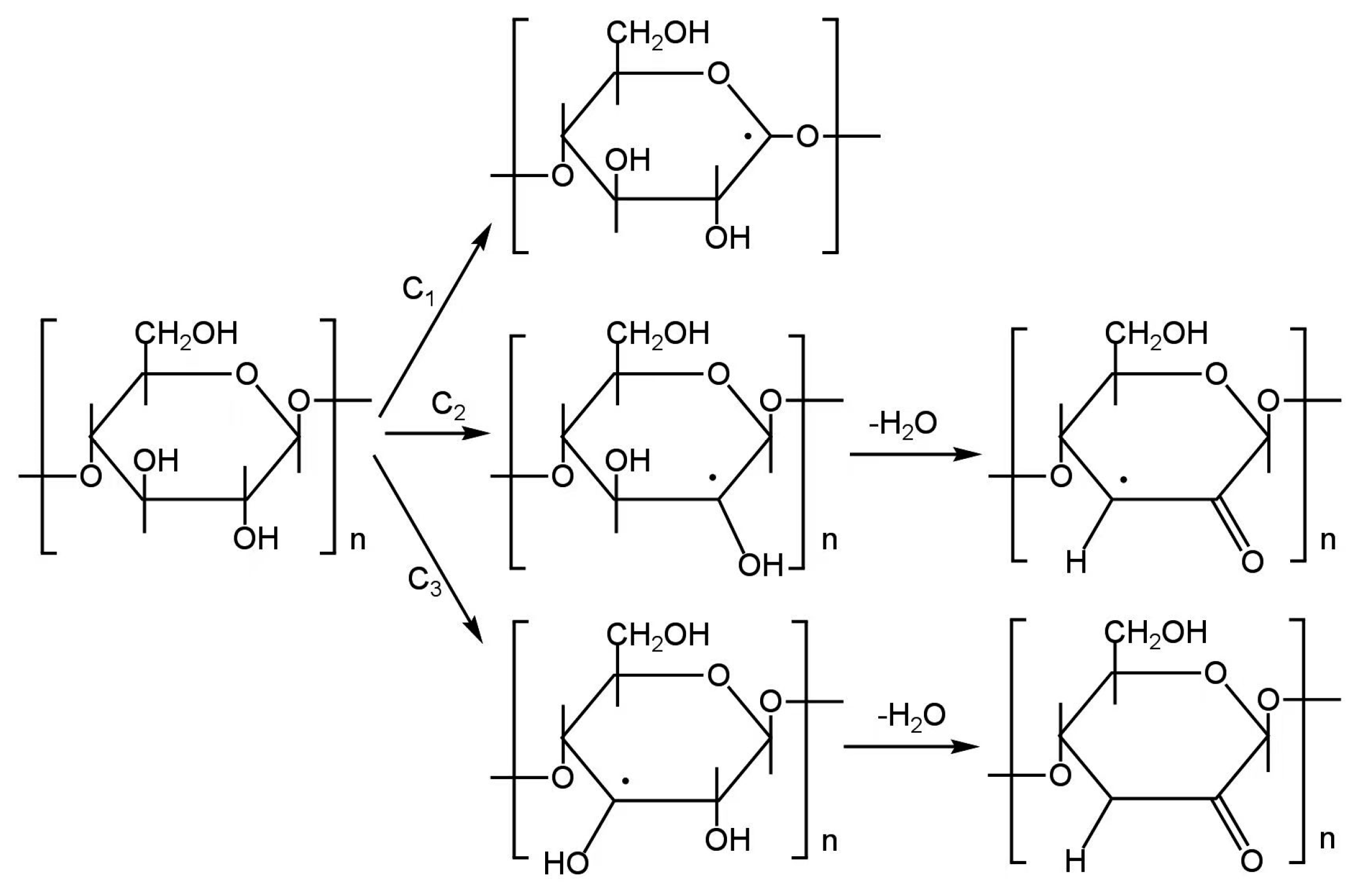
3.3. Cellulose
3.3.1. Cellulose In Situ EPR Pyrolysis
3.3.2. Radicals in Cellulose Pyrolysis Products
3.4. Hemicellulose
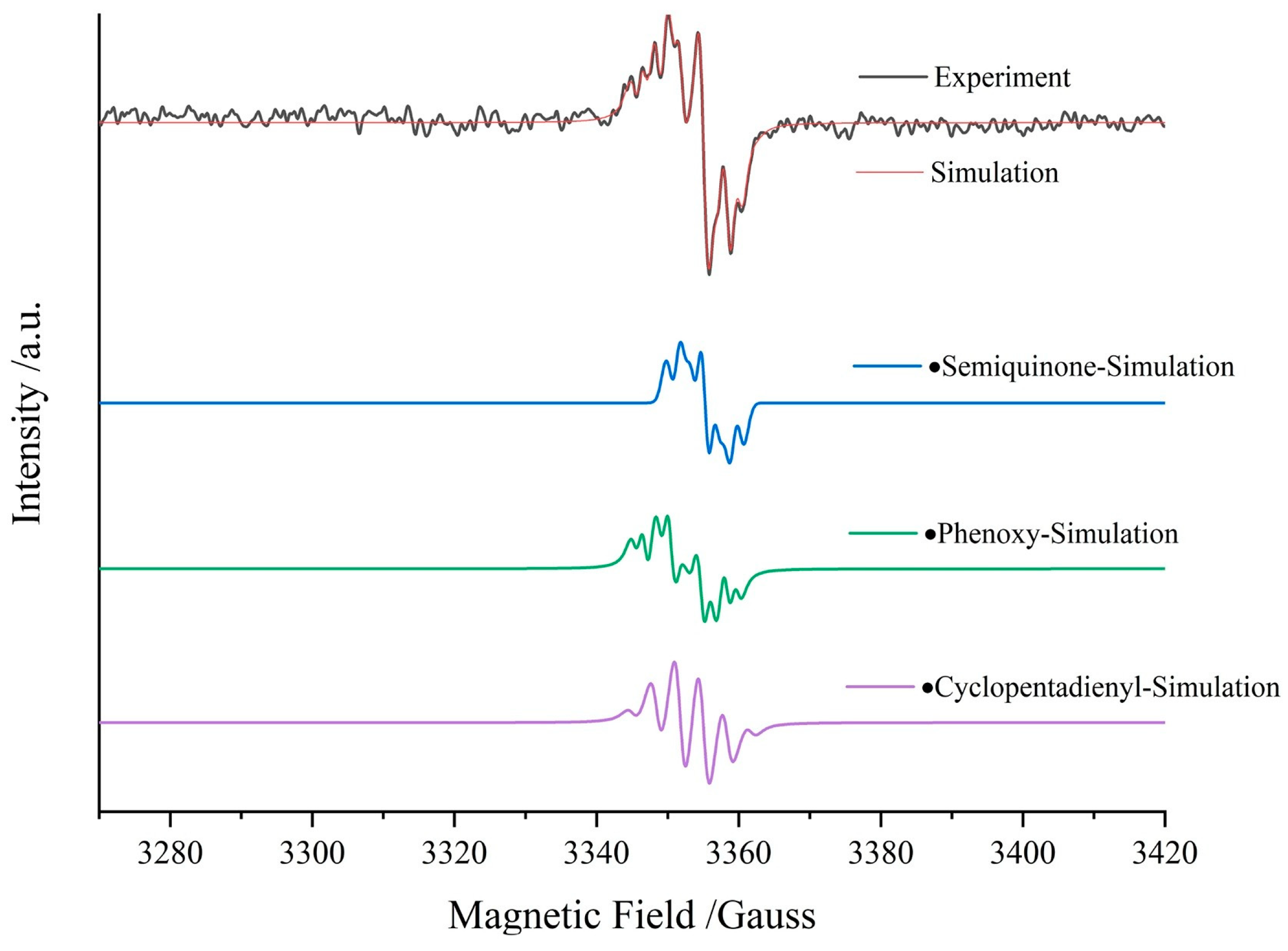
3.5. Lignin
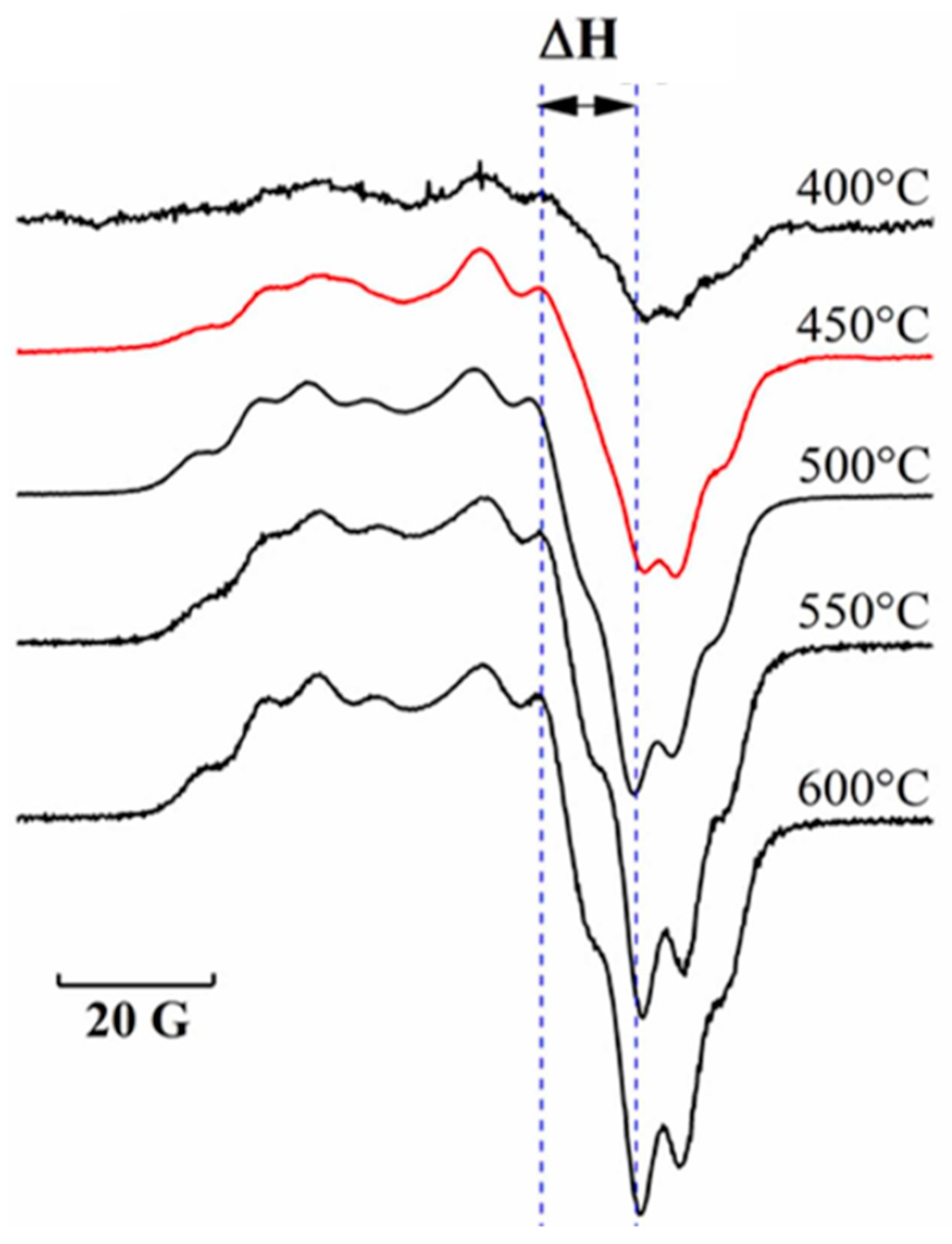
3.5.1. Lignin In Situ EPR Pyrolysis
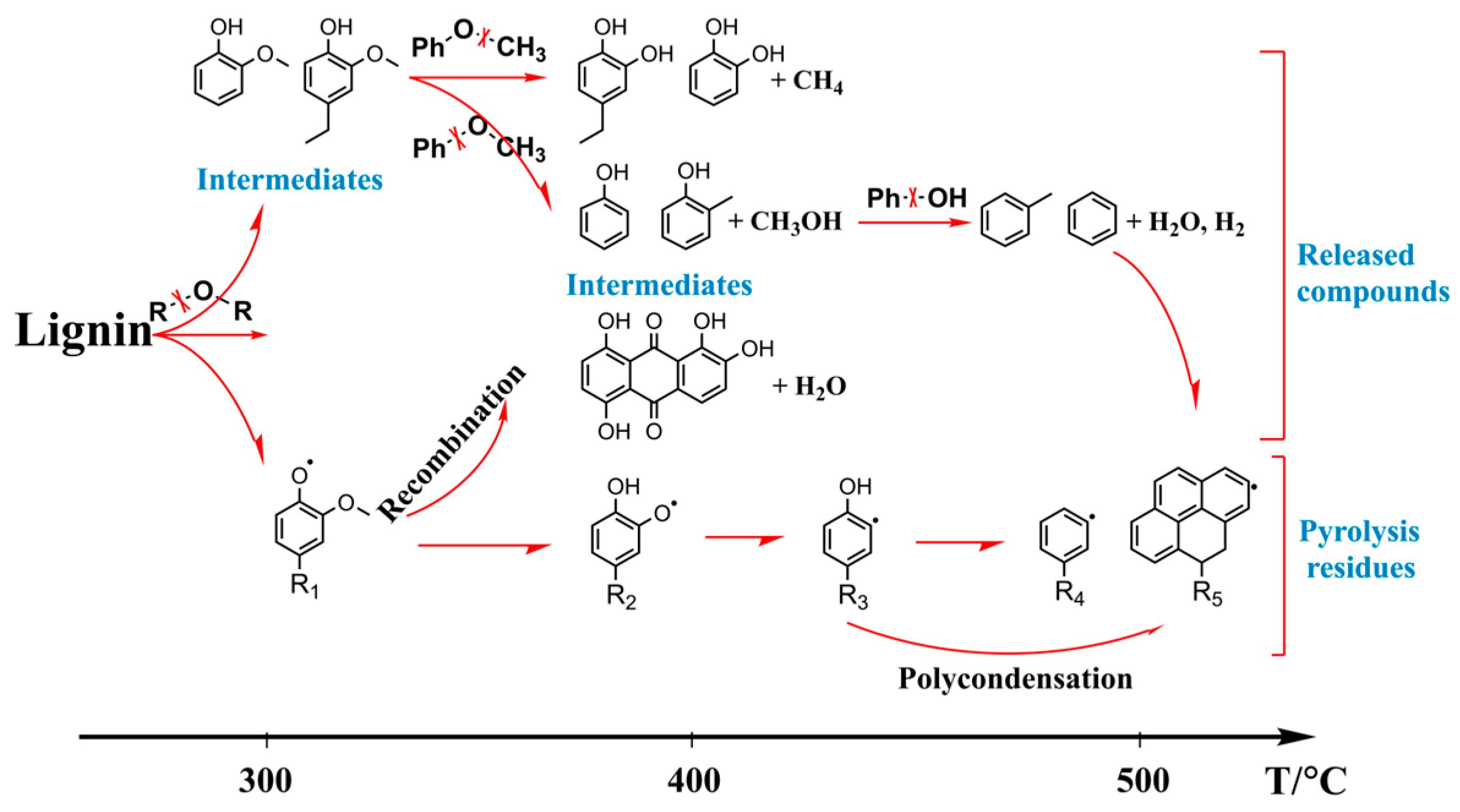
3.5.2. Radicals in Lignin Pyrolysis Products
4. EPR for Lignocellulosic Biomass Pyrolysis
4.1. Radical Behavior of Three Components
4.2. Radicals in Lignocellulosic Biomass Pyrolysis Products
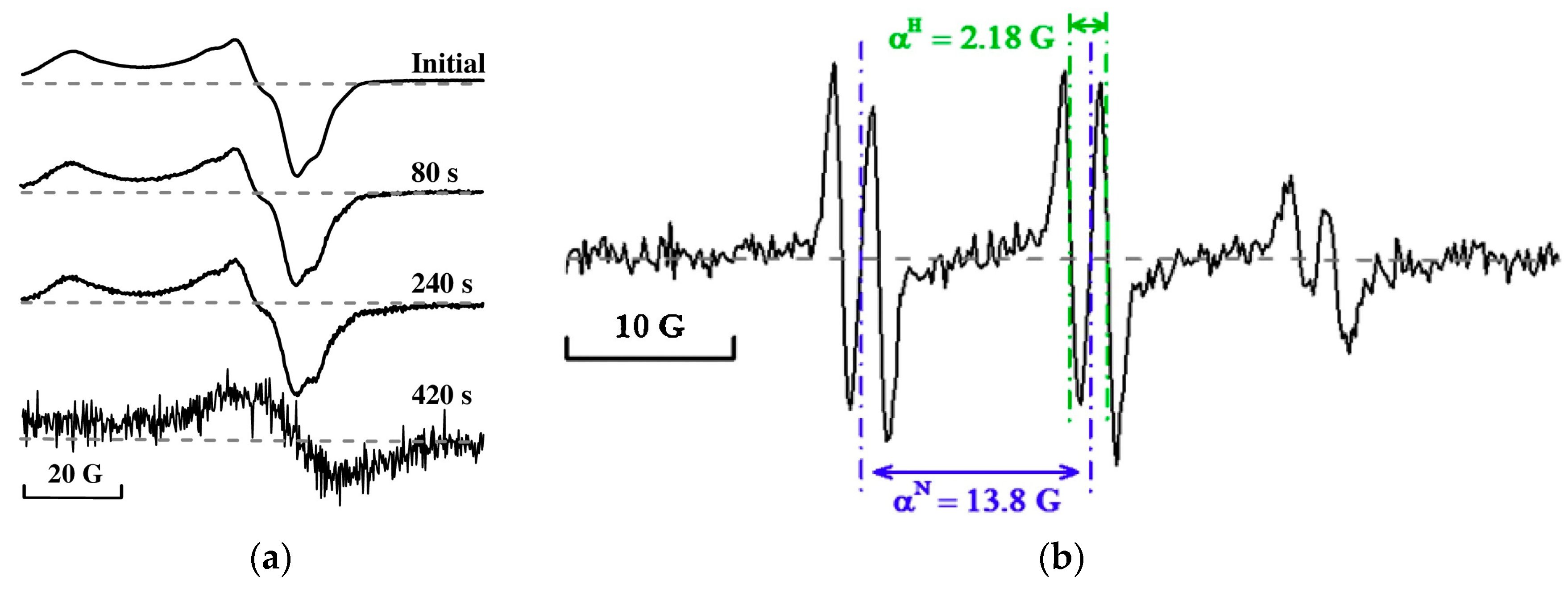
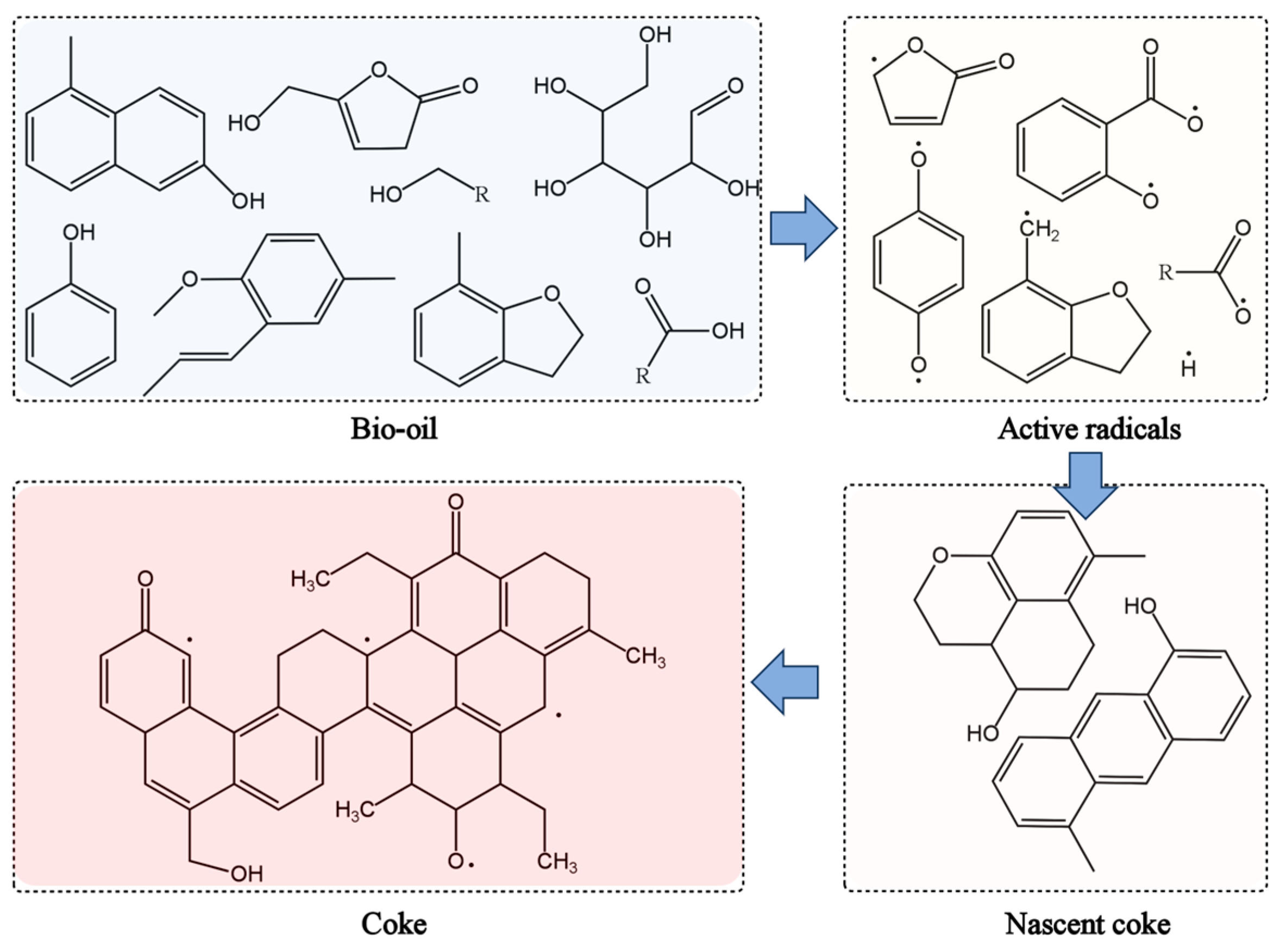
4.2.1. Radicals in Bio-Oil
4.2.2. Radicals in Biochar
4.2.3. Influencing Factors of EPFRs
5. EPR for Pretreatment of Biomass Pyrolysis
5.1. Effect of Lignin Extraction Method on Pyrolysis
5.2. Effect of Pre-Oxidation Method on Pyrolysis
5.3. Effect of Torrefaction Method on Pyrolysis
5.4. Effect of Plasma Method on Pyrolysis
6. Current Limitations and Future Prospects
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| CFA | Coniferyl alcohol |
| CNA | Cinnamyl alcohol |
| CPD | Cyclopentadienyl radical |
| DFT | Density functional theory |
| DHP | 9,10-Dihydrophenanthrene |
| DMPO | 5,5-Dimethyl-1-pyrroline-N-oxide |
| EPFRs | Environmental persistent free radicals |
| EPR | Electron paramagnetic resonance |
| h | Plank constant |
| LTMI | Low-temperature matrix isolation |
| MNP | 2-Methyl-2-nitrosppropane dimer |
| OVs | Oxygen vacancies |
| PBN | N-tert-butyl-α-phenylnitrone |
| PFT | Pyrolysis-frozen trapping |
| PPE | 2-Phenylethyl phenyl ether |
| PST | Pyrolysis-spin trapping |
| ge | g-factor of electron |
| μB | Bohr magneton |
| ∆H | Linewidth |
| αN | Hyperfine interaction of N atom |
| αH | Hyperfine interaction of H atom |
References
- Wang, S.; Dai, G.; Yang, H.; Luo, Z. Lignocellulosic Biomass Pyrolysis Mechanism: A State-of-the-Art Review. Prog. Energy Combust. Sci. 2017, 62, 33–86. [Google Scholar] [CrossRef]
- Bridgwater, A.V.; Peacocke, G.V.C. Fast Pyrolysis Processes for Biomass. Renew. Sustain. Energy Rev. 2000, 4, 1–73. [Google Scholar] [CrossRef]
- Bai, X.; Kim, K.H.; Brown, R.C.; Dalluge, E.; Hutchinson, C.; Lee, Y.J.; Dalluge, D. Formation of Phenolic Oligomers during Fast Pyrolysis of Lignin. Fuel 2014, 128, 170–179. [Google Scholar] [CrossRef]
- Hameed, S.; Sharma, A.; Pareek, V.; Wu, H.; Yu, Y. A Review on Biomass Pyrolysis Models: Kinetic, Network and Mechanistic Models. Biomass Bioenergy 2019, 123, 104–122. [Google Scholar] [CrossRef]
- Custodis, V.B.F.; Hemberger, P.; Ma, Z.; Van Bokhoven, J.A. Mechanism of Fast Pyrolysis of Lignin: Studying Model Compounds. J. Phys. Chem. B 2014, 118, 8524–8531. [Google Scholar] [CrossRef]
- Liu, C.; Deng, Y.; Wu, S.; Mou, H.; Liang, J.; Lei, M. Study on the Pyrolysis Mechanism of Three Guaiacyl-Type Lignin Monomeric Model Compounds. J. Anal. Appl. Pyrolysis 2016, 118, 123–129. [Google Scholar] [CrossRef]
- Lei, M.; Wu, S.; Liu, C.; Liang, J.; Xiao, R. Revealing the Pyrolysis Behavior of 5-5′ Biphenyl-Type Lignin Fragment. Part I: A Mechanistic Study on Fragmentation via Experiments and Theoretical Calculation. Fuel Process. Technol. 2021, 217, 106812. [Google Scholar] [CrossRef]
- Kan, T.; Strezov, V.; Evans, T.J. Lignocellulosic Biomass Pyrolysis: A Review of Product Properties and Effects of Pyrolysis Parameters. Renew. Sustain. Energy Rev. 2016, 57, 1126–1140. [Google Scholar] [CrossRef]
- Cai, W.; Luo, Z.; Zhou, J.; Wang, Q. A Review on the Selection of Raw Materials and Reactors for Biomass Fast Pyrolysis in China. Fuel Process. Technol. 2021, 221, 106919. [Google Scholar] [CrossRef]
- Várhegyi, G.; Antal, M.J.; Jakab, E.; Szabó, P. Kinetic Modeling of Biomass Pyrolysis. J. Anal. Appl. Pyrolysis 1997, 42, 73–87. [Google Scholar] [CrossRef]
- Koufopanos, C.A.; Lucchesi, A.; Maschio, G. Kinetic Modelling of the Pyrolysis of Biomass and Biomass Components. Can. J. Chem. Eng. 1989, 67, 75–84. [Google Scholar] [CrossRef]
- Vinu, R.; Broadbelt, L.J. Unraveling Reaction Pathways and Specifying Reaction Kinetics for Complex Systems. Annu. Rev. Chem. Biomol. Eng. 2012, 3, 29–54. [Google Scholar] [CrossRef]
- Du, L.; Luo, Z.; Wang, K.; Miao, F.; Qian, Q. Catalytic Co-Conversion of Poplar Pyrolysis Vapor and Methanol for Aromatics Production via Ex-Situ Configuration. J. Anal. Appl. Pyrolysis 2022, 165, 105571. [Google Scholar] [CrossRef]
- Gea, S.; Hutapea, Y.A.; Piliang, A.F.R.; Pulungan, A.N.; Rahayu, R.; Layla, J.; Tikoalu, A.D.; Wijaya, K.; Saputri, W.D. A Comprehensive Review of Experimental Parameters in Bio-Oil Upgrading from Pyrolysis of Biomass to Biofuel Through Catalytic Hydrodeoxygenation. Bioenerg. Res. 2023, 16, 325–347. [Google Scholar] [CrossRef]
