29Si NMR Chemical Shifts in Crystalline and Amorphous Silicon Nitrides


Abstract
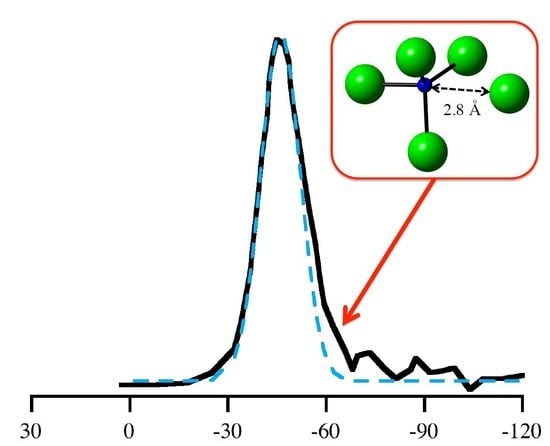
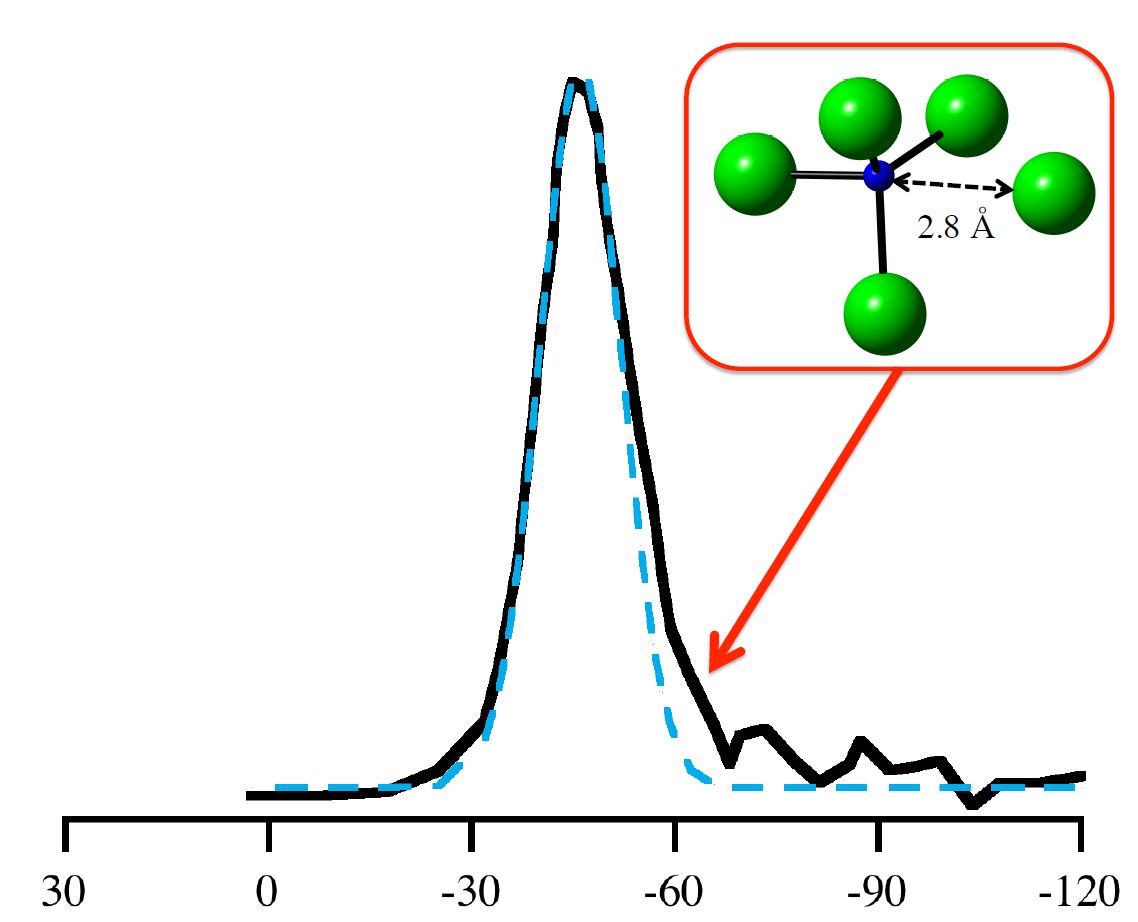
:
1. Introduction
2. Computational Methods
3. Results and Discussion
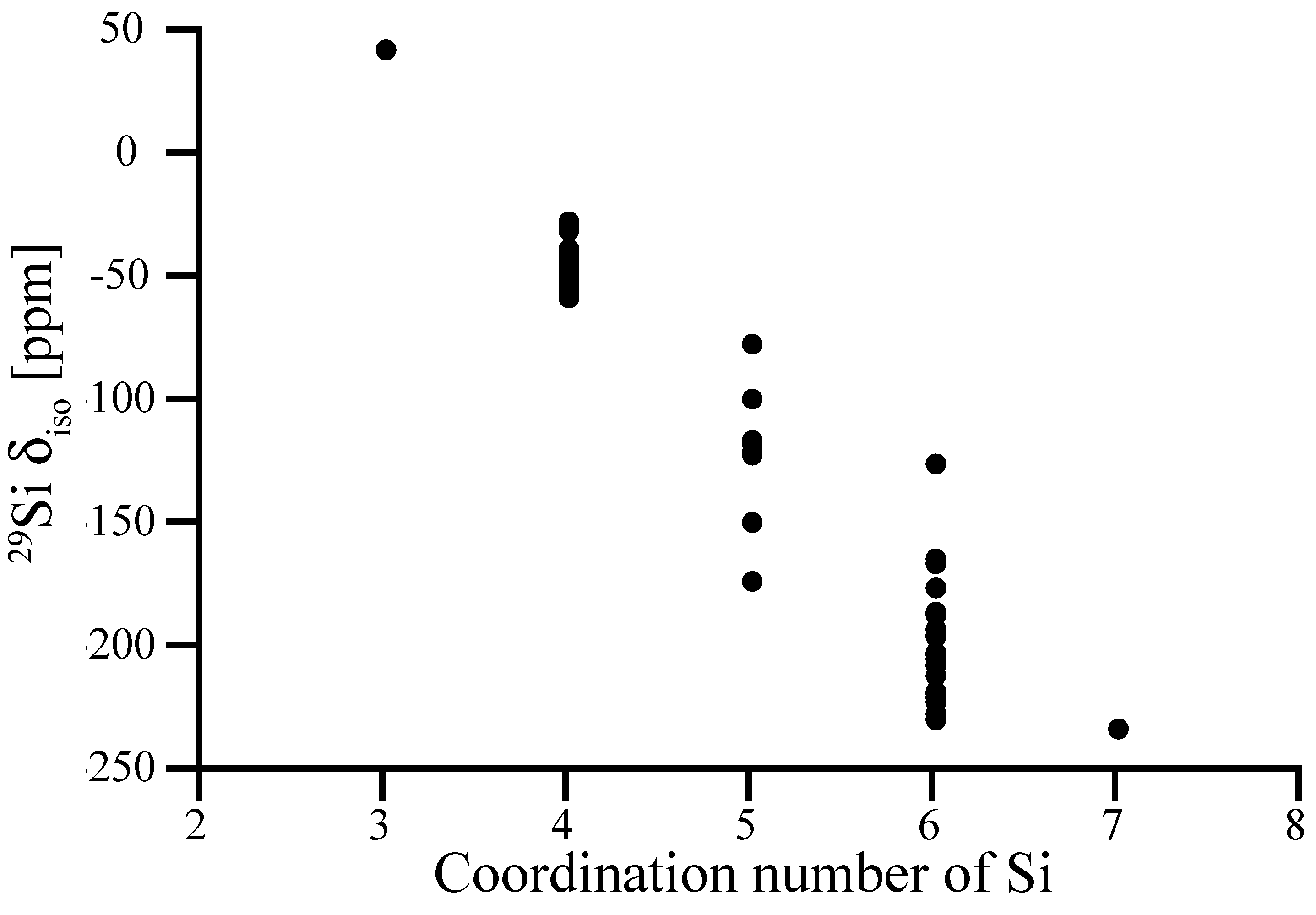
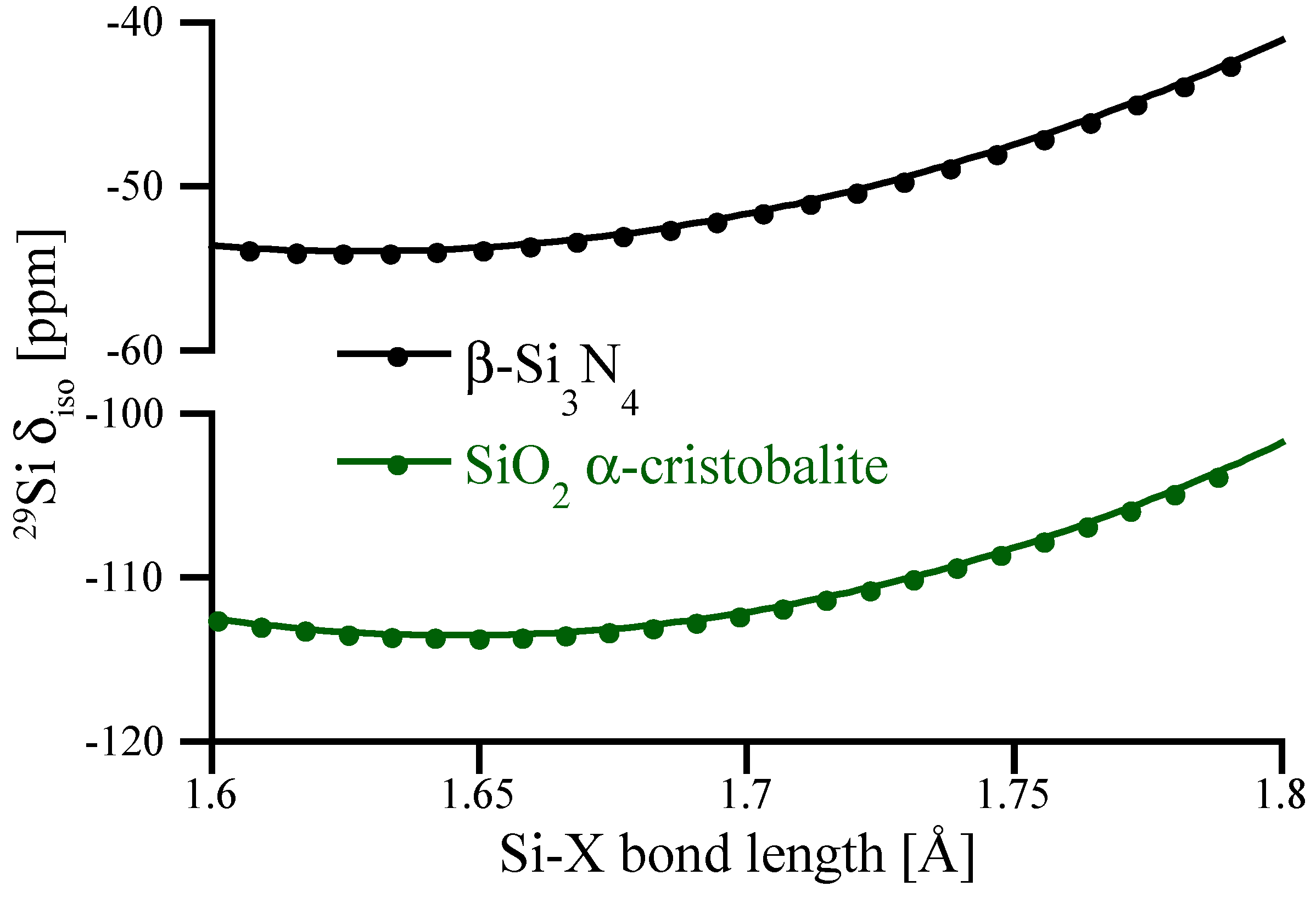
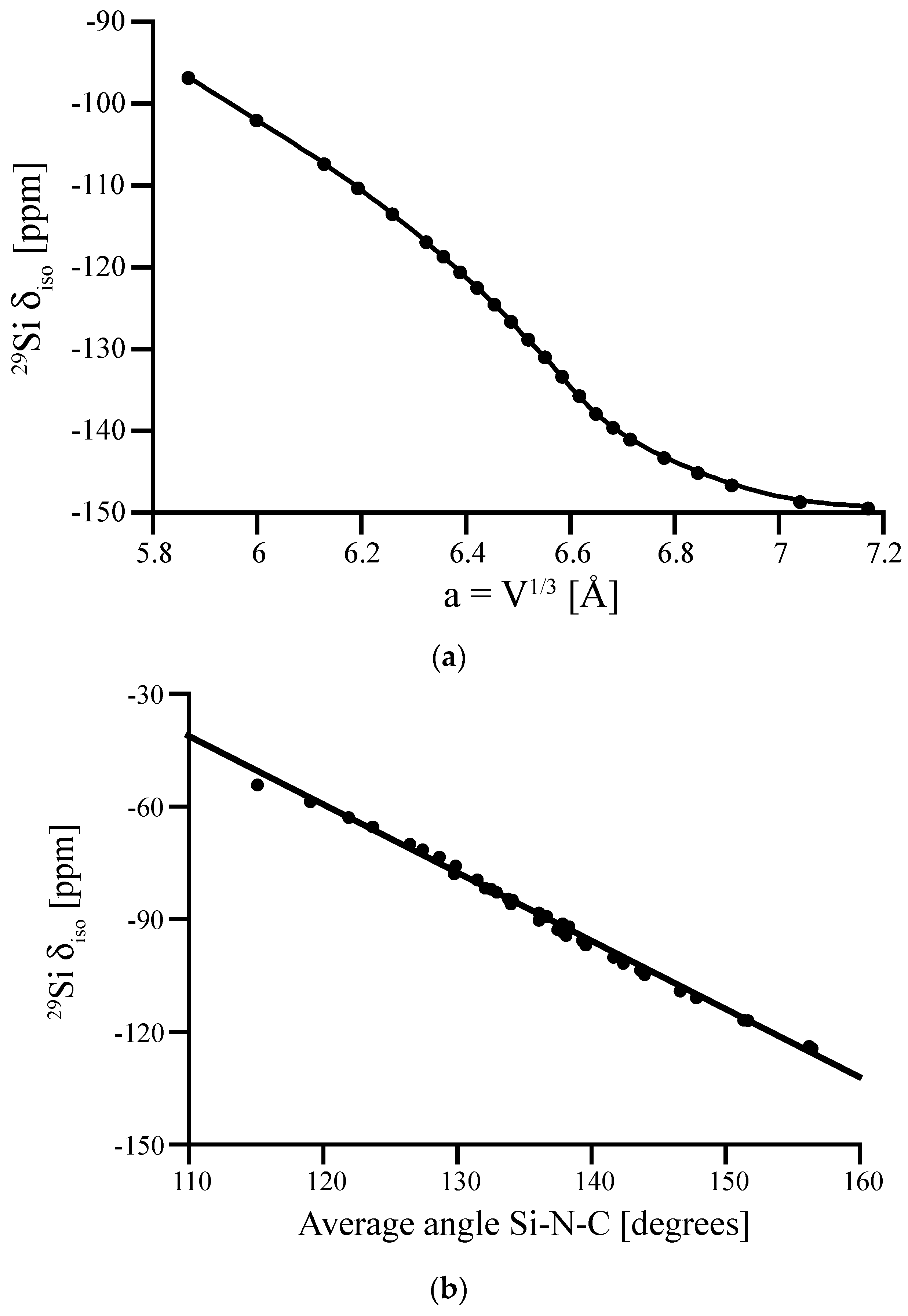
3.1. Effects of Coordination Number—Known and Hypothetical Crystalline Si3N4 Structures
3.2. Impact of Local Structure—Distortions of Crystalline Si3N4 Models
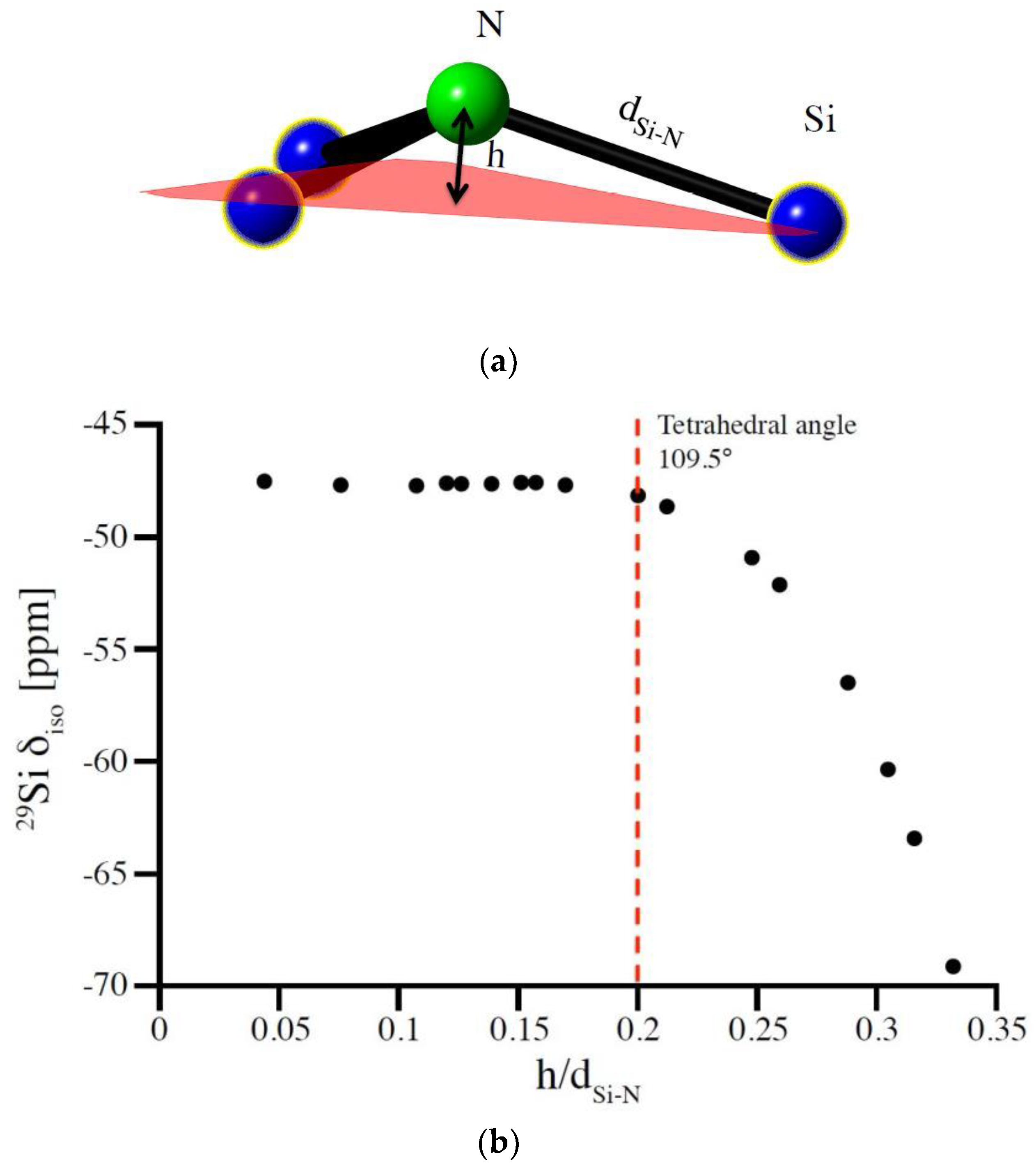
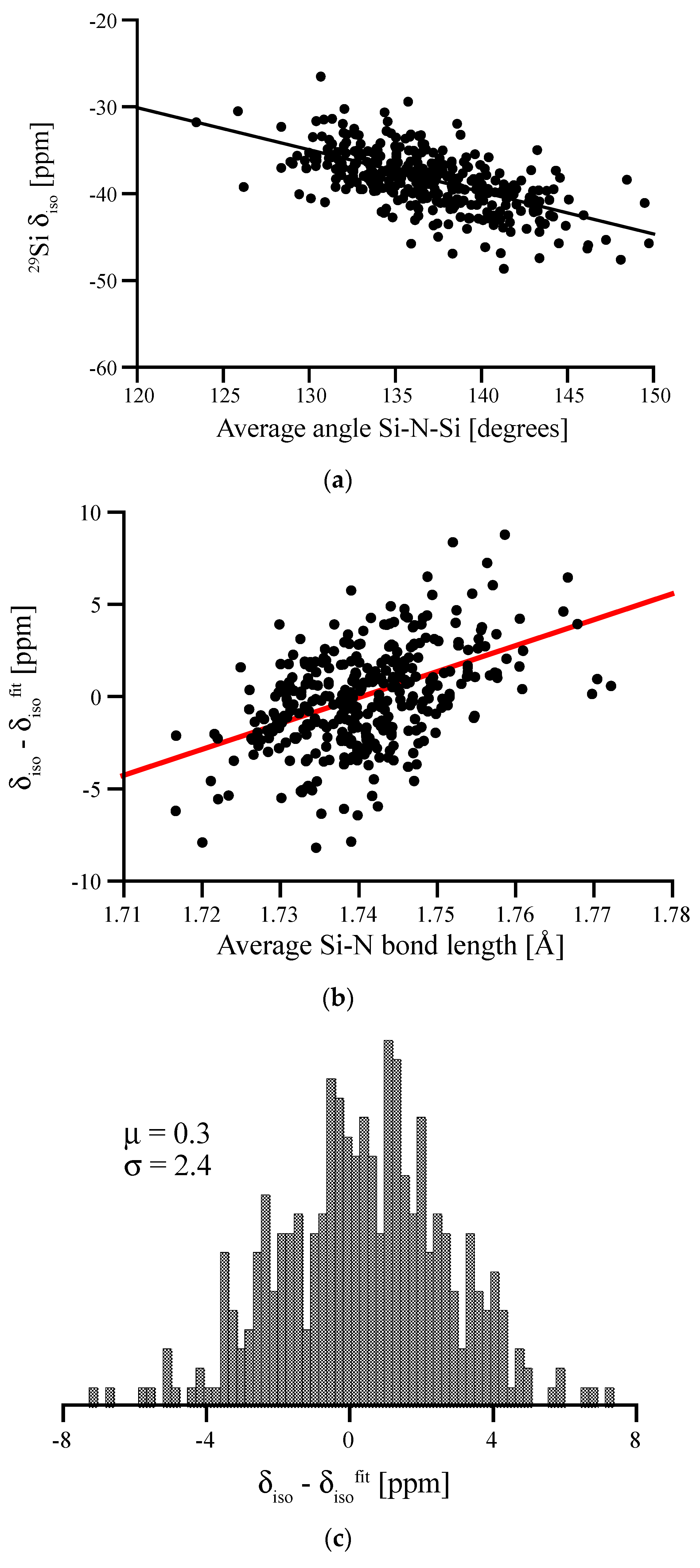
3.3. Special Case of the Silicon Dicarbodiimide—A Bond Angle Dependence for a SiN4 Environment
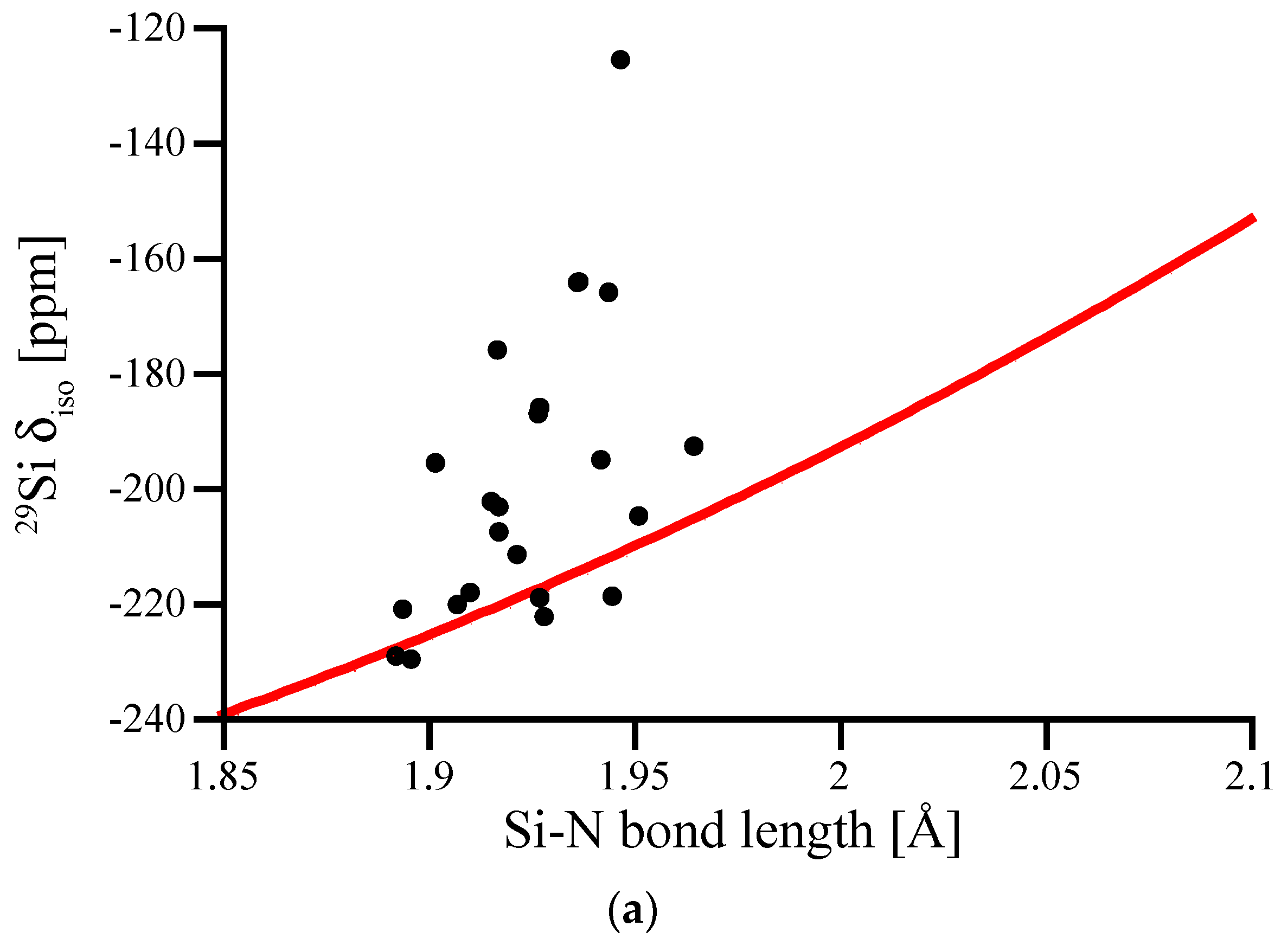
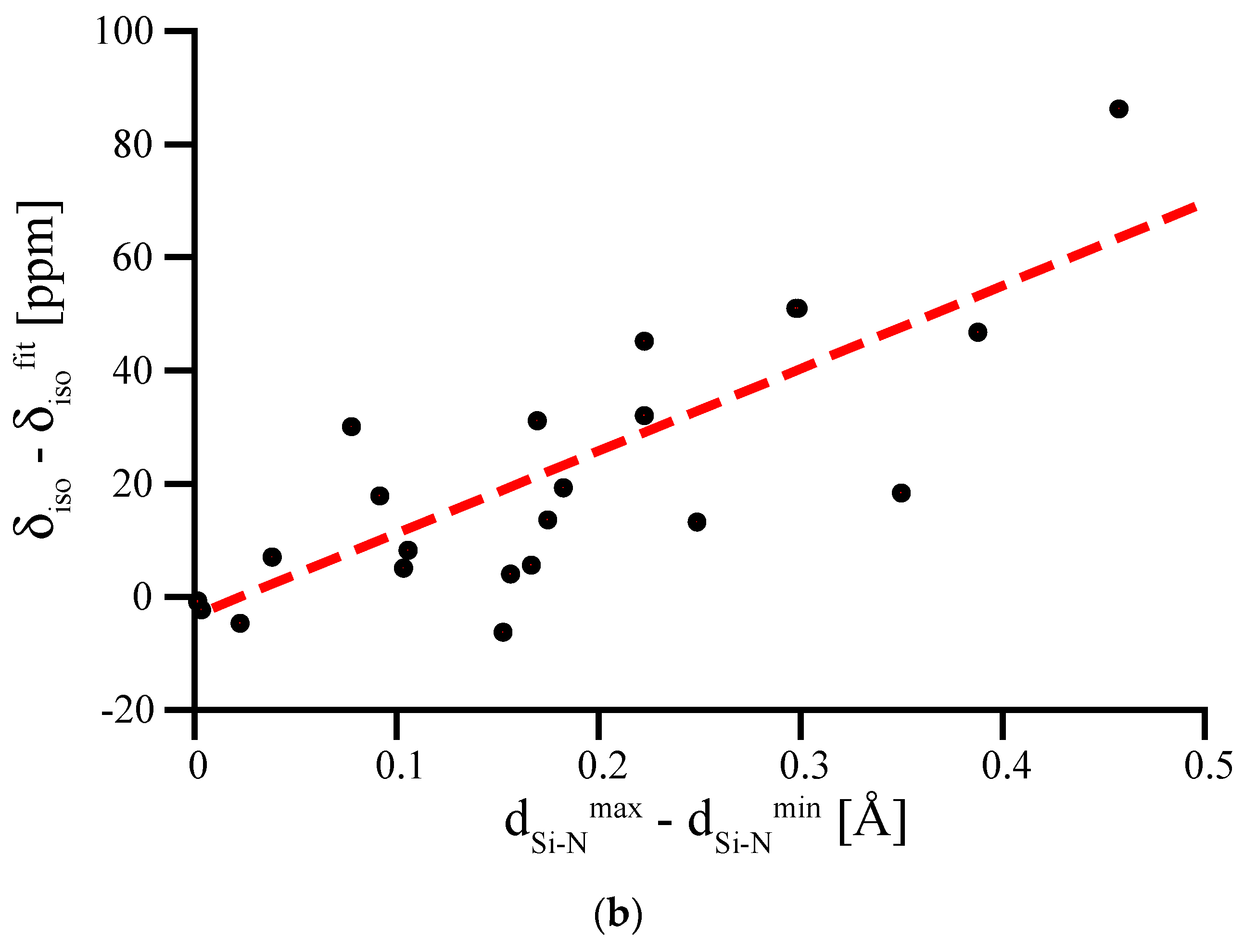
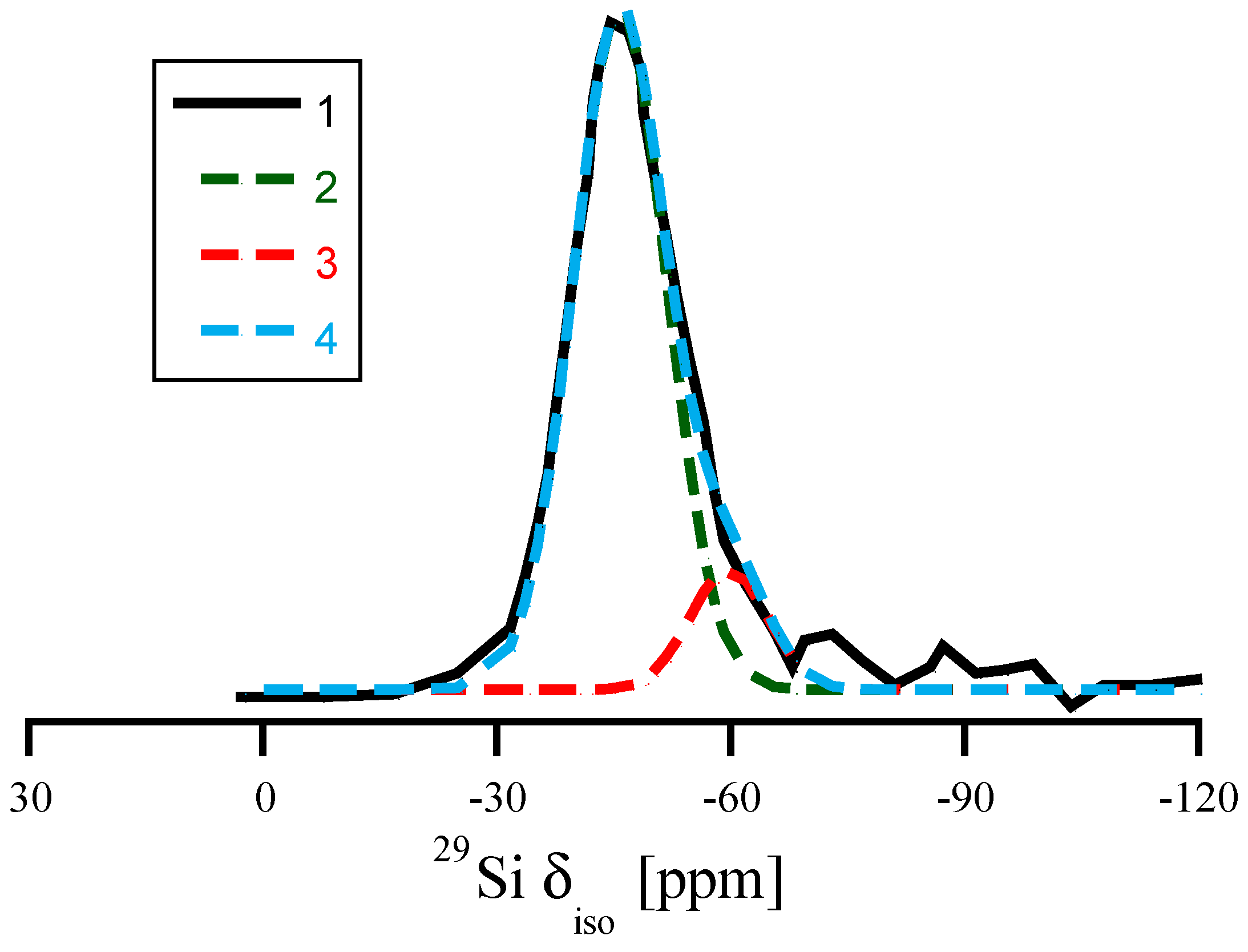
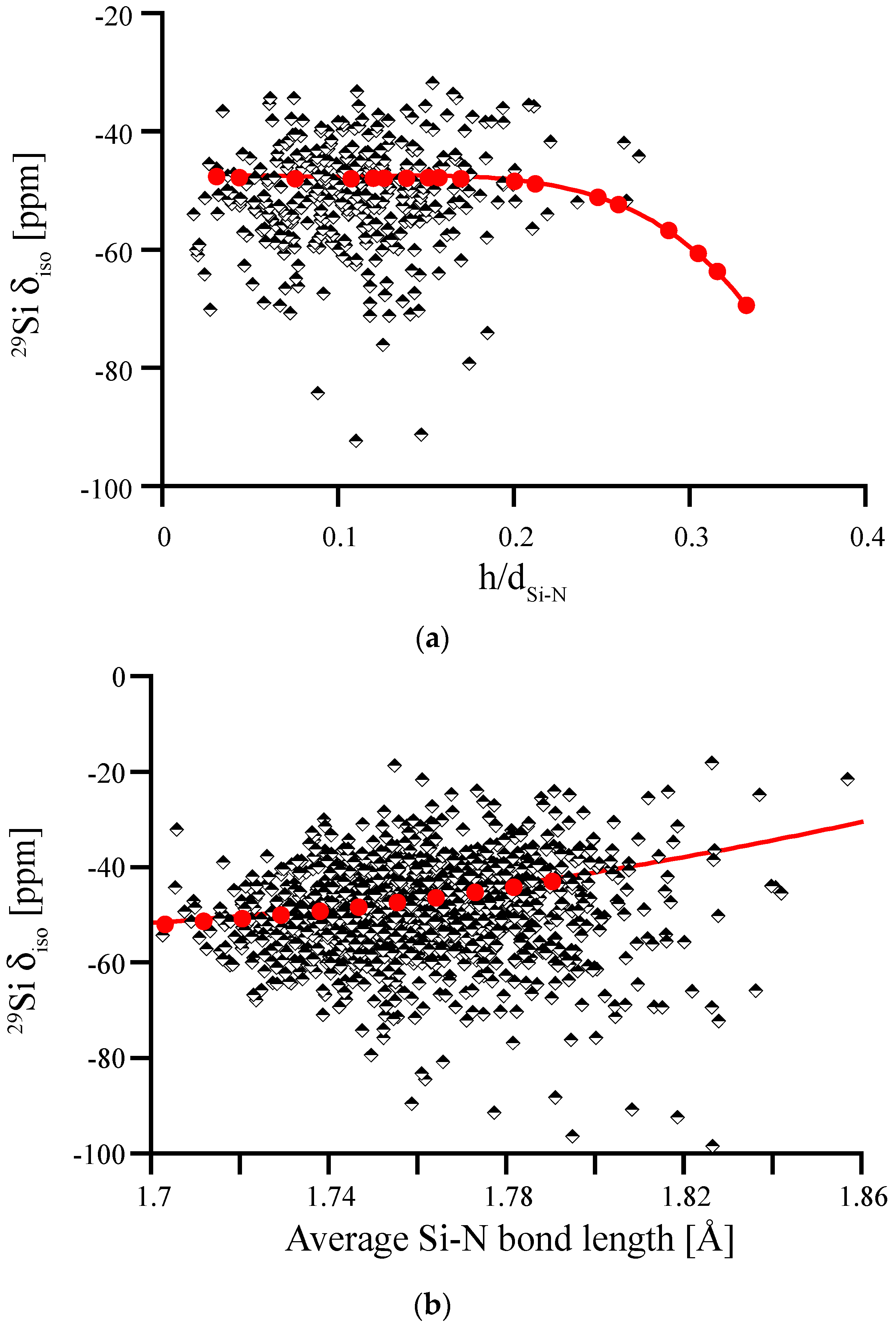
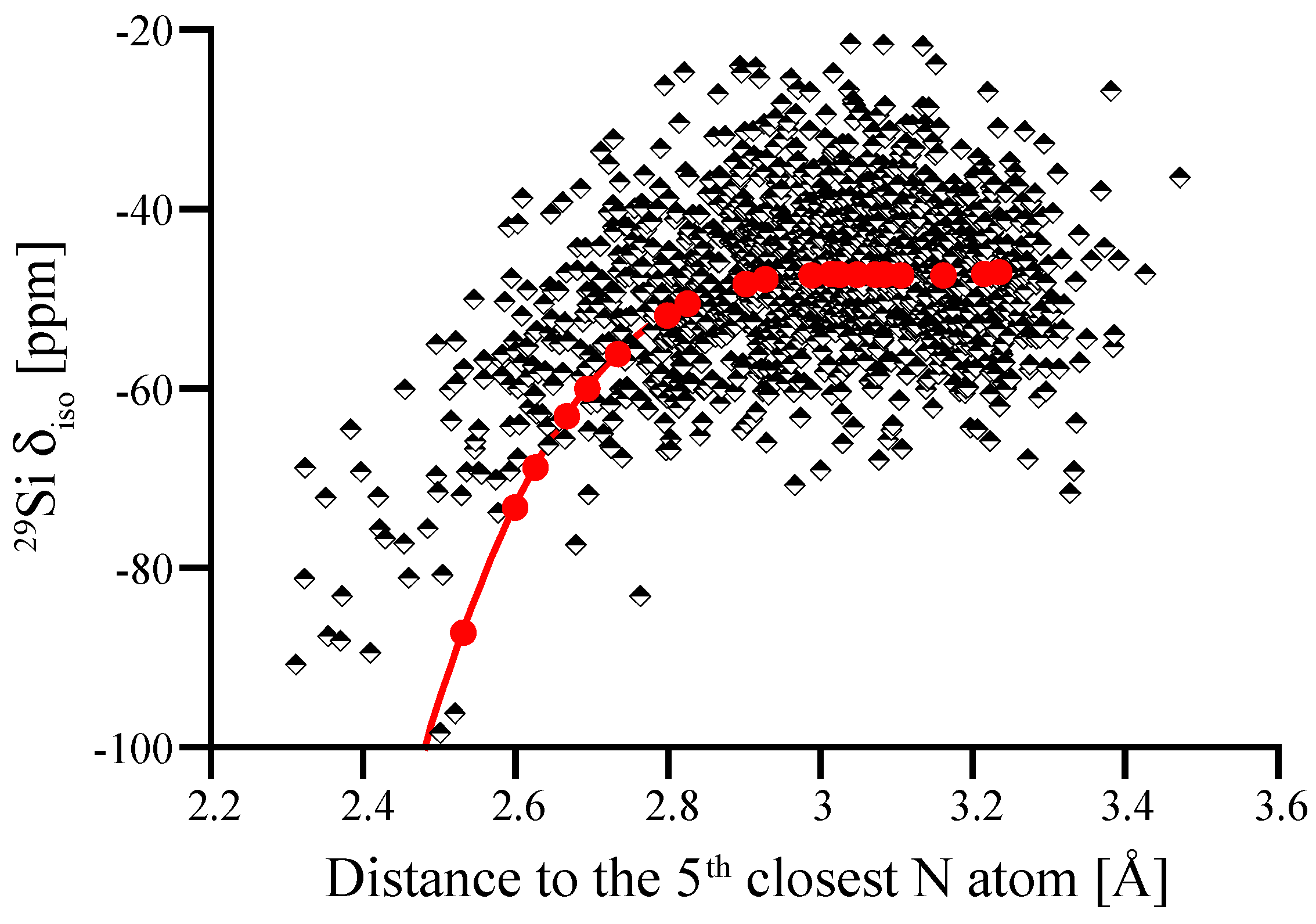
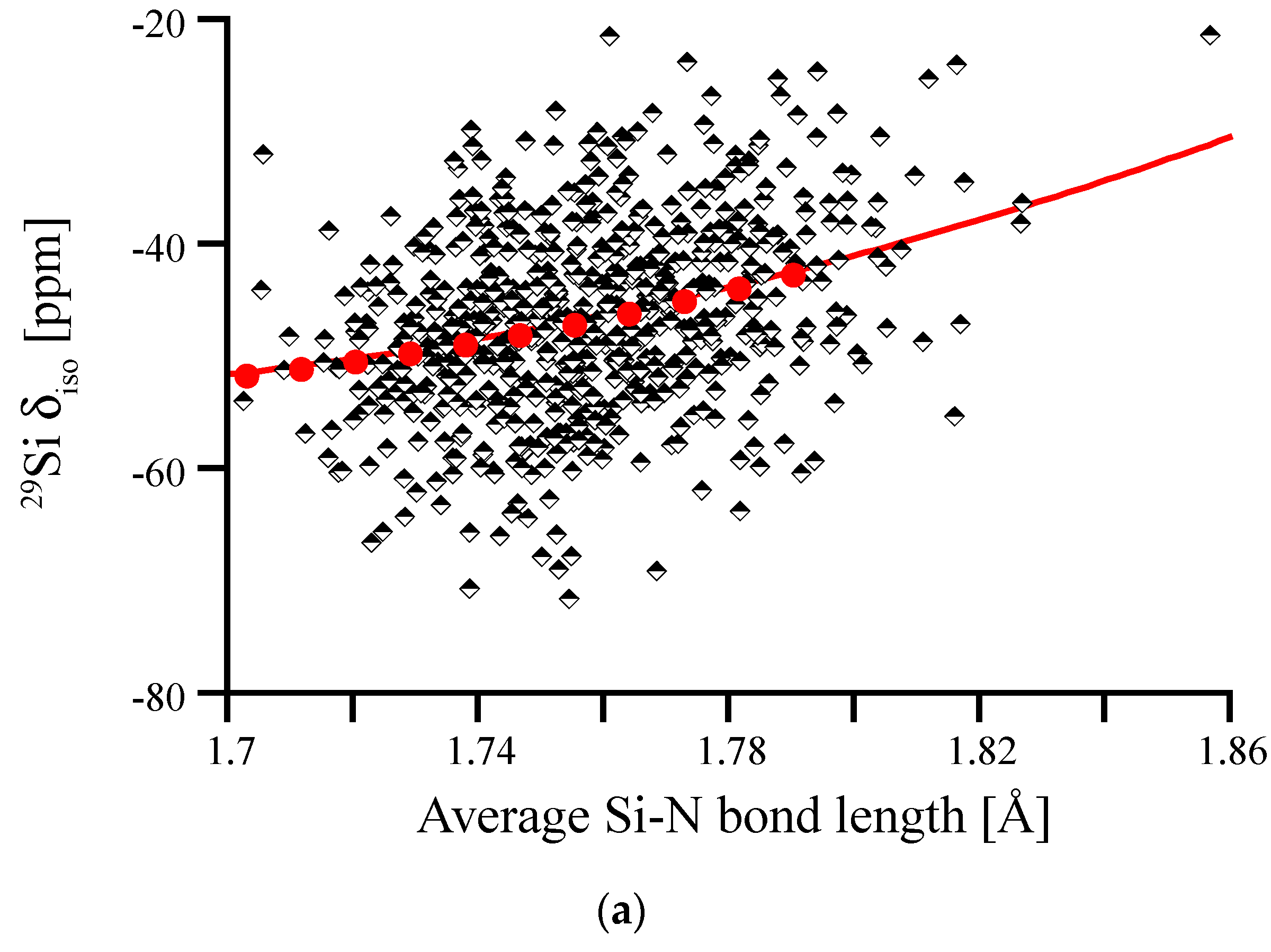
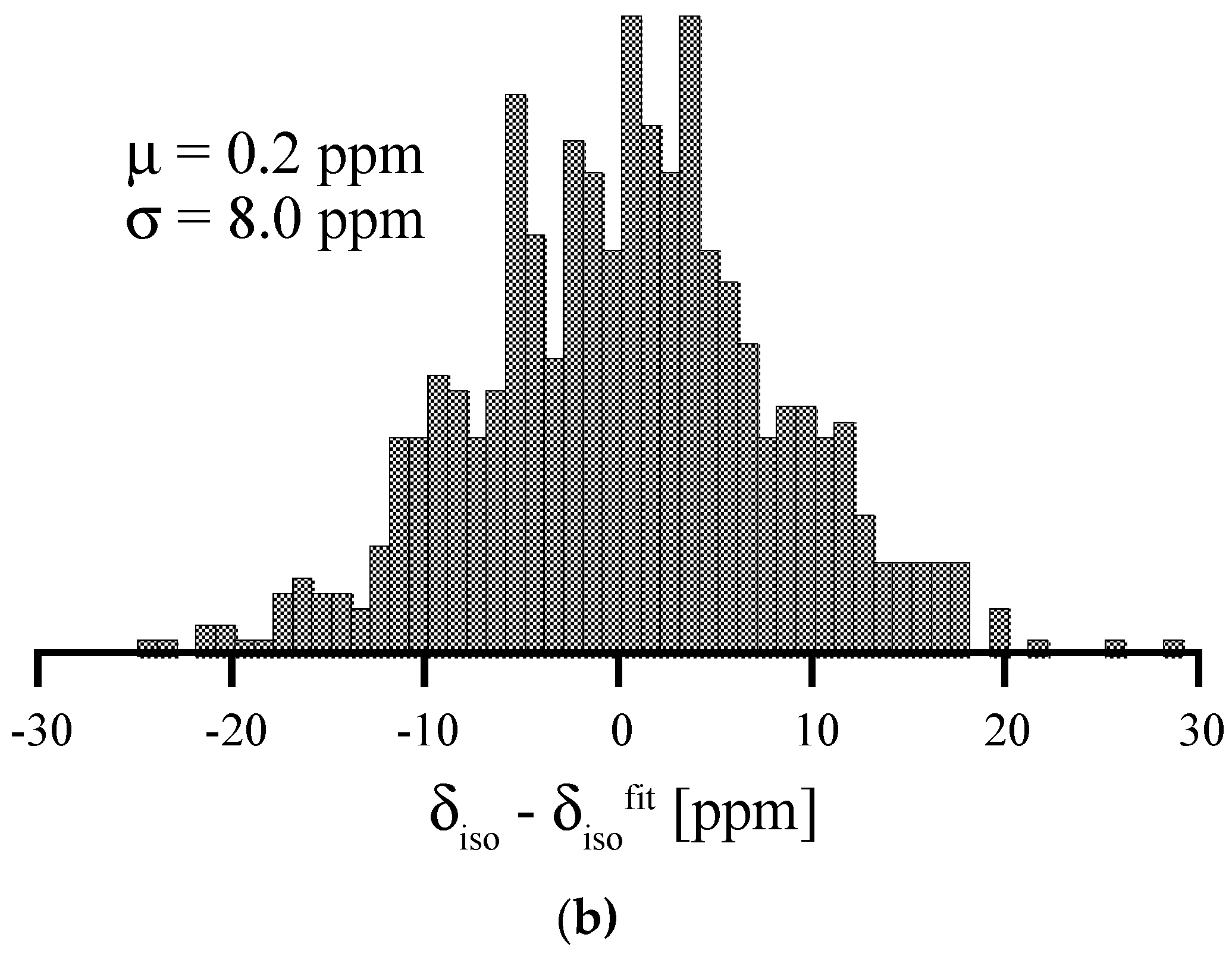
3.4. Amorphous Si3N4
3.5. Amorphous Hydrogenated Silicon Nitride
4. Summary and Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zerr, A.; Kempf, M.; Schwarz, M.; Kroke, E.; Goken, M.; Riedel, R. Elastic moduli and hardness of cubic silicon nitride. J. Am. Ceram. Soc. 2002, 85, 86–90. [Google Scholar] [CrossRef]
- Riley, F.L. Silicon nitride and related materials. J. Am. Ceram. Soc. 2000, 83, 245–265. [Google Scholar] [CrossRef]
- Hector, A.L. Synthesis and processing of silicon nitride and related materials using preceramic polymer and non-oxide sol-gel approaches. Coord. Chem. Rev. 2016, 323, 120–137. [Google Scholar] [CrossRef] [Green Version]
