Abstract
Polymethylsiloxane (PMS) and fumed silica, alone and in a blended form (1:1 w/w), differently pretreated, hydrated, and treated again, were studied using TEM and SEM, nitrogen adsorption–desorption, 1H MAS and 29Si CP/MAS NMR spectroscopy, infrared spectroscopy, and methods of quantum chemistry. Analysis of the effects of adding water (0–0.5 g of water per gram of solids) to the blends while they are undergoing different mechanical treatment (stirring with weak (~1–2 kg/cm2) and strong (~20 kg/cm2) loading) show that both dry and wetted PMS (as a soft material) can be grafted onto a silica surface, even with weak mechanical loading, and enhanced mechanical loading leads to enhanced homogenization of the blends. The main evidence of this effect is strong nonadditive changes in the textural characteristics, which are 2–3 times smaller than additive those expected. All PMS/nanosilica blends, demonstrating a good distribution of nanosilica nanoparticles and their small aggregates in the polymer matrix (according to TEM and SEM images), are rather meso/microporous, with the main pore-size distribution peaks at R > 10 nm in radius and average <RV> values of 18–25 nm. The contributions of nanopores (R < 1 nm), mesopores (1 nm < R < 25 nm), and macropores (25 nm < R < 100 nm), which are of importance for studied medical sorbents and drug carriers, depend strongly on the types of the materials and treatments, as well the amounts of water added. The developed technique (based on small additions of water and controlled mechanical loading) allows one to significantly change the morphological and textural characteristics of fumed silica (hydrocompaction), PMS (drying–wetting–drying), and PMS/A-300 blends (wetting–drying under mechanical loading), which is of importance from a practical point of view.
1. Introduction
Silicas can easily be modified to significantly change many important characteristics of the materials [1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16]. Various functionalized silicas (fumed nanosilica, silica gels, precipitated silicas, ordered mesoporous silicas, etc.) and siloxane-based polymers (e.g., linear polydimethylsiloxane, PDMS, 3D-cross-linked polymethylsiloxane (PMS)) are widely used in industry and medicine as well in various branches of science [1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23]. One of the main characteristics of these materials is their degree of hydrophobicity, which allows their interactions with various liquids, polymers, and solids to be controlled. PMS, as a 3D-cross-linked polymer with one CH3 group attached to each Si atom and residual silanols [21,22,23], may be considered as an intermediate between PDMS and methylated nanosilica containing residual silanols [1,2,24,25,26,27,28]. In a series of nanostructured silicas with surface functionalities, such as –O)3SiCH3, –O)2Si(CH3)2, and –O–Si(CH3)3, the degree of hydrophobicity increases, i.e., PMS is less hydrophobic than PDMS. There is a commercial hydrogel with PMS incompletely cross-linked and containing a certain amount of residual silanols (a medicinal sorbent entitled Enterosgel, it contains 7–8 wt.% of PMS and 93–92 wt.% of water, is produced by Kreoma-Pharm, Ukraine, and it is also used as a drug carrier), which represents a gel-like soft material [29,30]. However, pure PDMS is hydrophobic (there are no silanols, and the siloxane bonds tend to be low-polar bonds) and cannot form hydrogels. Note that the Si–O–Si bonds are relatively flexible due to small changes in the energy which follow upon changes in the ∠SiOSi angle in the 140 to 180° range [1,2]. Amorphous silicas are therefore relatively stable despite certain differences in 3–8-member siloxane rings (characterized various values of ∠SiOSi) formed upon the synthesis. Clear, PMS is less flexible than PDMS, but PMS is soft because only three bonds from four ones of each Si atom can take part in cross-linking [29,30].
Clear, the properties and characteristic of any nanostructured composites strongly depend on the distributions (uniform – nonuniform, nanolayers – nanoclusters – nanodomains – microdomains – monolith fragments) of different components in the composites. These effects could be stronger (especially upon interaction with water [31,32,33,34,35,36,37]) for the composites with nanostructured hydrophilic and hydrophobic components [38,39]. Using specific treatments and certain (rather small) amounts of water, it is possible to prepare hydrophilic blends with hydrophobic and hydrophilic components. Subsequent treatments (e.g., drying) of these blends allow one to obtain hydrophobic composites, the hydrophilic properties of which can renew after additional treatment (e.g., wetting and stirring or grinding at greater mechanical loading) [38,39].
Fumed nanosilica and Enterosgel are used in medical applications not only as sorbents but also as drug carriers [17,29,30,40]. Therefore, changes in the textural characteristics of the drug carriers can strongly affect the drug release upon the medical applications of the drug delivery systems. The textural and hydrophilic/hydrophobic characteristics of the blends of nanosilica and PMS could be strongly changed and better controlled than those of individual sorbents due to their morphological, textural, and structural features.
Fumed nanosilica and PMS are soft-powder materials because their aggregates of nanoparticles and agglomerates of aggregates may be easily rearranged under any treatment [36]. The results of these treatments depend on several factors such as the (i) hydrational, thermal, and mechanical history of the components; (ii) the weight ratio of the components; (iii) the amounts of added water; (iv) the mechanical loading; (v) the time of treatment; and (vi) the temperature [1,2,24,25,26,36,37,38,39,40,41]. In this study, two important factors, namely, a small amount of added water and the type of mechanical loading upon stirring of the PMS/nanosilica blends, are analyzed to control the characteristics of the final blends.