- Zhu, J.; Yang, H.; Hu, H.; Zhou, Y.; Li, J.; Jin, L. Novel Insight into Pyrolysis Behaviors of Lignin Using In-Situ Pyrolysis-Double Ionization Time-of-Flight Mass Spectrometry Combined with Electron Paramagnetic Resonance Spectroscopy. Bioresour. Technol. 2020, 312, 123555. [Google Scholar] [CrossRef]
- Fan, Y.; Lei, M.; Zhang, Z.; Kong, X.; Xu, W.; Han, Y.; Li, M.; Liu, C.; Xiao, R. Unmasking Radical-Mediated Lignin Pyrolysis after Benzyl Hydroxyl Shielding. Bioresour. Technol. 2021, 342, 125944. [Google Scholar] [CrossRef]
- Lei, M.; Wu, S.; Liang, J.; Liu, C. Comprehensive Understanding the Chemical Structure Evolution and Crucial Intermediate Radical in Situ Observation in Enzymatic Hydrolysis/Mild Acidolysis Lignin Pyrolysis. J. Anal. Appl. Pyrolysis 2019, 138, 249–260. [Google Scholar] [CrossRef]
- Truong, H.; Lomnicki, S.; Dellinger, B. Mechanisms of Molecular Product and Persistent Radical Formation from the Pyrolysis of Hydroquinone. Chemosphere 2008, 71, 107–113. [Google Scholar] [CrossRef]
- Kim, K.H.; Bai, X.; Cady, S.; Gable, P.; Brown, R.C. Quantitative Investigation of Free Radicals in Bio-Oil and Their Potential Role in Condensed-Phase Polymerization. ChemSusChem 2015, 8, 894–900. [Google Scholar] [CrossRef]
- He, W.; Liu, Q.; Shi, L.; Liu, Z.; Ci, D.; Lievens, C.; Guo, X.; Liu, M. Understanding the Stability of Pyrolysis Tars from Biomass in a View Point of Free Radicals. Bioresour. Technol. 2014, 156, 372–375. [Google Scholar] [CrossRef]
- Trubetskaya, A.; Larsen Andersen, M.; Talbro Barsberg, S. The Nature of Stable Char Radicals: An ESR and DFT Study of Structural and Hydrogen Bonding Requirements. ChemPlusChem 2018, 83, 780–786. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Feng, J. The Reactivity of Free Radicals in Biomass Char Studied by EPR Spectroscopy. Mini-Rev. Org. Chem. 2011, 8, 438–447. [Google Scholar] [CrossRef]
- Van Der Est, A. Transient EPR: Using Spin Polarization in Sequential Radical Pairs to Study Electron Transfer in Photosynthesis. Photosynth. Res》 2009, 102, 335–347. [Google Scholar] [CrossRef] [PubMed]
- Bährle, C.; Custodis, V.; Jeschke, G.; van Bokhoven, J.A.; Vogel, F. In Situ Observation of Radicals and Molecular Products During Lignin Pyrolysis. ChemSusChem 2014, 7, 2022–2029. [Google Scholar] [CrossRef]
- Ma, L.; Syed-Hassan, S.S.A.; Xiong, Z.; Chen, Y.; Xu, J.; Jiang, L.; Su, S.; Hu, S.; Wang, Y.; Xiang, J. Evolution of Stable Free Radicals during Bio-Oil Pyrolysis and Its Relation to Coke Formation: An in Situ EPR Study. Energy Fuels 2022, 36, 7608–7616. [Google Scholar] [CrossRef]
- Wang, Z.; Ma, W.; Chen, C.; Ji, H.; Zhao, J. Probing Paramagnetic Species in Titania-Based Heterogeneous Photocatalysis by Electron Spin Resonance (ESR) Spectroscopy—A Mini Review. Chem. Eng. J. 2011, 170, 353–362. [Google Scholar] [CrossRef]
- Li, G.; Luo, Z.; Wang, W.; Cen, J. A Study of the Mechanisms of Guaiacol Pyrolysis Based on Free Radicals Detection Technology. Catalysts 2020, 10, 295. [Google Scholar] [CrossRef]
- Mile, B. Free Radical Studies at Low Temperatures. Angew. Chem. Int. Ed. Engl. 1968, 7, 507–519. [Google Scholar] [CrossRef]
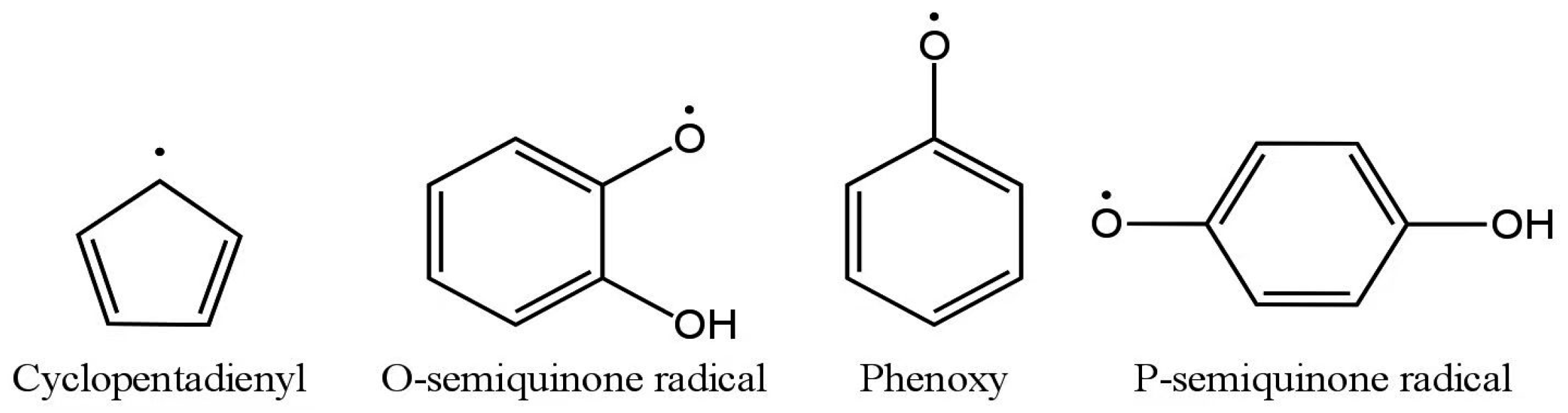
- Khachatryan, L.; Adounkpe, J.; Maskos, Z.; Dellinger, B. Formation of Cyclopentadienyl Radical from the Gas-Phase Pyrolysis of Hydroquinone, Catechol, and Phenol. Environ. Sci. Technol. 2006, 40, 5071–5076. [Google Scholar] [CrossRef]
- Khachatryan, L.; Adounkpe, J.; Dellinger, B. Formation of Phenoxy and Cyclopentadienyl Radicals from the Gas-Phase Pyrolysis of Phenol. J. Phys. Chem. A 2008, 112, 481–487. [Google Scholar] [CrossRef]
- Davies, M.J. Detection and Characterisation of Radicals Using Electron Paramagnetic Resonance (EPR) Spin Trapping and Related Methods. Methods 2016, 109, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Modic, K.A.; Smidt, T.E.; Kimchi, I.; Breznay, N.P.; Biffin, A.; Choi, S.; Johnson, R.D.; Coldea, R.; Watkins-Curry, P.; McCandless, G.T.; et al. Realization of a Three-Dimensional Spin–Anisotropic Harmonic Honeycomb Iridate. Nat. Commun. 2014, 5, 4203. [Google Scholar] [CrossRef] [PubMed]
- Dack, S.W.; Hobday, M.D.; Smith, T.D.; Pilbrow, J.R.E.p.r. Study of Organic Free Radicals in Victorian Brown Coal. Fuel 1985, 64, 219–221. [Google Scholar] [CrossRef]
- Lancaster, G. Electron Paramagnetic Resonance (a Review). J. Mater. Sci. 1967, 2, 489–495. [Google Scholar] [CrossRef]
- Pilawa, B.; Więckowski, A.B.; Pietrzak, R.; Wachowska, H. Oxidation of Demineralized Coal and Coal Free of Pyrite Examined by EPR Spectroscopy. Fuel 2002, 81, 1925–1931. [Google Scholar] [CrossRef]
- Liu, J.; Jiang, X.; Shen, J.; Zhang, H. Chemical Properties of Superfine Pulverized Coal Particles. Part 1. Electron Paramagnetic Resonance Analysis of Free Radical Characteristics. Adv. Powder Technol. 2014, 25, 916–925. [Google Scholar] [CrossRef]