- Sardar, K.; Bounds, R.; Carravetta, M.; Cutts, G.; Hargreaves, J.S.J.; Hector, A.L.; Hriljac, J.A.; Levason, W.; Wilson, F. Sol-gel preparation of low oxygen content, high surface area silicon nitride and imidonitride materials. Dalton Trans. 2016, 45, 5765–5774. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Liu, H.T.; Cheng, H.F. Oxidation behavior of silicon nitride fibers obtained from polycarbosilane fibers via electron beam irradiation curing. RSC Adv. 2017, 7, 47833–47839. [Google Scholar] [CrossRef] [Green Version]
- Chung, Y.K.; Koo, J.H.; Kim, S.A.; Chi, E.O.; Cho, J.Y.; Sohn, W.B.; Kim, M.Y.; Park, C. Growth mechanism of Si3N4 nanowires from amorphous Si3N4 powders synthesized by low-temperature vapor-phase reaction. Crystengcomm 2016, 18, 3247–3255. [Google Scholar] [CrossRef]
- Charpentier, T.; Ispas, S.; Profeta, M.; Mauri, F.; Pickard, C.J. First-principles calculation of O-17, Si-29, and Na-23 NMR spectra of sodium silicate crystals and glasses. J. Phys. Chem. B 2004, 108, 4147–4161. [Google Scholar] [CrossRef]
- Charpentier, T.; Kroll, P.; Mauri, F. First-principles nuclear magnetic resonance structural analysis of vitreous silica. J. Phys. Chem. C 2009, 113, 7917–7929. [Google Scholar] [CrossRef]
- Ispas, S.; Charpentier, T.; Mauri, F.; Neuville, D.R. Structural properties of lithium and sodium tetrasilicate glasses: Molecular dynamics simulations versus NMR experimental and first-principles data. Solid State Sci. 2010, 12, 183–192. [Google Scholar] [CrossRef]