2. Materials and Methods
2.1. Materials
Commercial polymethylsiloxane (PMS) hydrogel (Enterosgel, hPMS, Kreoma-Pharm, Ukraine, ~7–8 wt.% of PMS and 93–92 wt.% of water) and dry PMS (dPMS) were used as the initial materials. Note that after drying of Enterosgel at room temperature for a week, the amount of residual water bound in PMS is small (~0.7 wt.%), i.e., the material becomes rather hydrophobic. Fumed silica (nanosilica) A-300 (Pilot plant of Chuiko Institute of Surface Chemistry, Kalush, Ukraine) was mixed with water (1:5) and dried at 160 °C for several hours, resulting in hydrocompacted nanosilica (cA-300) [41], with increased bulk density (ρb) toward 0.25 g/cm3, since the initial A-300 has ρb ≈ 0.05 g/cm3.
Dry PMS (dPMS) and dry cA-300 (1:1 w/w) powders were mixed in a porcelain mortar for 5 min without strong mechanical loading (sample 1, Bl). Distilled water (h = 0.1 g per gram of solids) was then added and the blend was stirred without (5 min, sample 2, Bh1l) or with (10 min, sample 3, Bh1s) strong mechanical loading. Water (h = 0.2 g/g) was added to S1, and the sample was stirred without strong mechanical loading (sample 4, Bh2l). Bh2l was additionally stirred with strong mechanical loading for 10 min (sample 5, Bh2s). Sample 6 (Adl) corresponds to dried and stirred cA-300 (Adl). Sample 7 (Pdl) corresponds to stirred dPMS. Sample 8 (Phdl) is hPMS-dried for a week (in sample labels: A is cA-300, B is the blend of dPMS and cA-300, l is low mechanical loading (~1 kg/cm2) upon simple mixing of samples, s is strong mechanical loading (~20 kg/cm2) upon mixing, d is dried samples, and h is hydrated samples at h = 0.1 g/g (h1) or 0.2 g/g (h2)).
2.2. Transmission (TEM) and Scanning (SEM) Electron Microscopy
TEM (TECNAI G2 F30 microscope (FEI–Philips, Amsterdam, The Netherlands), operating voltage 300 kV) was used to analyze the particulate morphology of samples (Figure 1 and Figures S1 and S2 in Electronic Supplementary Material (ESM) file). The powder samples were added to acetone (chromatographic grade) and sonicated. A suspension drop was then deposited onto a copper grid covered by a thin carbon film. After acetone evaporation, the dry sample remaining on the film was studied.
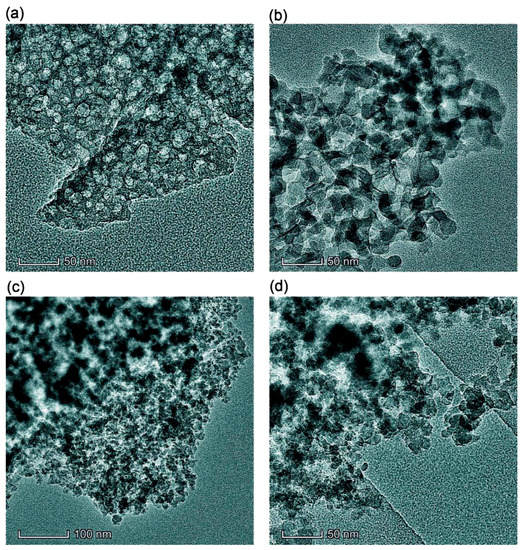
Figure 1.
TEM images of samples (a) Bl, (b) Bh2s, (c) Adl, and (d) Pdl (scale bar 50 nm (a, b, d) and 100 nm (c)).
SEM (FE–SEM, Hitachi S–4700, Tokyo, Japan, operating voltage of 15 kV, and magnification of ×5000–100000) (Figure 2, Figures S3 and S4) was used to analyze the morphological features of the dried powder samples.

Figure 2.
SEM images of (a) Bl, (b) Bh2s, (c) Adl, and (d) Pdl (scale bar 500 nm).
2.3. Textural Characteristics
To analyze the textural characteristics of individual and mixed samples degassed at 110 °C for 12 h (Table 1), low-temperature (77.4 K) nitrogen adsorption–desorption isotherms (Figure S5 in ESM file) were recorded using a Micromeritics ASAP 2460 adsorption analyzer (Micromeritics, Norcross, GA, USA). The specific surface area (Table 1, SBET) was calculated according to the standard BET method at 0.05 < p/p0 < 0.3 (using Micromeritics software), where p and p0 denote the equilibrium and saturation pressure of nitrogen at 77.4 K, respectively [42]. The total pore volume (Table 1, Vp) was estimated from the nitrogen adsorption at p/p0 ≈ 0.98-0.99 [43]. The nitrogen desorption data were used to compute the pore-size distributions (PSD) (differential fV(R) ~ dVp/dR and fS(R) ~ dS/dR) using a self-consistent regularization (SCR) procedure under non-negativity condition (fV(R) ≥ 0 at any pore radius R) at a fixed regularization parameter α = 0.01 (Figure 3) [44]. A complex pore model including slit-shaped (S) and cylindrical (C) pores and voids (V) between spherical particles packed in random aggregates (SCV/SCR method) was applied [44]. The differential PSD with respect to the pore volume fV(R) ~ dV/dR, ∫fV(R)dR ~ Vp were recalculated to incremental PSD (IPSD) at ΦV(Ri) = (fV(Ri+1) + fV(Ri))(Ri+1 − Ri)/2 at ∑ ΦV(Ri) = Vp) for a better view of the PSD at large R values. The fV(R) and fS(R) functions were used to calculate contributions of nanopores (Vnano and Snano at 0.35 nm < R < 1 nm), mesopores (Vmeso and Smeso at 1 nm < R < 25 nm), and macropores (Vmacro and Smacro at 25 nm < R < 100 nm) [44]. The average values of the pore radii (Table 1) were determined with respect to the pore volume (X = V) and specific surface area (X = S) as the corresponding moments of the distribution functions:

Table 1.