- Adounkpe, J.; Khachatryan, L.; Dellinger, B. Radicals from the Gas-Phase Pyrolysis of Hydroquinone: 1. Temperature Dependence of the Total Radical Yield. Energy Fuels 2008, 22, 2986–2990. [Google Scholar] [CrossRef]
- Khachatryan, L.; Adounkpe, J.; Asatryan, R.; Dellinger, B. Radicals from the Gas-Phase Pyrolysis of Catechol: 1. o -Semiquinone and Ipso -Catechol Radicals. J. Phys. Chem. A 2010, 114, 2306–2312. [Google Scholar] [CrossRef]
- Liu, C.; Ye, L.; Yuan, W.; Zhang, Y.; Zou, J.; Yang, J.; Wang, Y.; Qi, F.; Zhou, Z. Investigation on Pyrolysis Mechanism of Guaiacol as Lignin Model Compound at Atmospheric Pressure. Fuel 2018, 232, 632–638. [Google Scholar] [CrossRef]
- Nowakowska, M.; Herbinet, O.; Dufour, A.; Glaude, P.A. Kinetic Study of the Pyrolysis and Oxidation of Guaiacol. J. Phys. Chem. A 2018, 122, 7894–7909. [Google Scholar] [CrossRef]
- Furutani, Y.; Dohara, Y.; Kudo, S.; Hayashi, J.; Norinaga, K. Theoretical Study on the Kinetics of Thermal Decomposition of Guaiacol and Catechol. J. Phys. Chem. A 2017, 121, 8495–8503. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Luo, Z.; Li, G.; Li, S. EPR Detection of Key Radicals during Coking Process of Lignin Monomer Pyrolysis. J. Anal. Appl. Pyrolysis 2020, 152, 104948. [Google Scholar] [CrossRef]
- Asatryan, R.; Hudzik, J.M.; Bozzelli, J.W.; Khachatryan, L.; Ruckenstein, E. OH-Initiated Reactions of p- Coumaryl Alcohol Relevant to the Lignin Pyrolysis. Part I. Potential Energy Surface Analysis. J. Phys. Chem. A 2019, 123, 2570–2585. [Google Scholar] [CrossRef]
- Khachatryan, L.; Xu, M.; Wu, A.; Pechagin, M.; Asatryan, R. Radicals and Molecular Products from the Gas-Phase Pyrolysis of Lignin Model Compounds. Cinnamyl Alcohol. J. Anal. Appl. Pyrolysis 2016, 121, 75–83. [Google Scholar] [CrossRef]
- Xu, M.; Khachatryan; Baev, A.; Asatryan, R. Radicals from the Gas-Phase Pyrolysis of a Lignin Model Compound: P-Coumaryl Alcohol. RSC Adv. 2016, 6, 62399–62405. [Google Scholar] [CrossRef]
- Kawamoto, H. Lignin Pyrolysis Reactions. J. Wood Sci. 2017, 63, 117–132. [Google Scholar] [CrossRef]
- Barekati-Goudarzi, M.; Khachatryan, L.; Boldor, D.; Xu, M.; Ruckenstein, E.; Asatryan, R. Radicals and Molecular Products from the Gas-Phase Pyrolysis of Lignin Model Compounds: Coniferyl Alcohol, Theory and Experiment. J. Anal. Appl. Pyrolysis 2022, 161, 105413. [Google Scholar] [CrossRef]
- Pu, Y.; Zhang, D.; Singh, P.M.; Ragauskas, A.J. The New Forestry Biofuels Sector. Biofuels Bioprod. Biorefining 2008, 2, 58–73. [Google Scholar] [CrossRef]
- Britt, P.F.; Buchanan, A.C.; Cooney, M.J.; Martineau, D.R. Flash Vacuum Pyrolysis of Methoxy-Substituted Lignin Model Compounds. J. Org. Chem. 2000, 65, 1376–1389. [Google Scholar] [CrossRef]
- Britt, P.F.; Buchanan, A.C.I.; Malcolm, E.A. Thermolysis of Phenethyl Phenyl Ether: A Model for Ether Linkages in Lignin and Low Rank Coal. J. Org. Chem. 1995, 60, 6523–6536. [Google Scholar] [CrossRef]
- He, T.; Zhang, Y.; Zhu, Y.; Wen, W.; Pan, Y.; Wu, J.; Wu, J. Pyrolysis Mechanism Study of Lignin Model Compounds by Synchrotron Vacuum Ultraviolet Photoionization Mass Spectrometry. Energy Fuels 2016, 30, 2204–2208. [Google Scholar] [CrossRef]
- Jarvis, M.W.; Daily, J.W.; Carstensen, H.-H.; Dean, A.M.; Sharma, S.; Dayton, D.C.; Robichaud, D.J.; Nimlos, M.R. Direct Detection of Products from the Pyrolysis of 2-Phenethyl Phenyl Ether. J. Phys. Chem. A 2011, 115, 428–438. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Bai, X.; Brown, R.C. Pyrolysis Mechanisms of Methoxy Substituted α-O-4 Lignin Dimeric Model Compounds and Detection of Free Radicals Using Electron Paramagnetic Resonance Analysis. J. Anal. Appl. Pyrolysis 2014, 110, 254–263. [Google Scholar] [CrossRef]
- Ragauskas, A.J.; Williams, C.K.; Davison, B.H.; Britovsek, G.; Cairney, J.; Eckert, C.A.; Frederick, W.J.; Hallett, J.P.; Leak, D.J.; Liotta, C.L.; et al. The Path Forward for Biofuels and Biomaterials. Science 2006, 311, 484–489. [Google Scholar] [CrossRef]
- Wang, S.; Guo, X.; Liang, T.; Zhou, Y.; Luo, Z. Mechanism Research on Cellulose Pyrolysis by Py-GC/MS and Subsequent Density Functional Theory Studies. Bioresour. Technol. 2012, 104, 722–728. [Google Scholar] [CrossRef]
- Xiao, Y.; Yan, Y.; Do, H.; Rankin, R.; Zhao, H.; Qian, P.; Song, K.; Wu, T.; Pang, C.H. Understanding Cellulose Pyrolysis via Ab Initio Deep Learning Potential Field. Bioresour. Technol. 2024, 399, 130590. [Google Scholar] [CrossRef]
- Ma, L.; Syed-Hassan, S.S.A.; Tong, Y.; Xiong, Z.; Chen, Y.; Xu, J.; Jiang, L.; Su, S.; Hu, S.; Wang, Y.; et al. Interactions of Cellulose- and Lignin-Derived Radicals during Pyrolysis: An in-Situ Electron Paramagnetic Resonance (EPR) Study. Fuel Process. Technol. 2023, 239, 107536. [Google Scholar] [CrossRef]