- Ponomarev, I.; Kroll, P. Impact of transition metal cations on the Si-29 NMR signal in metal oxide glasses: A dft case study of hafnia silica glass. J. Phys. Chem. C 2017, 121, 24152–24158. [Google Scholar] [CrossRef]
- Charpentier, T. The paw/gipaw approach for computing NMR parameters: A new dimension added to nmr study of solids. Solid State Nucl. Magn. Reson. 2011, 40, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Bonhomme, C.; Gervais, C.; Babonneau, F.; Coelho, C.; Pourpoint, F.; Azais, T.; Ashbrook, S.E.; Griffin, J.M.; Yates, J.R.; Mauri, F.; et al. First-principles calculation of NMR parameters using the gauge including projector augmented wave method: A chemist’s point of view. Chem. Rev. 2012, 112, 5733–5779. [Google Scholar] [CrossRef] [PubMed]
- Ferlat, G.; Charpentier, T.; Seitsonen, A.P.; Takada, A.; Lazzeri, M.; Cormier, L.; Calas, G.; Mauri, F. Boroxol rings in liquid and vitreous B2O3 from first principles. Phys. Rev. Lett. 2008, 101, 065504. [Google Scholar] [CrossRef] [PubMed]
- Pickard, C.J.; Mauri, F. All-electron magnetic response with pseudopotentials: NMR chemical shifts. Phys. Rev. B 2001, 63, 245101. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Inhomogeneous electron gas. Phys. Rev. B 1964, 136, B864. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab-initio molecular-dynamics simulation of the liquid-metal amorphous-semiconductor transition in germanium. Phys. Rev. B 1994, 49, 14251–14269. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmuller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Blochl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [Green Version]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Belsky, A.; Hellenbrandt, M.; Karen, V.L.; Luksch, P. New developments in the inorganic crystal structure database (icsd): Accessibility in support of materials research and design. Acta Crystallogr. B-Struct. Sci. 2002, 58, 364–369. [Google Scholar] [CrossRef]
- Toraya, H. Crystal structure refinement of alpha-Si3N4 using synchrotron radiation powder diffraction data: Unbiased refinement strategy. J. Appl. Crystallogr. 2000, 33, 95–102. [Google Scholar] [CrossRef]
- du Boulay, D.; Ishizawa, N.; Atake, T.; Streltsov, V.; Furuya, K.; Munakata, F. Synchrotron x-ray and ab initio studies of beta-Si3N4. Acta Crystallogr. B 2004, 60, 388–405. [Google Scholar] [CrossRef] [PubMed]
- Zerr, A.; Miehe, G.; Serghiou, G.; Schwarz, M.; Kroke, E.; Riedel, R.; Fuess, H.; Kroll, P.; Boehler, R. Synthesis of cubic silicon nitride. Nature 1999, 400, 340–342. [Google Scholar] [CrossRef]
- Schwarz, M.; Miehe, G.; Zerr, A.; Kroke, E.; Poe, B.T.; Fuess, H.; Riedel, R. Spinel-Si3N4: Multi-anvil press synthesis and structural refinement. Adv. Mater. 2000, 12, 883–887. [Google Scholar] [CrossRef]
- Stadler, F.; Schnick, W. The reduced nitridosilicate baSi6N8. Z. Anorg. Allg. Chem. 2007, 633, 589–592. [Google Scholar] [CrossRef]
- Pagano, S.; Zeuner, M.; Hug, S.; Schnick, W. Single-crystal structure determination and solid-state NMR investigations of lithium nitridosilicate Li2SiN2 synthesized by a precursor approach employing amorphous “Si(CN2)2”. Eur. J. Inorg. Chem. 2009, 1579–1584. [Google Scholar] [CrossRef]