Textural characteristics of PMS, A-300, and their blends (SCV/SCR method).
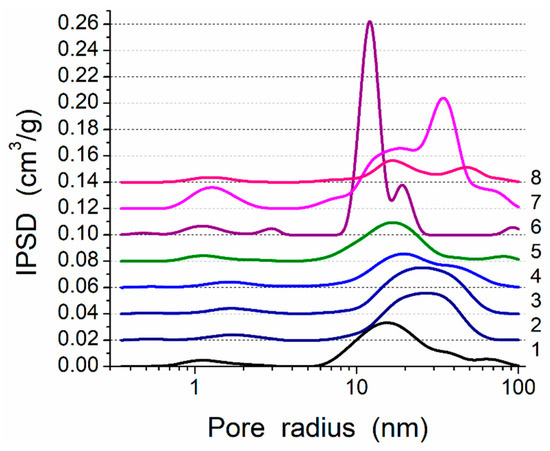
Figure 3.
Incremental pore-size distributions (PSD) (SCV/SCR method) for PMS, A-300 and their blends (curve numbers correspond to sample numbers in Table 1).
Additionally, the nonlocal density functional theory (NLDFT, Quantachrome software (Quantachrome Instruments, Boynton Beach, FL, USA), with a model of cylindrical pores in silica [45]) method was used to calculate the differential PSD (Figure S6).
The main error in the evaluation of the textural characteristics such as the SBET value based on the nitrogen adsorption isotherms is due to variation in the area (σ) occupied by nitrogen molecules. It is assumed that σ = 0.162 nm2. This value is correct for graphite, but in the case of silica, it is smaller [46] due nonparallel location of N2 molecules with respect to the surface plane. Therefore, overestimation of the SBET value could be 16% for silica. Despite this fact, in firm software σ = 0.162 nm2, and for simplicity, one can use this value because the effective σ value for various materials is different and unknown.
2.4. 1H MAS and 29Si CP/MAS NMR Spectroscopy
Solid-state 1H MAS NMR spectra (Figure 4a) were recorded using an Agilent DD2 600 MHz NMR spectrometer (Agilent, Santa Clara, CA, USA, magnetic field strength 14.157 T). A powder sample was placed in a pencil-type zirconia rotor of 4.0 mm o.d. The spectra were recorded at a spinning speed of 8 kHz with a recycle delay of 5 s. The adamantane was used as the reference of the 1H chemical shift of proton resonance (δH).
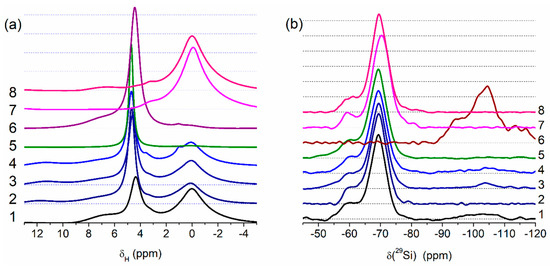
Figure 4.
Solid state NMR spectra of (a) 1H (MAS) and (b) 29Si (CP/MAS) for PMS, A-300 and their blends (curve numbers correspond to sample numbers) (Qn corresponds to Si(OH)4−n(OSi≡)n at n = 2 (−91~−93 ppm), 3 (−99~−102 ppm), and 4 (−109~−111 ppm); T3 (−70 ppm) corresponds to (≡SiO)3SiCH3) and T2 (−60 ppm) corresponds to (≡SiO)2Si(OH)CH3), see Table S1).
Solid-state 29Si CP/MAS NMR spectra (Figure 4b) were recorded using the same NMR spectrometer at a resonance frequency of 199.13 MHz for 29Si using the cross-polarization (CP), magic-angle spinning (MAS), and high-power 1H decoupling. The spectra were recorded at a spinning speed of 8 kHz (4 µs 90° pulses), 2 ms CP pulse, and a recycle delay of 3 s. The Si signal of tetramethylsilane (TMS) at 0 ppm was used as the reference of the 29Si chemical shift (δ(29Si)) [2,47].
2.5. Infrared Spectroscopy
The infrared (IR) spectra (Figure 5 and Figure S7) were recorded in the range of 1800 to 300 cm−1 using a Specord M80 (Carl Zeiss AG, Oberkochen, Germany, 4 cm−1 step, integration time of 3 s, the transmission spectra were converted into absorbance ones). Samples were carefully ground (5 min) with KBr (Sigma-Aldrich, Saint Louis, MO, USA for spectroscopy) as 1:400 and pressed into thin pellets.
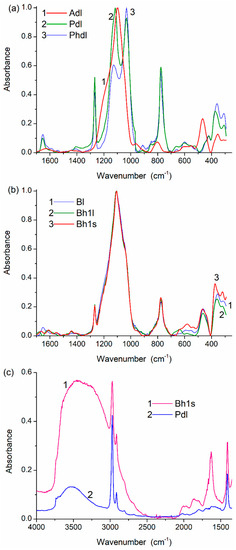
Figure 5.