- Tao, W.; Zhang, P.; Li, H.; Yang, Q.; Oleszczuk, P.; Pan, B. Generation Mechanism of Persistent Free Radicals in Lignocellulose-Derived Biochar: Roles of Reducible Carbonyls. Environ. Sci. Technol. 2022, 56, 10638–10645. [Google Scholar] [CrossRef]
- Liang, J.; Chen, J.; Wu, S.; Liu, C.; Lei, M. Comprehensive Insights into Cellulose Structure Evolution via Multi-Perspective Analysis during a Slow Pyrolysis Process. Sustain. Energy Fuels 2018, 2, 1855–1862. [Google Scholar] [CrossRef]
- Bi, D.; Yin, M.; Huang, F.; Zhang, J.; Yi, W. Evolution and Prediction Model of Environmentally-Persistent Free Radicals in Biomass Three-Component Pyrolytic Carbon with Pyrolysis Temperature. Ind. Crops Prod. 2023, 206, 117643. [Google Scholar] [CrossRef]
- Wang, S.; Ru, B.; Lin, H.; Luo, Z. Degradation Mechanism of Monosaccharides and Xylan under Pyrolytic Conditions with Theoretic Modeling on the Energy Profiles. Bioresour. Technol. 2013, 143, 378–383. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Li, W.; Mabon, R.; Broadbelt, L.J. A Mechanistic Model of Fast Pyrolysis of Hemicellulose. Energy Environ. Sci. 2018, 11, 1240–1260. [Google Scholar] [CrossRef]
- Liang, J.; Chen, J.; Wu, S.; Liu, C.; Lei, M. Comprehensive Insights into Xylan Structure Evolution via Multi-Perspective Analysis during Slow Pyrolysis Process. Fuel Process. Technol. 2019, 186, 1–7. [Google Scholar] [CrossRef]
- Song, G.; Huang, D.; Ren, Q.; Hu, S.; Xu, J.; Xu, K.; Jiang, L.; Wang, Y.; Su, S.; Xiang, J. Inner-Particle Reaction Mechanism of Cellulose, Hemicellulose and Lignin during Photo-Thermal Pyrolysis Process: Evolution Characteristics of Free Radicals. Energy 2024, 297, 131201. [Google Scholar] [CrossRef]
- Moreno, B.M.; Quach, A.L.; Merves, M.N.; Klein, M.T. Discrimination between Free-Radical and Concerted Pyrolysis Mechanisms. Energy Fuels 2014, 28, 4256–4259. [Google Scholar] [CrossRef]
- Hu, J.; Shen, D.; Xiao, R.; Wu, S.; Zhang, H. Free-Radical Analysis on Thermochemical Transformation of Lignin to Phenolic Compounds. Energy Fuels 2013, 27, 285–293. [Google Scholar] [CrossRef]
- Kibet, J.; Khachatryan, L.; Dellinger, B. Molecular Products and Radicals from Pyrolysis of Lignin. Environ. Sci. Technol. 2012, 46, 12994–13001. [Google Scholar] [CrossRef]
- Zhao, J.; Xiuwen, W.; Hu, J.; Liu, Q.; Shen, D.; Xiao, R. Thermal Degradation of Softwood Lignin and Hardwood Lignin by TG-FTIR and Py-GC/MS. Polym. Degrad. Stab. 2014, 108, 133–138. [Google Scholar] [CrossRef]
- Fan, Y.; Zhang, Z.; Wang, Z.; Yu, H.; Kong, X.; Li, P.; Li, M.; Xiao, R.; Liu, C. Radical Footprinting and Regularity Revealing during the Pyrolysis of Technical Lignins. Bioresour. Technol. 2022, 360, 127648. [Google Scholar] [CrossRef]
- Tao, W.; Yang, X.; Li, Y.; Zhu, R.; Si, X.; Pan, B.; Xing, B. Components and Persistent Free Radicals in the Volatiles during Pyrolysis of Lignocellulose Biomass. Environ. Sci. Technol. 2020, 54, 13274–13281. [Google Scholar] [CrossRef]
- Patil, S.V.; Argyropoulos, D.S. Stable Organic Radicals in Lignin: A Review. ChemSusChem 2017, 10, 3284–3303. [Google Scholar] [CrossRef] [PubMed]
- Collard, F.-X.; Blin, J. A Review on Pyrolysis of Biomass Constituents: Mechanisms and Composition of the Products Obtained from the Conversion of Cellulose, Hemicelluloses and Lignin. Renew. Sustain. Energy Rev. 2014, 38, 594–608. [Google Scholar] [CrossRef]
- Zhu, J.; Xu, J.; Hu, H.; Wang, X.; Zhou, Y.; Jin, L. Novel Detection of Primary and Secondary Volatiles from Cedar Pyrolysis Using In-Situ Pyrolysis Double Ionization Time-of-Flight Mass Spectrometry. Chem. Eng. Sci. 2021, 236, 116545. [Google Scholar] [CrossRef]
- Ma, L.; Syed-Hassan, S.S.A.; Zhou, J.; Deng, W.; Xiong, Y.; Wang, X.; Hu, X.; Xu, J.; Jiang, L.; Su, S.; et al. Effect of Alkali and Alkali Earth Metals on Reactions of Stable Free Radicals during Biomass Pyrolysis: An in-Situ EPR Study. Fuel Process. Technol. 2023, 250, 107916. [Google Scholar] [CrossRef]
- Sharma, A.; Pareek, V.; Zhang, D. Biomass Pyrolysis—A Review of Modelling, Process Parameters and Catalytic Studies. Renew. Sustain. Energy Rev. 2015, 50, 1081–1096. [Google Scholar] [CrossRef]
- Meng, J.; Smirnova, T.I.; Song, X.; Moore, A.; Ren, X.; Kelley, S.; Park, S.; Tilotta, D. Identification of Free Radicals in Pyrolysis Oil and Their Impact on Bio-Oil Stability. RSC Adv. 2014, 4, 29840–29846. [Google Scholar] [CrossRef]
- No, S.-Y. Application of Bio-Oils from Lignocellulosic Biomass to Transportation, Heat and Power Generation—A Review. Renew. Sustain. Energy Rev. 2014, 40, 1108–1125. [Google Scholar] [CrossRef]