- Stadler, F.; Oeckler, O.; Senker, J.; Hoppe, H.A.; Kroll, P.; Schnick, W. Sr Si6N8—A reduced nitridosilicate with a Si-Si bond. Angew. Chem. Int. Ed. 2005, 44, 567–570. [Google Scholar] [CrossRef] [PubMed]
- Carduner, K.R.; Blackwell, C.S.; Hammond, W.B.; Reidinger, F.; Hatfield, G.R. 29Si NMR characterization of alpha-silicon and beta-silicon nitride. J. Am. Chem. Soc. 1990, 112, 4676–4679. [Google Scholar] [CrossRef]
- Sekine, T.; Tansho, M.; Kanzaki, M. Si-29 magic-angle-spinning nuclear-magnetic-resonance study of spinel-type Si3N4. Appl. Phys. Lett. 2001, 78, 3050–3051. [Google Scholar] [CrossRef]
- Kroll, P. Modelling polymer-derived ceramics. J. Eur. Ceram. Soc. 2005, 25, 163–174. [Google Scholar] [CrossRef]
- Kroll, P. Structure and reactivity of amorphous silicon nitride investigated with density-functional methods. J. Non-Cryst. Solids 2001, 293, 238–243. [Google Scholar] [CrossRef]
- Kroll, P. A dft study of amorphous silicon oxynitride. J. Non-Cryst. Solids 2005, 351, 1127–1132. [Google Scholar] [CrossRef]
- Garofalini, S.H.; Luo, W.W. Molecular dynamics simulations of calcium silicate intergranular films between silicon nitride crystals. J. Am. Ceram. Soc. 2003, 86, 1741–1752. [Google Scholar] [CrossRef]
- Plimpton, S. Fast parallel algorithms for short-range molecular-dynamics. J. Comput. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef]
- Dasmahapatra, A.; Kroll, P. Modeling amorphous silicon nitride: A comparative study of empirical potentials. Comput. Mater. Sci. 2018, 148, 165–175. [Google Scholar] [CrossRef]
