A Literature Review of Metagenomics and Culturomics of the Peri-implant Microbiome: Current Evidence and Future Perspectives

 ,
,  ,
,  ,
, 

{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
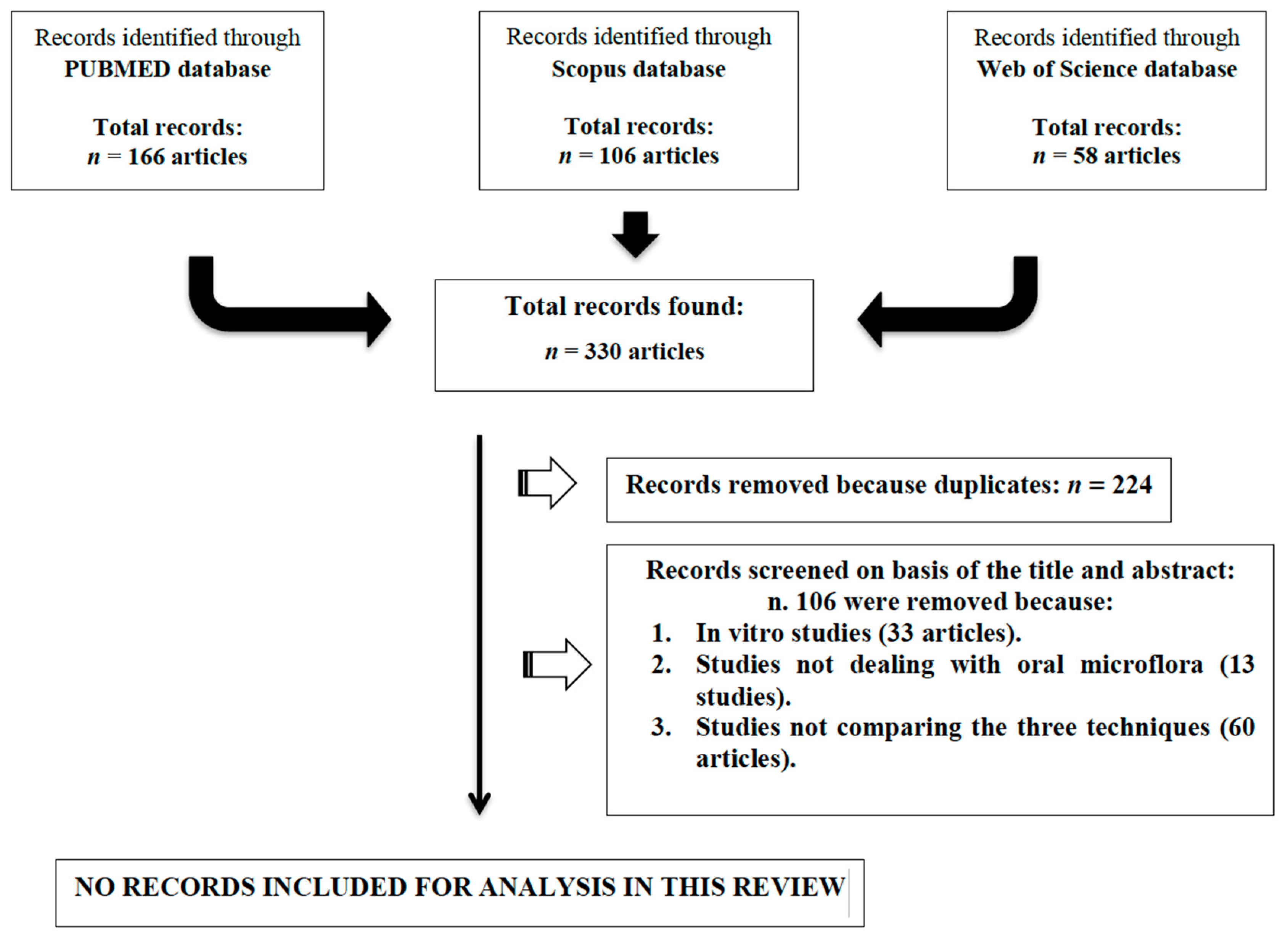
3. Results
4. Literature Review
4.1. Metagenomics
4.2. The Peri-Implant and Periodontal Microbiome
4.3. Future Perspectives: Culturomics
Author Contributions
Funding
Conflicts of Interest
References
- The Human Microbiome Project: Extending the Definition of What Constitutes a Human. Available online: https://www.genome.gov/27549400/the-human-microbiome-project-extending-the-definition-of-what-constitutes-a-human (accessed on 29 July 2019).