IR spectra of (a,c) PMS, A-300 and (b,c) their blends in the range of (a,b) 1700–300 cm−1 and (c) 4000–1300 cm−1.
3. Results and Discussion
Simple stirring (without strong mechanical loading) of the dry PMS and dry cA-300 powders (Bl) results in good distribution of small nanosilica aggregates in the composite (Figure 1a, Figure 2a, Figures S1a, S3a and S4).
This composite is characterized by strong interactions between the PMS and silica nanoparticles because significant nonadditivity is observed for the textural characteristics of Bl in comparison to those of cA-300 (sample Adl) and dPMS (sample Pdl) (Table 1, Figure 3, Figures S5 and S6): SBET = 186 m2/g instead of the expected (SBET,S6 + SBET,S7)/2 = 365 m2/g and Vp = 0.788 cm3/g instead of the expected 1.556 cm3/g. This result could be explained by the embedding of small aggregates of silica nanoparticles into soft PMS (as light structures observed in larger PMS structures, Figure 1a and Figure S1a,b), and the grafting of PMS onto solid silica particles (similar to butter grafting onto macroporous bread resulting in a flat nonporous surface). If the silica aggregates could be remained at a surface of PMS particles that the strong decrease in the textural characteristics could be absent. The treatment changes the PSD in the total pore size range (Table 1, Snano, Vnano, Smeso, Vmeso, Smacro, and Vmacro); and the contributions of pores of different shapes change too (Table 1, cslit, ccyl, and cvoid) in comparison to those of dry components (Adl and Pdl). A certain compaction of the blend is observed as a diminution of the <Rv> value (Table 1).
The nitrogen adsorption isotherms (Figure S5) correspond to type II according to the IUPAC classification [48]. The desorption hysteresis shape provides clear evidence for formation of constricted, textural mesopores in sample 6 (various voids between particles). However, this is less visible for other samples having a simple shape of the hysteresis loops. The fact that the hysteresis loop for sample 7 (dPMS) does not close, probably indicates irreversible swelling of this material during nitrogen adsorption step, as well the presence of long pores with a narrow throat formed upon drying of the PMS hydrogel that results in additional cross-linking of the polymers due to condensation of residual silanols.
The addition of a small amount of water (h = 0.1 g/g) to the dry blend and stirring without strong mechanical loading results in slightly better homogenization (better embedding of A-300 aggregates into PMS and better PMS grafting onto silica) of the blend (Figures S1b and S3b), because water can play the role of a lubricant thus reducing the interactions of silica–silica and PMS–PMS. However, silica aggregates are visible in Bh1l as in Bl (Figure 1 and Figure S1).
There are certain changes in the textural characteristics of the Bh1l blend compared to Bl (Table 1, Figure 3, Figures S5 and S6), since both nanoporosity and mesoporosity decrease, but macroporosity increases due to reorganization of the secondary particles.
An increase in mechanical loading (Bh1s) leads to significant homogenization of the blend (Figure S1c), since silica aggregates (light structures) are not clearly visible as in Bl and Bh1l (Figure 1 and Figure S1a,b). This can be explained by decomposition of the aggregates upon stirring and stronger embedding of individual silica nanoparticles into the PMS matrix. There is a certain decrease in the nanoporosity (Table 1, Snano and Vnano) as well an increase in the macroporosity (Smacro, Vmacro, and <Rv>). The contributions of pores of different shapes slightly change (Table 1, cslit, ccyl, and cvoid).
An increase in the amount of added water (h = 0.2 g/g) without strong mechanical loading (Bh2l) results in decreased porosity (water can fill the narrow pores and merge the particles together upon drying and degassing) (Table 1, Figure 3, Figures S5 and S6). However, the morphological changes are small (see Figures S1b,d, S2, S3b,d and S4) in comparison to Bh1l. Quantitative analysis of SEM images of samples Bl and Bh2s as representatives (Figure S4) shows that the mechanical treatment of a wetted blend results in decomposition of aggregates. This is appropriate for compaction of the blend observed in the textural characteristics (Table 1).
Enhanced mechanical loading (Bh2s) results in a strong diminution of the textural characteristics (Table 1), and the contributions of pores of different shapes change in comparison to Bh2l. Thus, upon stronger mechanical loading, the embedding-grafting effects increase in parallel to decomposition of silica aggregates, which are not visible similarly to Bh1s, but in contrast to Bl, Bh1l, and Bh2l.
Treatment of the hydrogel hPMS (drying, degassing) can give (Phdl) very strong compaction of the system (Table 1, Figures S5 and S6). However, this compaction occurs in domains (aggregates) because the relative contributions of the macropores (Smacro/SBET and Vmacro/Vp) are maximal for Phdl (this process is similar to the strong drying of wet soil with the appearance of checks).
The results of the treatment processes appear in the solid-state NMR (Figure 4, Table S1) and IR (Figure 5) spectra. Treatment with the addition of water (resulting in better PMS grafting onto silica surface) reduces the intensity of the Q2–Q4 lines, well observed for Adl (cA-300) (Table S1, Figure 4b).