- Xiong, Z.; Syed-Hassan, S.S.A.; Xu, J.; Wang, Y.; Hu, S.; Su, S.; Zhang, S.; Xiang, J. Evolution of Coke Structures during the Pyrolysis of Bio-Oil at Various Temperatures and Heating Rates. J. Anal. Appl. Pyrolysis 2018, 134, 336–342. [Google Scholar] [CrossRef]
- Zhu, C.; Guo, M.; Zhu, X.; Chen, J.; Su, J.-H. Probing the Catalytic Center of TiO2/SO4 2− Solid Superacid Catalyst by X-Band In Situ High-Temperature EPR Spectroscopy. Appl. Magn. Reson. 2012, 42, 313–320. [Google Scholar] [CrossRef]
- Ma, L.; Deng, W.; Hu, X.; Xu, K.; Xu, J.; Jiang, L.; Wang, Y.; Su, S.; Hu, S.; Xiang, J. Identifying the Coking of Bio-Oil in Pyrolysis: An in-Situ EPR Investigation. Fuel Process. Technol. 2024, 253, 108012. [Google Scholar] [CrossRef]
- Xiao, G.; Xiong, Z.; Syed-Hassan, S.S.A.; Ma, L.; Xu, J.; Jiang, L.; Su, S.; Hu, S.; Wang, Y.; Xiang, J. Coke Formation during the Pyrolysis of Bio-Oil: Further Understanding on the Evolution of Radicals. Appl. Energy Combust. Sci. 2022, 9, 100050. [Google Scholar] [CrossRef]
- Zhang, T.; Lu, Q.; Xie, X.; Zhao, Y.; Yuan, S. Study on the Catalytic Activity of Char-Based Fe–Co–K Catalyst for Volatiles Reforming during Biomass Pyrolysis. J. Energy Inst. 2022, 102, 289–301. [Google Scholar] [CrossRef]
- Avni, E.; Suib, S.L. Free Radical Formation in Lignin During Pyrolysis. Holzforschung 1985, 39, 33–40. [Google Scholar] [CrossRef]
- Curran, G.P.; Struck, R.T.; Gorin, E. Mechanism of Hydrogen-Transfer Process to Coal and Coal Extract. Ind. Eng. Chem. Proc. Des. Dev. 1967, 6, 166–173. [Google Scholar] [CrossRef]
- Zhou, Q.; Luo, Z.; Miao, F.; Wang, K.; Du, L.; Qian, Q. Study on Free Radical Mechanism and Experimental Verification in Biomass Hydropyrolysis Conversion to Liquid Fuels. J. Anal. Appl. Pyrolysis 2024, 177, 106362. [Google Scholar] [CrossRef]
- Zhang, Q.; Chang, J.; Wang, T.; Xu, Y. Review of Biomass Pyrolysis Oil Properties and Upgrading Research. Energy Convers. Manag. 2007, 48, 87–92. [Google Scholar] [CrossRef]
- Lu, Q.; Li, W.-Z.; Zhu, X.-F. Overview of Fuel Properties of Biomass Fast Pyrolysis Oils. Energy Convers. Manag. 2009, 50, 1376–1383. [Google Scholar] [CrossRef]
- Lin, J.; Sun, S.; Luo, J.; Cui, C.; Ma, R.; Fang, L.; Liu, X. Effects of Oxygen Vacancy Defect on Microwave Pyrolysis of Biomass to Produce High-Quality Syngas and Bio-Oil: Microwave Absorption and in-Situ Catalytic. Waste Manag. 2021, 128, 200–210. [Google Scholar] [CrossRef]
- Jiang, S.; Shu, R.; Wang, A.; Deng, Z.; Xiao, Y.; Li, J.; Meng, Q.; Zhang, Q. Efficient Hydrodeoxygenation of Lignin-Derived Phenolic Compounds under Acid-Free Conditions over Carbon-Supported NiMo Catalysts. Green Chem. 2024, 26, 9330–9345. [Google Scholar] [CrossRef]
- Xu, X.; Yang, H.; Tu, R.; Liu, S.; Hu, J.; Li, Y.; Sun, Y. Quenching Method to Prepare Ultra-Low Loading High-Entropy Catalyst for Furfural Selectively Hydrogenation at Ambient Temperature. Appl. Catal. B Environ. 2024, 342, 123358. [Google Scholar] [CrossRef]
- Bährle, C.; Custodis, V.; Jeschke, G.; van Bokhoven, J.A.; Vogel, F. The Influence of Zeolites on Radical Formation During Lignin Pyrolysis. ChemSusChem 2016, 9, 2397–2403. [Google Scholar] [CrossRef] [PubMed]
- Phiciato, P.; Monika, I.; Hardian, A. Heat Treatment of Pitch Obtained from Atmospheric Fixed-Bed Coal Gasification. Indones. J. Chem. 2018, 18, 560. [Google Scholar] [CrossRef]
- Liao, S.; Pan, B.; Li, H.; Zhang, D.; Xing, B. Detecting Free Radicals in Biochars and Determining Their Ability to Inhibit the Germination and Growth of Corn, Wheat and Rice Seedlings. Environ. Sci. Technol. 2014, 48, 8581–8587. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Zhang, Y.; Huang, Z.; Hu, X.; Gao, Y.; Shao, Z.; Qi, X.; Yang, Y. Influence of Temperature Change on the Change Law of Free Radicals in Coal. ACS Omega 2021, 6, 33685–33693. [Google Scholar] [CrossRef]
- Qin, L.; Yang, L.; Liu, X.; Li, C.; Lin, B.; Zheng, M.; Liu, G. Formation of Environmentally Persistent Free Radicals from Thermochemical Reactions of Catechol. Sci. Total Environ. 2021, 772, 145313. [Google Scholar] [CrossRef]
- Dellinger, B.; Lomnicki, S.; Khachatryan, L.; Maskos, Z.; Hall, R.W.; Adounkpe, J.; McFerrin, C.; Truong, H. Formation and Stabilization of Persistent Free Radicals. Proc. Combust. Inst. 2007, 31, 521–528. [Google Scholar] [CrossRef]
- Lomnicki, S.; Truong, H.; Vejerano, E.; Dellinger, B. Copper Oxide-Based Model of Persistent Free Radical Formation on Combustion-Derived Particulate Matter. Environ. Sci. Technol. 2008, 42, 4982–4988. [Google Scholar] [CrossRef]
- Dela Cruz, A.L.N.; Gehling, W.; Lomnicki, S.; Cook, R.; Dellinger, B. Detection of Environmentally Persistent Free Radicals at a Superfund Wood Treating Site. Environ. Sci. Technol. 2011, 45, 6356–6365. [Google Scholar] [CrossRef]