- Smith, J.V.; Blackwell, C.S. Nuclear magnetic-resonance of silica polymorphs. Nature 1983, 303, 223–225. [Google Scholar] [CrossRef]
- Bonhomme, C.; Gervais, C.; Coelho, C.; Pourpoint, F.; Azais, T.; Bonhomme-Coury, L.; Babonneau, F.; Jacob, G.; Ferrari, M.; Canet, D.; et al. New perspectives in the paw/gipaw approach: Jp-o-si coupling constants, antisymmetric parts of shift tensors and nqr predictions. Magn. Reson. Chem. 2010, 48, S86–S102. [Google Scholar] [CrossRef] [PubMed]
- Oestrike, R.; Yang, W.H.; Kirkpatrick, R.J.; Hervig, R.L.; Navrotsky, A.; Montez, B. High-resolution Na-23, Al-27, and Si-29 NMR-spectroscopy of framework aluminosilicate glasses. Geochim. Cosmochim. Acta 1987, 51, 2199–2209. [Google Scholar] [CrossRef]
- Mauri, F.; Pasquarello, A.; Pfrommer, B.G.; Yoon, Y.G.; Louie, S.G. Si-O-Si bond-angle distribution in vitreous silica from first-principles 29Si NMR analysis. Phys. Rev. B 2000, 62, R4786–R4789. [Google Scholar] [CrossRef]
- Nimmo, J.P.; Kroll, P. First-principles calculations and analysis of Si-29 nuclear magnetic resonance chemical shifts in silicon oxycarbide ceramics. J. Phys. Chem. C 2014, 118, 29952–29961. [Google Scholar] [CrossRef]
- Marumo, F.; Syono, Y. Crystal structure of Zn2SiO4-ii, a high-pressure phase of willemite. Acta Crystallogr. B-Struct. Sci. 1971, 27, 1868–1870. [Google Scholar] [CrossRef]
- Kroll, P. Pathways to metastable nitride structures. J. Solid State Chem. 2003, 176, 530–537. [Google Scholar] [CrossRef]
- Teter, D.M.; Hemley, R.J. Low-compressibility carbon nitrides. Science 1996, 271, 53–55. [Google Scholar] [CrossRef]
