Single Step, Electrochemical Preparation of Copper-Based Positive Electrode for Lithium Primary Cells

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
3. Results and Discussion
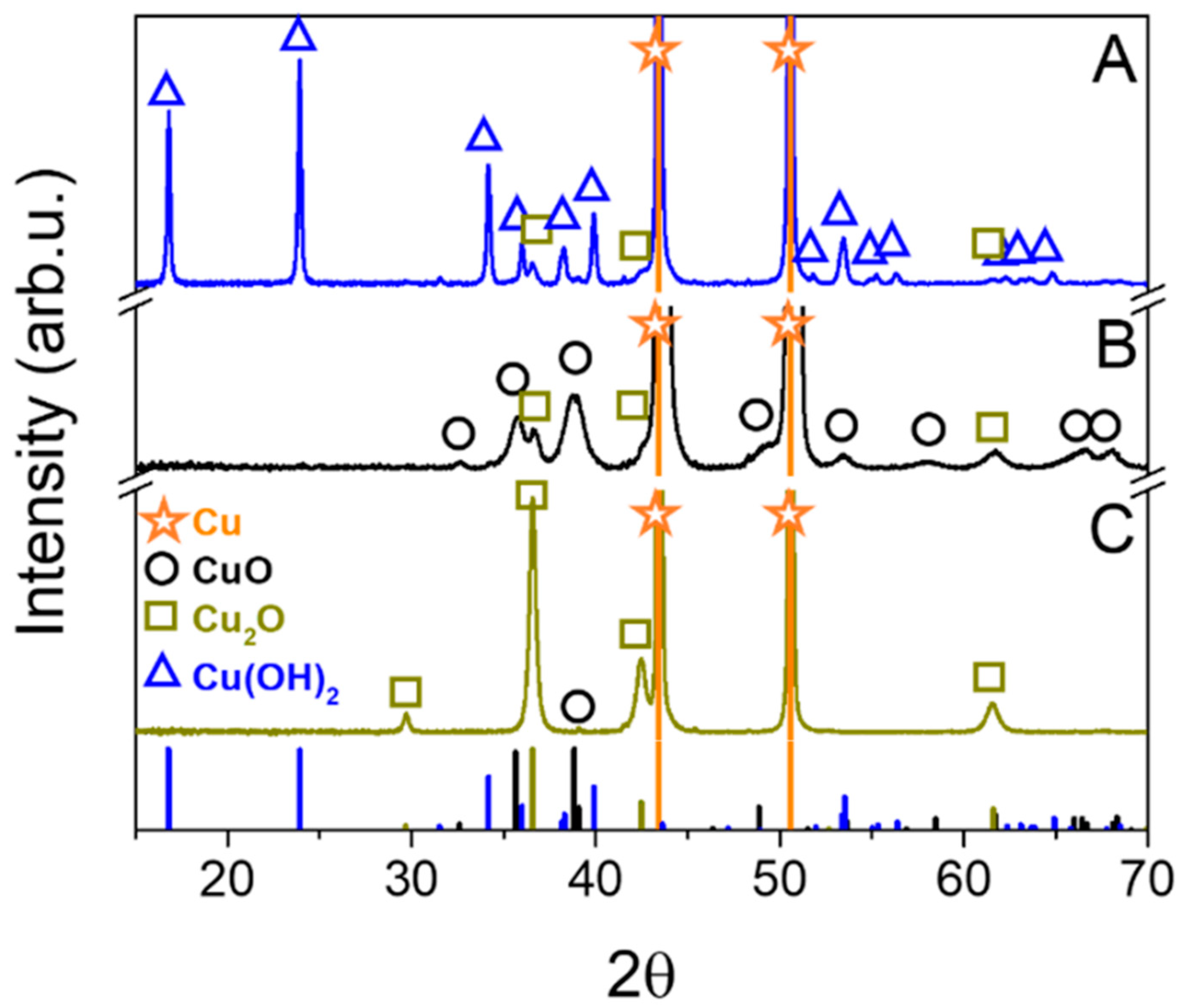
3.1. XRD Measurements
3.2. XPS Measurements
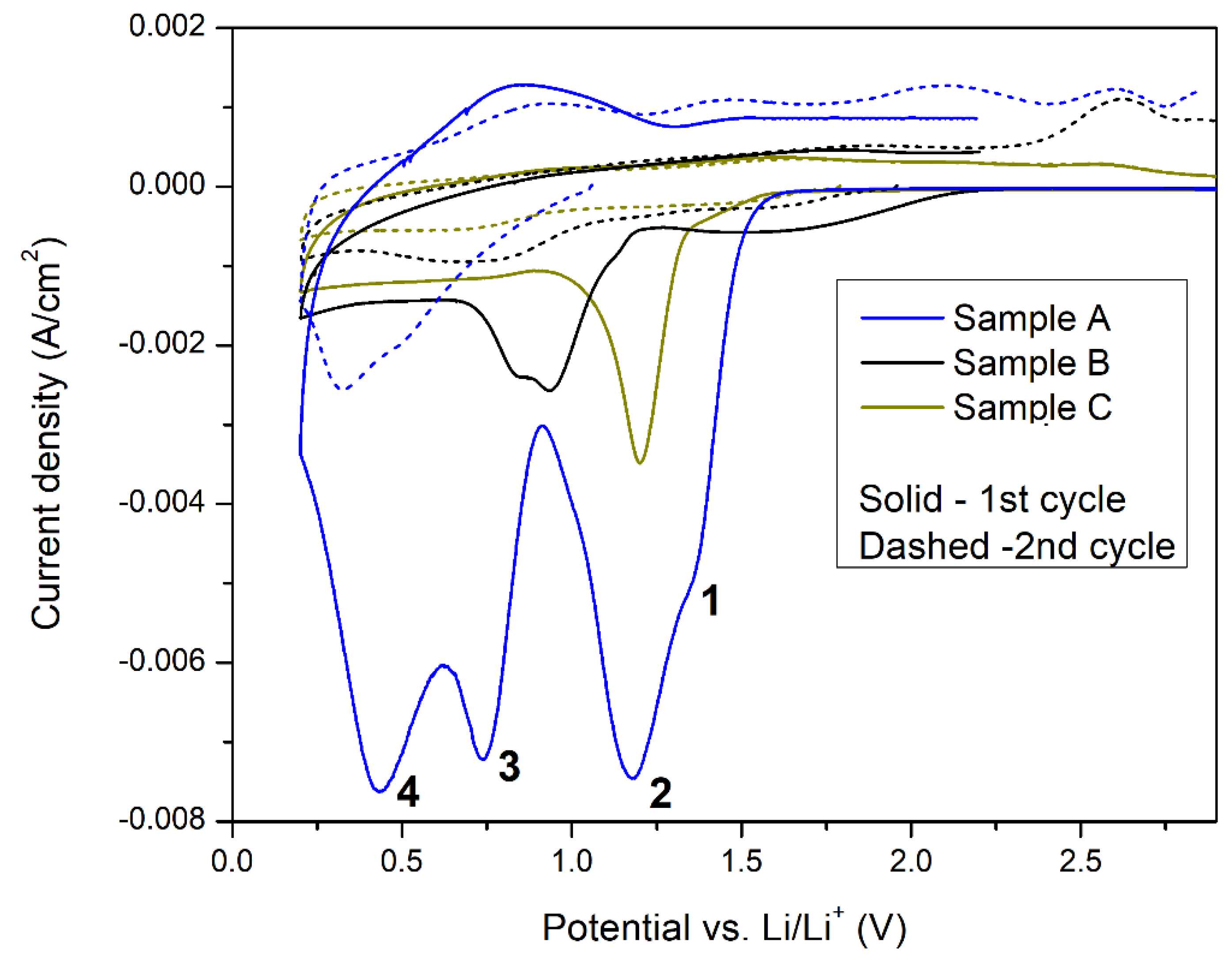
3.3. Electrochemical Measurements
3.3.1. Cyclic Voltammetry
3.3.2. Galvanostatic Discharge
4. Conclusions
5. Patents
Author Contributions
Funding
Conflicts of Interest
References
- Lewis, G.N.; Keyes, F.G. The potential of the lithium electrode. J. Am. Chem. Soc. 1913, 35, 340–344. [Google Scholar] [CrossRef]
- Huston, R.; Butler, J.N. The Standard Potential of the Lithium Electrode in Aqueous Solutions. J. Phys. Chem. 1968, 72, 4263–4264. [Google Scholar] [CrossRef]
- Reddy, T.B.; Linden, D. Linden’s Handbook of Batteries; McGraw-Hill: New York, NY, USA, 2011. [Google Scholar]
- Garche, J. Encyclopedia of Electrochemical Power Sources; Elsevier B.V.: Amsterdam, The Netherlands, 2009. [Google Scholar]
- Manane, Y.; Yazami, R. Accurate state of charge assessment of lithium-manganese dioxide primary batteries. J. Power Sources 2017, 359, 422–426. [Google Scholar] [CrossRef]
- Hamwi, A.; Guérin, K.; Dubois, M. Fluorinated Materials for Energy Conversion; Elsevier: Amsterdam, The Netherlands, 2005; Chapter 17. [Google Scholar]
- Ahmad, Y.; Dubois, M.; Guerin, K.; Hamwia, A.; Flahaut, E. High energy density of primary lithium batteries working with sub-fluorinated few walled carbon nanotubes cathode. J. Alloys Compd. 2017, 726, 852–859. [Google Scholar] [CrossRef] [Green Version]
- Mar, M.; Ahmad, Y.; Guérin, K.; Dubois, M.; Batisse, N. Fluorinated exfoliated graphite as cathode materials for enhanced performances in primary lithium battery. Electrochim. Acta 2017, 227, 18–23. [Google Scholar] [CrossRef]
- Frysz, C.A.; Shui, X.; Chung, D.D.L. Carbon filaments and carbon black as a conductive additive to the manganese dioxide cathode of a lithium electrolytic cell. J. Power Sources 1996, 58, 41–54. [Google Scholar] [CrossRef]