The NMR spectra of water (δH = 4–5 ppm, Figure 4a) bound to PMS and the residual SiOH groups attached to =Si(CH3) (T2 lines in Figure 4b) depend on the type of samples and their treatment conditions. The amounts of water bound to dry PMS (samples Pdl and Phdl, Figure 4a) are relatively small because the amounts of residual SiOH groups (Figure 4b, T2 at −60 ppm), which are the main adsorption sites for bound water, are relatively small (Table S1). The 1H MAS NMR spectrum of cA-300 (Adl) demonstrates three lines at δH ≈ 7 ppm (strongly disturbed silanols), δH ≈ 4–5 ppm (water and disturbed silanols), and 0–3 ppm (free silanols). Interactions of PMS with a silica surface lead to diminution of relative content of strongly disturbed silanols (e.g., for Bh2s, only one signal is observed at 4–5 ppm). For sample Bh2s, the lines of the CH3 groups (Figure 4a) and Q2–Q4 (Figure 4b) lines are not observed.
The spectral changes and differences for the samples studied are observed in the IR spectra (Figure 5 and Figure S6). For example, samples of dry PMS (Pdl and Phdl) are characterized by practically identical bands of the symmetrical Si–O stretching vibrations at 800–700 cm−1 (Figure 5a). However, the asymmetrical Si–O stretching vibrations are at 1200–1000 cm−1 (especially at 1130–1115 cm−1) due to the difference in the cross-linking degree (compaction), the relative numbers of ≡SiO)3SiCH3 (T3, Figure 4b, Table S1) and (≡SiO)2Si(OH)CH3 (T2), and the compaction type on drying (stronger check effects on drying of hPMS, Table 1). Water is stronger bound and in a larger amount in the blend than in dPMS alone (Figure 5c). The stirring effects more weakly appear in the IR spectra (Figure 5b) than in the textural characteristics or NMR spectra, because the IR spectra are transmission spectra of the total samples. The PMS-onto-silica grafting/embedding effect is therefore difficult to observe in the IR spectra.
Theoretical modeling (see Figures S8–S11 in ESM file) shows that PMS particles may be hydrated due to the presence of residual silanol groups appearing in the 29Si CP/MAS NMR spectra (Figure 4b) as a line at −60 ppm. This effect explains the good distribution of hydrated silica nanoparticles in the hydrated blends after significant mechanical loading (Figure 1 and Figures S2–S4). Note that the use of relatively small amounts of water (h = 0.1 or 0.2 g/g) can prevent the formation of separate phases of PMS and A-300 upon mechanical treatment of the blends.
4. Conclusions
The effects of adding amounts of water (0–0.5 g of water per gram of dry solids) to the polymethylsiloxane/fumed silica blends (1:1 w/w) undergoing different mechanical treatment (stirring with weak (~1–2 kg/cm2) and strong (~20 kg/cm2) loading) were analyzed using TEM and SEM images, nitrogen adsorption–desorption isotherms (pore size distribution and other textural characteristics), solid-state NMR (1H MAS and 29Si CP/MAS) spectra, and infrared spectra in the range of 1700 to 300 cm−1 (related to the Si–O stretching vibrations). Both dry and wetted PMS (with 3D cross-linking in the particles containing residual SiOH and CH3 group attached to each Si atoms) is a soft material, which can be grafted onto a silica surface, and silica aggregates can be embedded into the PMS matrix even upon weak mechanical loading. The main evidence of this effect is strongly nonadditive changes in the textural characteristics, which become two to three times smaller than additive those expected. All the PMS/nanosilica blends, demonstrating a good distribution of nanosilica nanoparticles and their small aggregates in the polymer matrix, are rather meso/microporous, with the main PSD peaks at R > 10 nm and average <RV> values of 18-25 nm. Contributions of nanopores (R < 1 nm), mesopores (1 nm < R < 25 nm), and macropores (25 nm < R < 100 nm) depend strongly on the type of the materials (A-300, cA-300, dPMS, dried hPMS, and dPMS/cA-300 blends dried or wetted) and the type of treatment and amounts of added water. Thus, the developed technique allows one to significantly change the morphological and textural characteristics of fumed silica (hydrocompaction), PMS (drying–wetting–drying), and PMS/A-300 blends (wetting–drying under controlled mechanical loading), something that is of importance from a practical point of view.
Supplementary Materials
The following are available online at https://www.mdpi.com/1996-1944/12/15/2409/s1. Figure S1: TEM images of (a) Bl, (b) Bh1l, (c) Bh1s, (d) Bh2l, (e) Bh2s, (f) Adl, (g) Pdl, and (h) Phdl (scale bar 100 nm (a–d,h) and 200 nm (e,f)), Figure S2: TEM image of Bh2l, Figure S3: SEM images of Bl, (b) Bh1l, (c) Bh1s, (d) Bh2l, (e) Bh2s, (f) Adl, (g) Pdl, and (h) Phdl (scale bar 1 μm), Figure S4: Particle size distributions (PaSD) of samples 1 (Bl) and 5 (Bh2s) calculated using SEM images (Figure S3) and Fiji software with local thickness plugin (https://imagej.net/Fiji), Figure S5: Nitrogen adsorption-desorption isotherms (77.4 K) for PMS, cA-300, and their blends (curve numbers correspond to sample numbers of in Table S1), Figure S6: NLDFT PSD (equilibrium model with cylindrical pores in silica) for PMS, cA-300, and their blends (curve numbers correspond to sample numbers of Table S1), Table S1: Contributions of various structures in PMS, cA-300, and their blends, Figure S7: IR spectra (in the range of 1700–300 cm−1) of dPMS/cA-300 blends differently hydrated before strong mechanical treatment (thin pallets were pressed using samples stirred with KBr as 1:400), Figure S8: Model of hydrated PMS nanoparticle (PM7 method), Figure S9: Model of hydrated silica nanoparticle (PM7 method), Figure S10: Model of two hydrated silica nanoparticles as a simple aggregate (PM7 method), Figure S11: Model 1H NMR spectra (PM7 + correlation function) of hydrated PMS (curve 1) and two nanosilica particles (curve 2); and experimental 1H NMR spectra of static PMS sample (curve 3) and nanosilica (curve 4, 1H MAS NMR).