- Riedel, R.; Greiner, A.; Miehe, G.; Dressier, W.; Fuess, H.; Bill, J.; Aldinger, F. The first crystalline solids in the ternary Si-C-N system. Angew. Chem. Int. Ed. 1997, 36, 603–606. [Google Scholar] [CrossRef]
- Kroll, P.; Riedel, R.; Hoffmann, R. Silylated carbodiimides in molecular and extended structures. Phys. Rev. B 1999, 60, 3126–3139. [Google Scholar] [CrossRef]
- Kroll, P.; Andrade, M.; Yan, X.H.; Ionescu, E.; Miehe, G.; Riedel, R. Isotropic negative thermal expansion in beta-Si(NCN)2 and its origin. J. Phys. Chem. C 2012, 116, 526–531. [Google Scholar] [CrossRef]
- Riedel, R.; Horvath-Bordon, E.; Kroll, P.; Miehe, G.; Dzivenko, D.; Kleebe, H.J.; van Aken, P.A.; Lauterbach, S. Novel binary and ternary phases in the Si-C-N system. J. Ceram. Soc. J. 2008, 116, 674–680. [Google Scholar] [CrossRef] [Green Version]
- Pedone, A.; Charpentier, T.; Malavasi, G.; Menziani, M.C. New insights into the atomic structure of 45S5 bioglass by means of solid-state NMR spectroscopy and accurate first-principles simulations. Chem. Mater. 2010, 22, 5644–5652. [Google Scholar] [CrossRef]
- Carduner, K.R.; Carter, R.O.; Milberg, M.E.; Crosbie, G.M. Determination of phase-composition of silicon-nitride powders by Si-29 magic angle spinning nuclear-magnetic-resonance spectroscopy. Anal. Chem. 1987, 59, 2794–2797. [Google Scholar] [CrossRef]
- Yue, Y.; Li, D.H.; Ye, C.H. Characterization of laser synthesized silicon nitride powders with nanoscale by solid-state NMR. J. Mater. Sci. Lett. 1996, 15, 1079–1080. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Yokota, H.; Kojima, N.; Hayashi, S.; Kobayashi, T.; Sekine, T.; Sugahara, Y. Effects of structural differences in starting materials on the formation behavior of cubic silicon nitride by shock compression. J. Ceram. Soc. J. 2013, 121, 741–744. [Google Scholar] [CrossRef] [Green Version]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
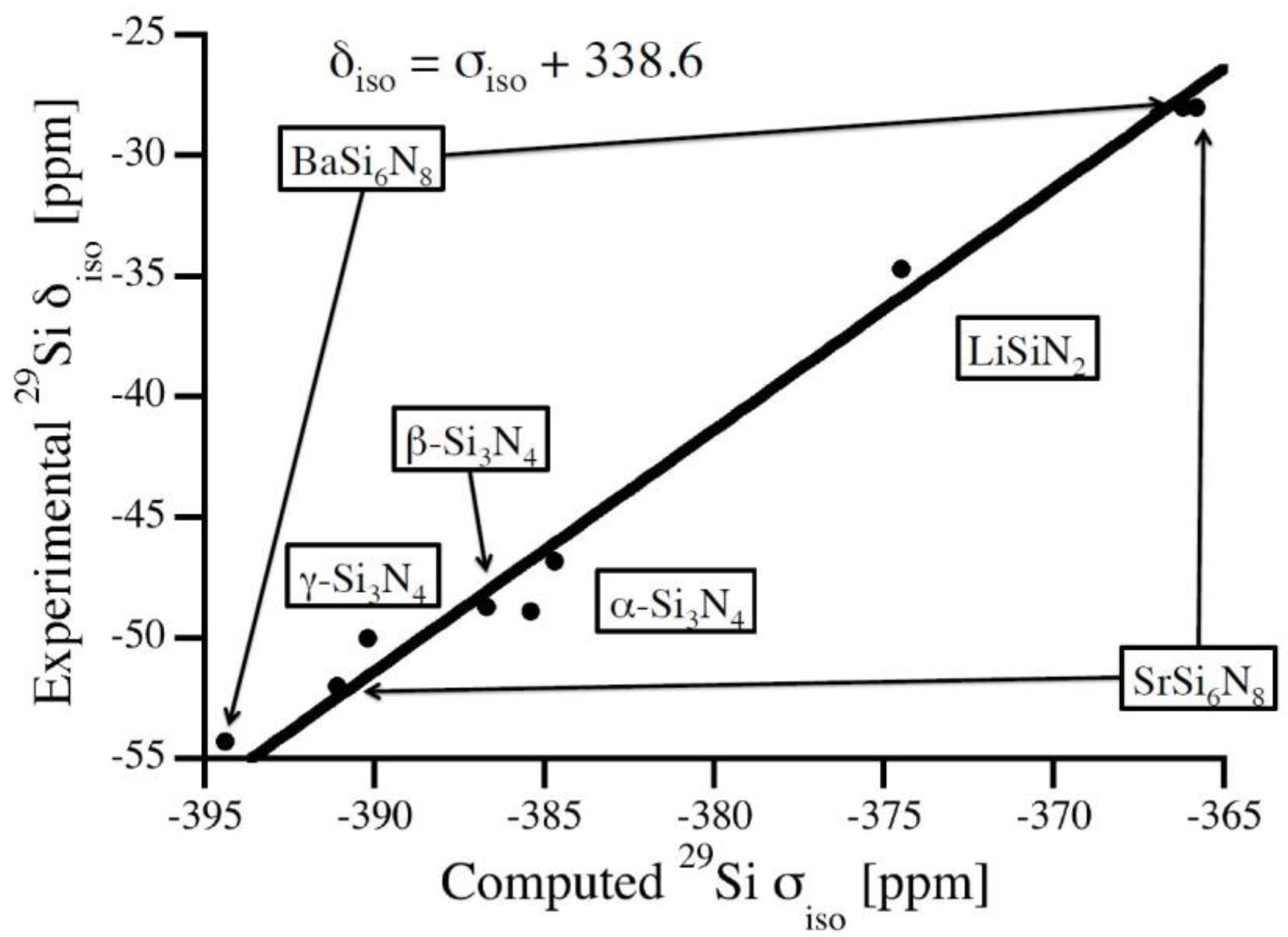
| Structure | Site | δisoexp (ppm) | σisocomp (ppm) | δisocomp (ppm) |
|---|---|---|---|---|
| BaSi6N8 [25] | 1 | −54.3 | −394.4 | −56.2 |
| 2 | −28.0 | −366.2 | −28.0 | |
| Li2SiN2 [26] | 1 | −34.7 | −374.5 | −36.3 |
| SrSi6N8 [27] | 1 | −52.0 | −391.1 | −52.9 |
| 2 | −28.0 | −365.8 | −27.6 | |
| α-Si3N4 [21,28] | 1 | −48.9 | −385.4 | −47.2 |
| 2 | −46.8 | −384.7 | −46.5 | |
| β-Si3N4 [22,28] | 1 | −48.7 | −386.7 | −48.5 |
| γ-Si3N4 [23,24,29] | 1 (Si[4]) | −50.0 | −390.2 | −52.0 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ponomarev, I.; Kroll, P. 29Si NMR Chemical Shifts in Crystalline and Amorphous Silicon Nitrides. Materials 2018, 11, 1646. https://doi.org/10.3390/ma11091646
Ponomarev I, Kroll P. 29Si NMR Chemical Shifts in Crystalline and Amorphous Silicon Nitrides. Materials. 2018; 11(9):1646. https://doi.org/10.3390/ma11091646
Chicago/Turabian StylePonomarev, Ilia, and Peter Kroll. 2018. "29Si NMR Chemical Shifts in Crystalline and Amorphous Silicon Nitrides" Materials 11, no. 9: 1646. https://doi.org/10.3390/ma11091646
APA StylePonomarev, I., & Kroll, P. (2018). 29Si NMR Chemical Shifts in Crystalline and Amorphous Silicon Nitrides. Materials, 11(9), 1646. https://doi.org/10.3390/ma11091646