- Doumer, M.E.; Arízaga, G.G.C.; Silva, D.A.; Yamamoto, C.I.; Novotny, E.H.; Santos, J.M.; Santos, L.O.; Wisniewski, A.; Andrade, J.B.; Mangrich, A.S. Slow Pyrolysis of Different Brazilian Waste Biomasses as Sources of Soil Conditioners and Energy, and for Environmental Protection. J. Anal. Appl. Pyrolysis 2015, 113, 434–443. [Google Scholar] [CrossRef]
- Khachatryan, L.; Barekati-Goudarzi, M.; Asatryan, R.; Ozarowski, A.; Boldor, D.; Lomnicki, S.M.; Cormier, S.A. Metal-Free Biomass-Derived Environmentally Persistent Free Radicals (Bio-EPFRs) from Lignin Pyrolysis. ACS Omega 2022, 7, 30241–30249. [Google Scholar] [CrossRef]
- Tian, L.; Koshland, C.P.; Yano, J.; Yachandra, V.K.; Yu, I.T.S.; Lee, S.C.; Lucas, D. Carbon-Centered Free Radicals in Particulate Matter Emissions from Wood and Coal Combustion. Energy Fuels 2009, 23, 2523–2526. [Google Scholar] [CrossRef] [PubMed]
- Kelley, M.A.; Hebert, V.Y.; Thibeaux, T.M.; Orchard, M.A.; Hasan, F.; Cormier, S.A.; Thevenot, P.T.; Lomnicki, S.M.; Varner, K.J.; Dellinger, B.; et al. Model Combustion-Generated Particulate Matter Containing Persistent Free Radicals Redox Cycle to Produce Reactive Oxygen Species. Chem. Res. Toxicol. 2013, 26, 1862–1871. [Google Scholar] [CrossRef] [PubMed]
- Fang, G.; Liu, C.; Wang, Y.; Dionysiou, D.D.; Zhou, D. Photogeneration of Reactive Oxygen Species from Biochar Suspension for Diethyl Phthalate Degradation. Appl. Catal. B Environ. 2017, 214, 34–45. [Google Scholar] [CrossRef]
- Odinga, E.S.; Waigi, M.G.; Gudda, F.O.; Wang, J.; Yang, B.; Hu, X.; Li, S.; Gao, Y. Occurrence, Formation, Environmental Fate and Risks of Environmentally Persistent Free Radicals in Biochars. Environ. Int. 2020, 134, 105172. [Google Scholar] [CrossRef]
- Fang, G.; Gao, J.; Liu, C.; Dionysiou, D.D.; Wang, Y.; Zhou, D. Key Role of Persistent Free Radicals in Hydrogen Peroxide Activation by Biochar: Implications to Organic Contaminant Degradation. Environ. Sci. Technol. 2014, 48, 1902–1910. [Google Scholar] [CrossRef]
- Huang, F.; Bi, D.; Liu, S.; Yi, W.; Zhang, J.; Yao, D. Formation and Regulation of Environmentally Persistent Free Radicals in Biochar and Removal of Diethyl-Phthalate in Water. J. Anal. Appl. Pyrolysis 2022, 168, 105770. [Google Scholar] [CrossRef]
- Gao, P.; Yao, D.; Qian, Y.; Zhong, S.; Zhang, L.; Xue, G.; Jia, H. Factors Controlling the Formation of Persistent Free Radicals in Hydrochar during Hydrothermal Conversion of Rice Straw. Environ. Chem. Lett. 2018, 16, 1463–1468. [Google Scholar] [CrossRef]
- Tang, Z.; Zhao, S.; Qian, Y.; Jia, H.; Gao, P.; Kang, Y.; Lichtfouse, E. Formation of Persistent Free Radicals in Sludge Biochar by Hydrothermal Carbonization. Environ. Chem. Lett. 2021, 19, 2705–2712. [Google Scholar] [CrossRef]
- Xiang, C.; Liu, Q.; Shi, L.; Liu, Z. A Study on the New Type of Radicals in Corncob Derived Biochars. Fuel 2020, 277, 118163. [Google Scholar] [CrossRef]
- Chen, Y.; Deng, Z.; Ren, Q.; Ren, D.; Su, S.; Hu, S.; Wang, Y.; Xiang, J. Evolution of Char Structure during the Pyrolysis of Biomass Pellet: Further Understanding on the Effects of Chars Two Phases. Fuel 2022, 312, 122994. [Google Scholar] [CrossRef]
- Wang, Y.; Gu, X.; Huang, Y.; Ding, Z.; Chen, Y.; Hu, X. Insight into Biomass Feedstock on Formation of Biochar-Bound Environmentally Persistent Free Radicals under Different Pyrolysis Temperatures. RSC Adv. 2022, 12, 19318–19326. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.-W.; Zheng, S.; Maciel, G.E. EPR Investigations of the Effects of Inorganic Additives on the Charring and Char/Air Interactions of Cellulose. Energy Fuels 2004, 18, 1049–1065. [Google Scholar] [CrossRef]
- Chintala, R.; Subramanian, S.; Fortuna, A.-M.; Schumacher, T.E. Examining Biochar Impacts on Soil Abiotic and Biotic Processes and Exploring the Potential for Pyrosequencing Analysis. In Biochar Application; Elsevier: Amsterdam, The Netherlands, 2016; ISBN 978-0-12-803433-0. [Google Scholar]
- Tao, W.; Duan, W.; Liu, C.; Zhu, D.; Si, X.; Zhu, R.; Oleszczuk, P.; Pan, B. Formation of Persistent Free Radicals in Biochar Derived from Rice Straw Based on a Detailed Analysis of Pyrolysis Kinetics. Sci. Total Environ. 2020, 715, 136575. [Google Scholar] [CrossRef]
- Zheng, S.; Feng, J.-W.; Maciel, G.E. In Situ High-Temperature EPR Investigation of the Charring of Tobacco and the O2-Induced and H2O-Induced Behavior of the Char. Energy Fuels 2005, 19, 2247–2253. [Google Scholar] [CrossRef]
- Sekiguchi, Y.; Frye, J.S.; Shafizadeh, F. Structure and Formation of Cellulosic Chars. J. Appl. Polym. Sci. 1983, 28, 3513–3525. [Google Scholar] [CrossRef]