Author Contributions
T.V.K., I.P., E.M.P., V.M.G., and V.V.T. conceived and designed the experiments; T.V.K., I.P., and D.Z. performed all the experiments; I.P., V.M.G., and V.V.T. analyzed and interpreted the data; I.P., V.G., and V.V.T. wrote the manuscript; Z.L. and W.D. contributed reagents/materials/analysis tools; all the authors revised and approved the final version of the manuscript.
Funding
I.P. is grateful to China Postdoctoral Science Foundation (grant No Z741020001), and Z.L. is grateful to Special Funding of the ‘Belt and Road’ International Cooperation of Zhejiang Province (grant No 2015C04005) for financial support.
Availability of Data and Materials
The datasets supporting the conclusions of this work are included within the article. Any raw data generated and/or analyzed in the present study are available from corresponding author on request.
Conflicts of Interest
The authors declare that they have no competing interests.
Notations
| Abbreviation | Full Name |
| SiO2 | Silica |
| PDMS | Poly(dimethylsiloxane) |
| PMS | Polymethylsiloxane |
| SBET | Surface area |
| δ | Chemical shift |
| CP/MAS NMR | Cross Polarization Magic-Angle Spinning Nuclear Magnetic Resonance |
| hPMS | hydrated polymethylsiloxane |
| dPMS | dehydrated polymethylsiloxane |
| ρb | bulk density |
| Bl | dPMS and dry cA-300 (1:1 w/w) mixed without strong mechanical loading |
| Bh1l | dPMS and dry cA-300 (1:1 w/w) mixed with an addition of distilled water without strong mechanical loading |
| Bh1s | dPMS and dry cA-300 (1:1 w/w) mixed with an addition of distilled water with strong mechanical loading |
| Bh2l | Water (h = 0.2 g/g) was added to S1 sample; and the sample was stirred without strong mechanical loading |
| Bh2s | Bh2l was additionally stirred with strong mechanical loading for 10 min; Adl, dried and stirred cA-300 |
| Pdl | Stirred dehydrated PMS |
| R | Pore radius |
| <RV> and <RS> | The average pore radii with respect to the pore volume and specific surface area, respectively |
| PSD | Pore size distributions |
| Vp | Pore volume |
| Vnano | Volume of nanopores (R < 1 nm) |
| Vmeso | Volume of mesopores (1 nm < R < 25 nm) |
| Vmacro | volume of macropores (25 nm < R < 100 nm) |
| Snano | Specific surface area of nanopores (R < 1 nm) |
| Smeso | Specific surface area of mesopores (1 nm < R < 25 nm) |
| Smacro | Specific surface area of macropores (25 nm < R < 100 nm) |
| SDFT/cyl | NLDFT specific surface area with a model of cylindrical pores in silica |
| cslit, ccyl, and cvoid | contributions of slit-shaped, cylindrical pores and voids between nonporous nanoparticles, the weight constants calculated using the SCV/SCR method |
| NLDFT | nonlocal density functional theory |
| TEM and SEM | Transmission and scanning electron microscopy; IR, infrared spectroscopy. |
References
- Iler, R.K. The Chemistry of Silica: Solubility, Polymerization, Colloid and Surface Properties and Biochemistry of Silica; Wiley: Chichester, UK, 1979; ISBN 978-0-471-02404-0. [Google Scholar]
- Legrand, A.P. The Surface Properties of Silicas, 1st ed.; Wiley: New York, NY, USA, 1998; ISBN 978-0471953326. [Google Scholar]
- Auner, N.; Weis, J. (Eds.) Oganosilicon Chemistry VI: From Molecules to Materials, 1st ed.; Wiley-VCH: Weinheim, Germany, 2005; ISBN 978-3527312146. [Google Scholar]
- Protsak, I.S.; Tertykh, V.A.; Pakhlov, E.M.; Derylo-Marczewska, A. Modification of fumed silica surface with mixtures of polyorganosiloxanes and dialkyl carbonates. Prog. Org. Coat. 2017, 106, 163–169. [Google Scholar] [CrossRef]
- Rao, A.V.; Kulkarni, M.; Amalnerkar, D.P.; Seth, T. Surface chemical modification of silica aerogels using various alkyl-alkoxy/chloro silanes. Appl. Surf. Sci. 2003, 206, 262–270. [Google Scholar] [CrossRef]
- Park, S.E.; Prasetyanto, E.A. Morphosynthesis and Catalysis by Organofunctionalized Mesoporous Materials. In Organosilanes Properties Performance and Applications; Wyman, E.B., Skief, M.C., Eds.; Nova Science Publishers: New York, NY, USA, 2010; pp. 101–131. ISBN 978-1-60876-452-5. [Google Scholar]
- Daoud, W.A.; Xin, J.H.; Xiaoming, T. Synthesis and characterization of hydrophobic silica nanocomposites. Appl. Surf. Sci. 2006, 252, 5368–5371. [Google Scholar] [CrossRef]
- Bernardoni, F.; Kouba, M.; Fadeev, A.Y. Effect of curvature on the packing and ordering of organosilane monolayers supported on solids. Chem. Mater. 2008, 20, 382–387. [Google Scholar] [CrossRef]
- Moitra, N.; Ichii, S.; Kamei, T.; Kanamori, K.; Zhu, Y.; Takeda, K.; Nakanishi, K.; Shimada, T. Surface Functionalization of Silica by Si–H Activation of Hydrosilanes. J. Am. Chem. Soc. 2014, 136, 11570–11573. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-F.; Xia, Y.-X.; Xu, D.-P.; Li, G.-L. Surface Reaction of Particulate Silica with Polydimethylsiloxanes. J. Polym. Sci. 1981, 19, 3069–3079. [Google Scholar] [CrossRef]