- Reddy, A.L.M.; Shaijumon, M.M.; Gowda, S.R.; Ajayan, P.M. Coaxial MnO2/Carbon nanotube array electrodes for high-performance lithium batteries. Nano Lett. 2009, 9, 1002–1006. [Google Scholar] [CrossRef] [PubMed]
- Park, S.-H.; Lee, W.-J. Hierarchically mesoporous CuO/carbon nanofiber coaxial shell-core nanowires for lithium ion batteries. Sci. Rep. 2015, 5, 9754–9767. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Wu, X.-L.; Shu, C.-Y.; Guo, Y.-G.; Wang, C.-R. Synthesis of CuO/grapheme nanocomposite as a high-performance anode material for lithium-ion batteries. J. Mater. Chem. 2010, 20, 10661–10664. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, B.; Zhou, J.; Xia, R.; Chu, Y.; Huang, J. Preparation of advanced CuO nanowires/functionalized graphene composite anode material for lithium ion batteries. Materials 2017, 10, 72. [Google Scholar] [CrossRef] [PubMed]
- Grimm, M. Lithium copper oxide lithium copper oxyphosphate battery systems. IEEE Trans. Consum. Electr. 1986, CE-32, 700–703. [Google Scholar] [CrossRef]
- Biesinger, M.C.; Lau, L.W.M.; Gerson, A.R.; Smart, R.S.C. Resolving surface chemical states in XPS analysis of first row transition metals, oxides and hydroxides: Sc, Ti, V, Cu and Zn. Appl. Surf. Sci. 2011, 257, 2717–2730. [Google Scholar] [CrossRef]
- NIST X-Ray Photoelectron Spectroscopy Database. Available online: https://srdata.nist.gov/xps/ (accessed on 25 October 2018).
- Wagner, C.D. Auger lines in x-ray photoelectron spectrometry. Anal. Chem. 1972, 44, 967–973. [Google Scholar] [CrossRef]
- Chen, W.; Zhang, W.; Chen, L.; Zeng, L.; Wei, M. Facile synthesis of Cu2O nanorod arrays on Cu foam as a self-supporting anode material for lithium ion batteries. J. Alloys Compd. 2017, 723, 172–178. [Google Scholar] [CrossRef]