- Fang, G.; Zhu, C.; Dionysiou, D.D.; Gao, J.; Zhou, D. Mechanism of Hydroxyl Radical Generation from Biochar Suspensions: Implications to Diethyl Phthalate Degradation. Bioresour. Technol. 2015, 176, 210–217. [Google Scholar] [CrossRef]
- Qin, J.; Cheng, Y.; Sun, M.; Yan, L.; Shen, G. Catalytic Degradation of the Soil Fumigant 1,3-Dichloropropene in Aqueous Biochar Slurry. Sci. Total Environ. 2016, 569–570, 1–8. [Google Scholar] [CrossRef]
- Ruan, X.; Sun, Y.; Du, W.; Tang, Y.; Liu, Q.; Zhang, Z.; Doherty, W.; Frost, R.L.; Qian, G.; Tsang, D.C.W. Formation, Characteristics, and Applications of Environmentally Persistent Free Radicals in Biochars: A Review. Bioresour. Technol. 2019, 281, 457–468. [Google Scholar] [CrossRef]
- Zhang, Y.; Lv, P.; Wang, J.; Wei, J.; Cao, P.; Bie, N.; Bai, Y.; Yu, G. Product Characteristics of Rice Straw Pyrolysis at Different Temperature: Role of Inherent Alkali and Alkaline Earth Metals with Different Occurrence Forms. J. Energy Inst. 2022, 101, 201–208. [Google Scholar] [CrossRef]
- Hu, A.; Xia, Q.; Wang, J.; Li, T.; Wang, K.; Wu, J.; Zhou, G.; Jiang, J. Influence of Alkali and Alkali Earth Metals on Pyrolysis of Tobacco Waste. Ind. Crops Prod. 2023, 206, 117636. [Google Scholar] [CrossRef]
- Trubetskaya, A.; Jensen, P.A.; Jensen, A.D.; Glarborg, P.; Larsen, F.H.; Andersen, M.L. Characterization of Free Radicals by Electron Spin Resonance Spectroscopy in Biochars from Pyrolysis at High Heating Rates and at High Temperatures. Biomass Bioenergy 2016, 94, 117–129. [Google Scholar] [CrossRef]
- Marsh, H.; Walker, P.L. The Effects of Impregnation of Coal by Alkali Salts upon Carbonization Properties. Fuel Process. Technol. 1979, 2, 61–75. [Google Scholar] [CrossRef]
- Butt, D.A.E. Formation of Phenols from the Low-Temperature Fast Pyrolysis of Radiata Pine (Pinus Radiata). J. Anal. Appl. Pyrolysis 2006, 76, 38–47. [Google Scholar] [CrossRef]
- Wang, H.; Srinivasan, R.; Yu, F.; Steele, P.; Li, Q.; Mitchell, B. Effect of Acid, Alkali, and Steam Explosion Pretreatments on Characteristics of Bio-Oil Produced from Pinewood. Energy Fuels 2011, 25, 3758–3764. [Google Scholar] [CrossRef]
- Jönsson, L.J.; Martín, C. Pretreatment of Lignocellulose: Formation of Inhibitory by-Products and Strategies for Minimizing Their Effects. Bioresour. Technol. 2016, 199, 103–112. [Google Scholar] [CrossRef]
- Alayont, Ş.; Kayan, D.B.; Durak, H.; Alayont, E.K.; Genel, S. The Role of Acidic, Alkaline and Hydrothermal Pretreatment on Pyrolysis of Wild Mustard (Sinapis Arvensis) on the Properties of Bio-Oil and Bio-Char. Bioresour. Technol. Rep. 2022, 17, 100980. [Google Scholar] [CrossRef]
- Oniki, T. Origin of Free Radicals Produced from the Syringyl End Groups in Lignins. J. Wood Sci. 1998, 44, 314–319. [Google Scholar] [CrossRef]
- Steelink, C. Free Radical Studies of Lignin, Lignin Degradation Products and Soil Humic Acids. Geochim. et Cosmochim. Acta 1964, 28, 1615–1622. [Google Scholar] [CrossRef]
- Dizhbite, T.; Telysheva, G.; Jurkjane, V.; Viesturs, U. Characterization of the Radical Scavenging Activity of Lignins––Natural Antioxidants. Bioresour. Technol. 2004, 95, 309–317. [Google Scholar] [CrossRef]
- Steelink, C. Stable Phenoxy Radicals Derived from Phenols Related to Lignin1. J. Am. Chem. Soc. 1965, 87, 2056–2057. [Google Scholar] [CrossRef]
- Bährle, C.; Nick, T.U.; Bennati, M.; Jeschke, G.; Vogel, F. High-Field Electron Paramagnetic Resonance and Density Functional Theory Study of Stable Organic Radicals in Lignin: Influence of the Extraction Process, Botanical Origin, and Protonation Reactions on the Radical g Tensor. J. Phys. Chem. A 2015, 119, 6475–6482. [Google Scholar] [CrossRef] [PubMed]
- Steelink, C.; Hansen, R.E. A Solid Phenoxy Radical from Disyringylmethane–A Model for Lignin Radicals. Tetrahedron Lett. 1966, 7, 105–111. [Google Scholar] [CrossRef]
- Fitzpatrick, J.D.; Steelink, C. Benzosemiquinone Radicals in Alkaline Solutions of Hardwood Lignins. Tetrahedron Lett. 1969, 10, 5041–5044. [Google Scholar] [CrossRef]
- Barekati-Goudarzi, M.; Boldor, D.; Marculescu, C.; Khachatryan, L. Peculiarities of Pyrolysis of Hydrolytic Lignin in Dispersed Gas Phase and in Solid State. Energy Fuels 2017, 31, 12156–12167. [Google Scholar] [CrossRef]
- Rahimi, A.; Ulbrich, A.; Coon, J.J.; Stahl, S.S. Formic-Acid-Induced Depolymerization of Oxidized Lignin to Aromatics. Nature 2014, 515, 249–252. [Google Scholar] [CrossRef]
- Mohan, D.; Pittman, C.U.J.; Steele, P.H. Pyrolysis of Wood/Biomass for Bio-Oil: A Critical Review. Energy Fuels 2006, 20, 848–889. [Google Scholar] [CrossRef]
- Prins, M.J.; Ptasinski, K.J.; Janssen, F.J.J.G. Torrefaction of Wood: Part 2. Analysis of Products. J. Anal. Appl. Pyrolysis 2006, 77, 35–40. [Google Scholar] [CrossRef]