- Xiao, D.; Zhang, H.; Wirth, M. Chemical Modification of the Surface of Poly(dimethylsiloxane) by Atom-Transfer Radical Polymerization of Acrylamide. Langmuir 2002, 18, 9971–9976. [Google Scholar] [CrossRef]
- Protsak, I.S.; Kuzema, P.O.; Tertykh, V.A.; Bolbukh, Y.M.; Kozakevich, R.B. Thermogravimetric analysis of silicas chemically modified with products of deoligomerization of polydimethylsiloxane. J. Therm. Anal. Calorim. 2015, 121, 547–557. [Google Scholar] [CrossRef]
- Barandeh, F.; Nguyen, P.; Kumar, R.; Iacobucci, G.J.; Kuznicki, M.L.; Kosterman, A.; Bergey, E.J.; Prasad, P.N.; Gunawardena, S. Organically modified silica nanoparticles are biocompatible and can be targeted to neurons in vivo. PLoS ONE 2012, 7, e29424. [Google Scholar] [CrossRef]
- Dash, S.; Mishra, S.; Patel, S.; Mishra, B.K. Organically modified silica: Synthesis and applications due to its surface interaction with organic molecules. Adv. Colloid Interface Sci. 2008, 140, 77–94. [Google Scholar] [CrossRef]
- Peng, P.; Lan, Y.; Luo, J. Modified silica incorporating into PDMS polymeric membranes for bioethanol selection. Adv. Polym. Technol. 2019, 2019, 5610282. [Google Scholar] [CrossRef]
- Jung, H.S.; Moon, D.S.; Lee, J.K. Quantitative analysis and efficient surface modification of silica nanoparticles. J. Nanomater. 2012, 2012, 593471. [Google Scholar] [CrossRef]
- Blitz, J.P.; Gun’ko, V.M. (Eds.) Surface Chemistry in Biomedical and Environmental Science; NATO Science Series II: Mathematics, Physics and Chemistry; Springer: Dordrecht, The Netherlands, 2006. [Google Scholar]
- Finiels, B.; Alonso, M.; Bousmina, D.; Brunel, A.; Kadib, E. Periodic mesoporous organosilicas derived from amphiphilic bulky polymethylsiloxane. New J. Chem. 2016, 40, 4132–4135. [Google Scholar] [CrossRef]
- Sharaf, M.A.; Mark, J.E. Modulus of randomly crosslinked polymethysiloxane networks. PMSE 1993, 68, 180–181. [Google Scholar] [CrossRef]
- Zhang, H.; Fidelis, L.C.; Serva, A.L.T.; Michaela, M.; Rezwan, K. Water-based freeze casting: Adjusting hydrophobic polymethylsiloxane for obtaining hierarchically ordered porous SiOC. J. Am. Ceram. Soc. 2017, 100, 1907–1918. [Google Scholar] [CrossRef]
- Clarson, S.J.; Semlyen, J.A. Studies of Cyclic and Linear Poly(dimethylsiloxanes): 21. High Temperature Thermal Behavior. Polymer 1986, 27, 91–95. [Google Scholar] [CrossRef]
- Krumpfer, J.W.; McCarthy, T.J. Rediscovering Silicones: “Unreactive” Silicones React with Inorganic Surfaces. Langmuir 2011, 27, 11514–11519. [Google Scholar] [CrossRef]
- Barthel, H.; Nikitina, E. INS and IR study of Intermolecular Interactions at the Fumed Silica-Polydimethylsiloxane Interphase, Part 3. Silica-Siloxane Adsorption Complexes. Silicon Chem. 2004, 1, 261–279. [Google Scholar] [CrossRef]
- Gun’ko, V.M.; Turov, V.V.; Zarko, V.I.; Goncharuk, E.V.; Gerashchenko, I.I.; Turova, A.A.; Mironyuk, I.F.; Leboda, R.; Skubiszewska-Zięba, J.; Janusz, W. Comparative characterization of polymethylsiloxane hydrogel and silylated fumed silica and silica gel. J. Colloid Interface Sci. 2007, 308, 142–156. [Google Scholar] [CrossRef]
- Gun’ko, V.M.; Turov, V.V.; Krupska, T.V.; Protsak, I.S.; Borysenko, M.V.; Pakhlov, E.M. Polymethylsiloxane alone and in composition with nanosilica under various conditions. J. Colloid Interface Sci. 2019, 541, 213–225. [Google Scholar] [CrossRef]
- Gun’ko, V.M.; Pakhlov, E.M.; Goncharuk, O.V.; Andriyko, L.S.; Marynin, A.I.; Ukrainets, A.I.; Charmas, B.; Skubiszewska-Zięba, J.; Blitz, J.P. Influence of hydrophobization of fumed oxides on interactions with polar and nonpolar adsorbates. Appl. Surf. Sci. 2017, 423, 855–868. [Google Scholar] [CrossRef]
- Protsak, I.S.; Henderson, I.M.; Tertykh, V.A.; Dong, W.; Le, Z. Cleavage of organosiloxanes with dimethyl carbonate: A mild approach to graft-to-surface modification. Langmuir 2018, 34, 9719–9730. [Google Scholar] [CrossRef] [PubMed]
- Protsak, I.S.; Morozov, Y.M.; Dong, W.; Le, Z.; Zhang, D.; Henderson, I.M. A 29Si, 1H, and 13C Solid-State NMR Study on the Surface Species of Various Depolymerized Organosiloxanes at Silica Surface. Nanoscale Res. Lett. 2019, 14, 160. [Google Scholar] [CrossRef] [PubMed]
- Nikolaev, V.G. Enterosgel: A Novel Organosilicon Enterosorbent with a Wide Range of Medical Applications; Mikhalovsky, S., Khajibaev, A., Eds.; Springer: Berlin, Germany, 2011; pp. 199–221. [Google Scholar]
- Shevchenko, Y.N.; Dushanin, B.M.; Yashinina, N.I. New Silicon Compounds Porous Organosilicon Matrices for Technology and Medicine. In Silicon for the Chemistry Industry III; The Norwegian University of Science and Technology: Sandefjord, Norway, 1996; ISBN 9788290265194. [Google Scholar]
- Chaplin, M. Water Structure and Science. Available online: http://www1.lsbu.ac.uk/water/ (accessed on 26 May 2019).