- Kilian, M.; Chapple, I.L.C.; Hannig, M.; Marsh, P.D.; Meuric, V.; Pedersen, A.M.L.; Tonetti, M.S.; Wade, W.G.; Zaura, E.; Pedersen, A.M.L. The oral microbiome—An update for oral healthcare professionals. Br. Dent. J. 2016, 221, 657–666. [Google Scholar] [CrossRef] [PubMed]
- Dewhirst, F.E.; Chen, T.; Izard, J.; Paster, B.J.; Tanner, A.C.; Yu, W.H.; Lakshmanan, A.; Wade, W.G. The human oral microbiome. J. Bacteriol. 2010, 192, 5002–5017. [Google Scholar] [CrossRef] [PubMed]
- Paster, B.J.; Boches, S.K.; Galvin, J.L.; Ericson, R.E.; Lau, C.N.; Levanos, V.A.; Sahasrabudhe, A.; Dewhirst, F.E. Bacterial Diversity in Human Subgingival Plaque. J. Bacteriol. 2001, 183, 3770–3783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paster, B.J.; Olsen, I.; Aas, J.A.; Dewhirst, F.E. The breadth of bacterial diversity in the human periodontal pocket and other oral sites. Periodontol 2000 2006, 42, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, K.; Chen, T.; Paster, B.J. A practical guide to the oral microbiome and its relation to health and disease. Oral Dis. 2017, 23, 276–286. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Gunsolley, J. Application of metagenomics in understanding oral health and disease. Virulence 2014, 5, 424–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Handelsman, J.; Rondon, M.R.; Brady, S.F.; Clardy, J.; Goodman, R.M. Molecular biological access to the chemistry of unknown soil microbes: A new frontier for natural products. Chem. Biol. 1998, 5, R245–R249. [Google Scholar] [CrossRef]
- Chen, K.; Pachter, L. Bioinformatics for Whole-Genome Shotgun Sequencing of Microbial Communities. PLoS Comput. Biol. 2005, 1, e24. [Google Scholar] [CrossRef] [PubMed]
- Daubin, V. Phylogenetics and the Cohesion of Bacterial Genomes. Science 2003, 301, 829–832. [Google Scholar] [CrossRef] [Green Version]
- Moon, J.-H.; Lee, J.-H. Probing the diversity of healthy oral microbiome with bioinformatics approaches. BMB Rep. 2016, 49, 662–670. [Google Scholar] [CrossRef] [Green Version]
- Szafranski, S.P.; Wos-Oxley, M.L.; Vilchez-Vargas, R.; Jáuregui, R.; Plumeier, I.; Klawonn, F.; Tomasch, J.; Meisinger, C.; Kühnisch, J.; Sztajer, H.; et al. High-Resolution Taxonomic Profiling of the Subgingival Microbiome for Biomarker Discovery and Periodontitis Diagnosis. Appl. Environ. Microbiol. 2015, 81, 1047–1058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quirynen, M.; Vogels, R.; Pauwels, M.; Haffajee, A.; Socransky, S.; Uzel, N.; Van Steenberghe, D. Initial Subgingival Colonization of ‘Pristine’ Pockets. J. Dent. Res. 2005, 84, 340–344. [Google Scholar] [CrossRef] [PubMed]
- Elter, C.; Heuer, W.; Demling, A.; Hannig, M.; Heidenblut, T.; Bach, F.W.; Stiesch-Scholz, M. Supra- and subgingival biofilm formation on implant abutments with different surface characteristics. Int. J. Oral Maxillofac. Implant. 2008, 23, 327–334. [Google Scholar]