- Zwart, R.W.R.; Boerrigter, H.; Van Der Drift, A. The Impact of Biomass Pretreatment on the Feasibility of Overseas Biomass Conversion to Fischer−Tropsch Products. Energy Fuels 2006, 20, 2192–2197. [Google Scholar] [CrossRef]
- Yan, W.; Hastings, J.T.; Acharjee, T.C.; Coronella, C.J.; Vásquez, V.R. Mass and Energy Balances of Wet Torrefaction of Lignocellulosic Biomass. Energy Fuels 2010, 24, 4738–4742. [Google Scholar] [CrossRef]
- Feng, Z.; Yang, J.; Ni, L.; Gao, Q.; Liu, Z. The Chemical and Structural Transformation of Bamboo Wastes during Torrefaction Process. Environ. Prog. Sustain. Energy 2021, 40, e13565. [Google Scholar] [CrossRef]
- Melkior, T.; Jacob, S.; Gerbaud, G.; Hediger, S.; Le Pape, L.; Bonnefois, L.; Bardet, M. NMR Analysis of the Transformation of Wood Constituents by Torrefaction. Fuel 2012, 92, 271–280. [Google Scholar] [CrossRef]
- Meng, J.; Park, J.; Tilotta, D.; Park, S. The Effect of Torrefaction on the Chemistry of Fast-Pyrolysis Bio-Oil. Bioresour. Technol. 2012, 111, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Meng, J.; Moore, A.; Tilotta, D.C.; Kelley, S.S.; Adhikari, S.; Park, S. Thermal and Storage Stability of Bio-Oil from Pyrolysis of Torrefied Wood. Energy Fuels 2015, 29, 5117–5126. [Google Scholar] [CrossRef]
- Flynn, C.N.; Byrne, C.P.; Meenan, B.J. Surface Modification of Cellulose via Atmospheric Pressure Plasma Processing in Air and Ammonia–Nitrogen Gas. Surf. Coat. Technol. 2013, 233, 108–118. [Google Scholar] [CrossRef]
- Benoit, M.; Rodrigues, A.; Zhang, Q.; Fourré, E.; De Oliveira Vigier, K.; Tatibouët, J.-M.; Jérôme, F. Depolymerization of Cellulose Assisted by a Nonthermal Atmospheric Plasma. Angew. Chem. Int. Ed. 2011, 50, 8964–8967. [Google Scholar] [CrossRef]
- Ravindran, R.; Sarangapani, C.; Jaiswal, S.; Cullen, P.J.; Jaiswal, A.K. Ferric Chloride Assisted Plasma Pretreatment of Lignocellulose. Bioresour. Technol. 2017, 243, 327–334. [Google Scholar] [CrossRef]
- Taghvaei, H.; Rahimpour, M.R. Catalytic Hydrodeoxygenation of Bio-Oil Using in Situ Generated Hydrogen in Plasma Reactor: Effects of Allumina Supported Catalysts and Plasma Parameters. Process Saf. Environ. Prot. 2019, 121, 221–228. [Google Scholar] [CrossRef]
- Kuzuya, M.; Morisaki, K.; Niwa, J.; Yamauchi, Y.; Xu, K. Spectrochemistry of Polycarbohydrate Free Radicals Generated by Argon Plasmolysis: Effect of Tertiary Structure on Free Radical Formation. J. Phys. Chem. 1994, 98, 11301–11307. [Google Scholar] [CrossRef]
- Lusi, A.; Hu, H.; Bai, X. Producing High Yield of Levoglucosan by Pyrolyzing Nonthermal Plasma-Pretreated Cellulose. Green Chem. 2020, 22, 2036–2048. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Biomass Type | Pyrolysis Temperature (°C) | g-Factor | Linewidth (G) | EPFR Concentration (1018 spins/g) | EPFR Type | References |
|---|---|---|---|---|---|---|
| Pine needles | 400 | 2.0037 | 6.8 | 15.2 ± 0.03 | Oxygenated carbon-centered | [117] |
| Pine needles | 550 | 2.0028 | 4.5 | 13.7 ± 0.06 | Carbon-centered | [105] |
| Wheat straw | 400 | 2.0030 | 5.0 | 16.5 ± 0.09 | Oxygenated carbon-centered | [105] |
| Wheat straw | 500 | 2.0029 | 4.8 | 28.6 ± 0.12 | Carbon-centered | [105] |
| Maize straw | 400 | 2.0031 | 6.2 | 6.25 ± 0.12 | Oxygenated carbon-centered | [105] |
| Maize straw | 500 | 2.0029 | 5.2 | 30.2 ± 0.09 | Carbon-centered | [105] |
| Rice husk | 300 | 2.0041 | 2.77 | 6.9 ± 0.1 | Oxygen-centered | [118] |
| Rice husk | 700 | 2.0036 | 0.16 | 1.8 ± 0.1 | Oxygenated carbon-centered | [118] |
| Corn straw | 500 | 2.0030 | 3.2 | 1.9 ± 0.03 | Oxygenated carbon-centered | [106] |
| Peanut husk | 500 | 2.0032 | 4.8 | 2.2 ± 0.02 | Oxygenated carbon-centered | [106] |
| Cotton stalk | 500 | 2.0032 | 4.1 | 2.2 ± 0.02 | Oxygenated carbon-centered | [106] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luo, Z.; Liu, L.; Miao, F.; Zhu, W.; Li, L.; Wang, Y. Electron Paramagnetic Resonance in Lignocellulosic Biomass Pyrolysis Mechanism: Advancements, Applications, and Prospects. Energies 2025, 18, 1598. https://doi.org/10.3390/en18071598
Luo Z, Liu L, Miao F, Zhu W, Li L, Wang Y. Electron Paramagnetic Resonance in Lignocellulosic Biomass Pyrolysis Mechanism: Advancements, Applications, and Prospects. Energies. 2025; 18(7):1598. https://doi.org/10.3390/en18071598
Chicago/Turabian StyleLuo, Zhongyang, Longyi Liu, Feiting Miao, Wanchen Zhu, Longfei Li, and Yuanlin Wang. 2025. "Electron Paramagnetic Resonance in Lignocellulosic Biomass Pyrolysis Mechanism: Advancements, Applications, and Prospects" Energies 18, no. 7: 1598. https://doi.org/10.3390/en18071598
APA StyleLuo, Z., Liu, L., Miao, F., Zhu, W., Li, L., & Wang, Y. (2025). Electron Paramagnetic Resonance in Lignocellulosic Biomass Pyrolysis Mechanism: Advancements, Applications, and Prospects. Energies, 18(7), 1598. https://doi.org/10.3390/en18071598