- Wang, C.; Higgins, D.; Wang, F.; Li, D.; Liu, R.; Xia, G.; Li, N.; Li, Q.; Xu, H.; Wu, G. Controlled synthesis of micro/nanostructured CuO anodes for lithium-ion batteries. Nano Energy 2014, 9, 334–344. [Google Scholar] [CrossRef]
- Dang, R.; Jia, X.; Wang, P.; Zhang, X.; Wang, D.; Wang, G. Hydrothermal synthesis of peony-like CuO micro/nanostructures for high-performance lithium-ion battery anodes. Chin. Chem. Lett. 2017, 28, 2263–2268. [Google Scholar] [CrossRef]
- Liu, X.; Liu, G.; Wang, L.; Li, Y.; Ma, Y.; Ma, J. Morphology- and facet-controlled synthesis of CuO micro/nanomaterials and analysis of their lithium ion storage properties. J. Power Sources 2016, 312, 199–206. [Google Scholar] [CrossRef]
- Pramanik, A.; Maiti, S.; Mahanty, S. Metal hydroxides as a conversion electrode for lithium-ion batteries: A case study with a Cu(OH)2 nanoflower array. J. Mater. Chem. A 2014, 2, 18515–18522. [Google Scholar] [CrossRef]
- Peng, H.-J.; Hao, G.-X.; Chu, Z.-H.; He, C.-L.; Lin, X.-M.; Cai, Y.-P. Mesoporous spindle-like hollow CuO/C fabricated from a Cu-based metal-organic framework as anodes for high-performance lithium storage. J. Alloys Compd. 2017, 727, 1020–1026. [Google Scholar] [CrossRef]
- Lu, L.Q.; Wang, Y. Facile synthesis of graphene-supported shuttle- and urchin-like CuO for high and fast Li-ion storage. Electrochem. Commun. 2012, 14, 82–85. [Google Scholar] [CrossRef]
- Xia, H.; Zhang, J.; Chen, Z.; Xu, Q. 1D Cu(OH)2 nanorod/2D SnO2 nanosheets core/shell structured array: Covering with graphene layer leads to excellent performances on lithium-ion battery. Appl. Sur. Sci. 2018, 440, 91–98. [Google Scholar] [CrossRef]
- Shi, L.; Fan, C.; Sun, C.; Ren, Z.; Fu, X.; Qian, G.; Wang, Z. Synthesis of different CuO nanostructures from Cu(OH)2 nanorods through changing drying medium for lithium-ion battery anodes. RSC Adv. 2015, 5, 28611–28618. [Google Scholar] [CrossRef]