- Mikheev, Y.A.; Guseva, L.N.; Davydov, E.Y.; Ershov, Y.A. The hydration of hydrophobic substances. Russ. J. Phys. Chem. A 2007, 81, 1897–1913. [Google Scholar] [CrossRef]
- Yaminsky, V.V.; Vogler, E.A. Hydrophobic hydration. Curr. Opin. Colloid Interface Sci. 2001, 6, 342–349. [Google Scholar] [CrossRef]
- Widom, B.; Bhimalapuram, P.; Koga, K. The hydrophobic effect. PCCP 2003, 5, 3085–3093. [Google Scholar] [CrossRef]
- Chandler, D. Interfaces and the driving force of hydrophobic assembly. Nature 2005, 437, 640–647. [Google Scholar] [CrossRef]
- Gun’ko, V.M.; Turov, V.V. Nuclear Magnetic Resonance Studies of Interfacial Phenomena; CRC Press: Boca Raton, FL, USA, 2013; ISBN 9781466551688. [Google Scholar]
- Gun’ko, V.M.; Zarko, V.I.; Turov, V.V.; Leboda, R.; Chibowski, E. Distribution effect of the second phase in disperse silica/X oxides (X=Al2O3, TiO2, GeO2) on their surface properties. Langmuir 1999, 15, 5694–5702. [Google Scholar] [CrossRef]
- Gun’ko, V.M.; Turov, V.V.; Pakhlov, E.M.; Krupska, T.V.; Borysenko, M.V.; Kartel, M.T.; Charmas, B. Water interactions with hydrophobic versus hydrophilic nanosilica. Langmuir 2018, 34, 12145–12153. [Google Scholar] [CrossRef]
- Gun’ko, V.M.; Turov, V.V.; Protsak, I.S.; Krupska, T.V.; Pakhlov, E.M.; Tsapko, M.D. Effects of pre-adsorbed water on methane adsorption onto blends with hydrophobic and hydrophilic nanosilicas. Colloids Surf. A Physicochem. Eng. Asp. 2019, 570, 471–480. [Google Scholar] [CrossRef]
- Chuiko, A.A. (Ed.) Medical Chemistry and Clinical Application of Silicon Dioxide; Naukova Dumka: Kiev, Ukraine, 2003; ISBN 966-00-0185-1. (In Russian) [Google Scholar]
- Gun’ko, V.M.; Turov, V.V.; Pakhlov, E.M.; Krupska, T.V.; Charmas, B. Effect of water content on the characteristics of hydro-compacted nanosilica. Appl. Surf. Sci. 2018, 459, 171–178. [Google Scholar] [CrossRef]
- Gregg, S.; Sing, K.S.W. Adsorption, Surface Area and Porosity; Academic Press: London, UK, 1982; ISBN 9780123009562. [Google Scholar]
- Adamson, A.W.; Gast, A.P. Physical Chemistry of Surface; Wiley: New York, NY, USA, 1997; ISBN 978-0-471-14873-9. [Google Scholar]
- Gun’ko, V.M. Composite materials: Textural characteristics. Appl. Surf. Sci. 2014, 307, 444–454. [Google Scholar] [CrossRef]
- Neimark, A.V.; Ravikovitch, P.I. Capillary condensation in MMS and pore structure characterization. Microporous Mesoporous Mater. 2001, 44, 697–707. [Google Scholar] [CrossRef]
- Thommes, M.; Köhn, R.; Fröba, M. Sorption and pore condensation behavior of pure fluids in mesoporous MCM-48 silica, MCM-41 silica, SBA-15 silica and controlled-pore glass at temperatures above and below the bulk triple point. Appl. Surf. Sci. 2002, 196, 239–249. [Google Scholar] [CrossRef]
- Corminbœuf, C.; Heine, T.; Weber, J. 29Si NMR chemical shifts of silane derivatives. Chem. Phys. Lett. 2002, 357, 1–7. [Google Scholar] [CrossRef]
- Kruk, M.; Jaroniec, M. Gas adsorption characterization of ordered organic-inorganic nanocomposite materials. Chem. Mater. 2001, 13, 3169–3183. [Google Scholar] [CrossRef]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).