- Chen, W.; Zhang, H.; Ma, Z.; Li, S.; Li, Z. Binder free Cu(OH)2/CuO electrodes fabricated directly on copper foils by facile large-scale production method. J. Alloys Compd. 2018, 762, 565–573. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Deposited Layer Phase Composition (%) | Crystal Size of Individual Compounds (Å) | ||||
|---|---|---|---|---|---|---|
| Cu(OH)2 | Cu2O | CuO | Cu(OH)2 | Cu2O | CuO | |
| A | 91 | 9 | - | 522 | 233 | - |
| B | - | 13 | 87 | - | 167 | 81 |
| C | - | 99 | 1 | - | 232 | bdl |
| Sample | Atomic Concentration (%) | O/Cu Ratio | |||
|---|---|---|---|---|---|
| Cu | O | C | Na | ||
| A | 23.1 | 51.8 | 25.1 | - | 2.2 |
| B | 30.1 | 43.2 | 26.7 | - | 1.4 |
| C | 22.1 | 38.3 | 37.0 | 2.6 | 1.7 |
| Sample | Binding Energy Cu 2p3/2 (eV) | % of Main Component | |
|---|---|---|---|
| Component 1 | Component 2 | ||
| A | 934.4 | 932.6 | 88 |
| B | 933.8 | - | 100 |
| C | 932.3 | 934.9 | 79 |
| Sample | Kinetic Energy (eV) Cu LMM | Auger Parameter (eV) | Assigned Chemistry | ||
|---|---|---|---|---|---|
| Component 1 | Component 2 | Component 1 | Component 2 | ||
| A | 961.4 | 1850.8 | 1849.0 | Cu(OH)2 | Cu2O |
| B | 917.6 | 1851.4 | - | CuO | |
| C | 916.6 | 1848.9 | 1851.5 | Cu2O | CuO |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ratynski, M.; Hamankiewicz, B.; Krajewski, M.; Boczar, M.; Ziolkowska, D.; Czerwinski, A. Single Step, Electrochemical Preparation of Copper-Based Positive Electrode for Lithium Primary Cells. Materials 2018, 11, 2126. https://doi.org/10.3390/ma11112126
Ratynski M, Hamankiewicz B, Krajewski M, Boczar M, Ziolkowska D, Czerwinski A. Single Step, Electrochemical Preparation of Copper-Based Positive Electrode for Lithium Primary Cells. Materials. 2018; 11(11):2126. https://doi.org/10.3390/ma11112126
Chicago/Turabian StyleRatynski, Maciej, Bartosz Hamankiewicz, Michal Krajewski, Maciej Boczar, Dominika Ziolkowska, and Andrzej Czerwinski. 2018. "Single Step, Electrochemical Preparation of Copper-Based Positive Electrode for Lithium Primary Cells" Materials 11, no. 11: 2126. https://doi.org/10.3390/ma11112126