- Persson, G.R.; Renvert, S. Cluster of bacteria associated with peri-implantitis. Clin. Implant Dent. Relat. Res. 2014, 16, 783–793. [Google Scholar] [CrossRef] [PubMed]
- Casado, P.L.; Otazu, I.B.; Balduino, A.; De Mello, W.; Barboza, E.P.; Duarte, M.E.L. Identification of Periodontal Pathogens in Healthy Periimplant Sites. Implant. Dent. 2011, 20, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Patini, R.; Staderini, E.; Lajolo, C.; Lopetuso, L.; Mohammed, H.; Rimondini, L.; Rocchetti, V.; Franceschi, F.; Cordaro, M.; Gallenzi, P. Relationship between oral microbiota and periodontal disease: A systematic review. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 5775–5788. [Google Scholar] [PubMed]
- Patini, R.; Gallenzi, P.; Spagnuolo, G.; Cordaro, M.; Cantiani, M.; Amalfitano, A.; Arcovito, A.; Callà, C.; Mingrone, G.; Nocca, G. Correlation Between Metabolic Syndrome, Periodontitis and Reactive Oxygen Species Production. A Pilot Study. Open Dent. J. 2017, 11, 621–627. [Google Scholar] [CrossRef] [Green Version]
- Mohammed, H.; Varoni, E.M.; Cochis, A.; Cordaro, M.; Gallenzi, P.; Patini, R.; Staderini, E.; Lajolo, C.; Rimondini, L.; Rocchetti, V. Oral Dysbiosis in Pancreatic Cancer and Liver Cirrhosis: A Review of the Literature. Biomedicines 2018, 6, 115. [Google Scholar] [CrossRef]
- Lucchese, A.; Matarese, G.; Manuelli, M.; Ciuffreda, C.; Bassani, L.; Isola, G.; Cordasco, G.; Gherlone, E. Reliability and efficacy of palifermin in prevention and management of oral mucositis in patients with acute lymphoblastic leukemia: A randomized, double-blind controlled clinical trial. Minerva Stomatol. 2016, 65, 43–50. [Google Scholar]
- Ferlazzo, N.; Currò, M.; Zinellu, A.; Caccamo, D.; Isola, G.; Ventura, V.; Carru, C.; Matarese, G.; Ientile, R. Influence of MTHFR genetic background on p16 and MGMT methylation in oral squamous cell cancer. Int. J. Mol. Sci. 2017, 18, 724. [Google Scholar] [CrossRef]
- Isola, G.; Ramaglia, L.; Cordasco, G.; Lucchese, A.; Fiorillo, L.; Matarese, G. The effect of a functional appliance in the management of temporomandibular joint disorders in patients with juvenile idiopathic arthritis. Minerva Stomatol. 2017, 66, 1–8. [Google Scholar]
- Lo Giudice, G.; Lo Giudice, R.; Matarese, G.; Isola, G.; Cicciù, M.; Terranova, A.; Palaia, G.; Romeo, U. Evaluation of magnification systems in restorative dentistry. An in-vitro study. Dent. Cadmos 2015, 83, 296–305. [Google Scholar] [CrossRef]
- Cutroneo, G.; Piancino, M.G.; Ramieri, G.; Bracco, P.; Vita, G.; Isola, G.; Vermiglio, G.; Favaloro, A.; Anastasi, G.; Trimarchi, F. Expression of muscle-specific integrins in masseter muscle fibers during malocclusion disease. Int. J. Mol. Med. 2012, 30, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Zaura, E.; Keijser, B.J.; Huse, S.M.; Crielaard, W. Defining the healthy “core microbiome” of oral microbial communities. BMC Microbiol. 2009, 9, 259. [Google Scholar] [CrossRef]
- Dabdoub, S.; Tsigarida, A.A.; Kumar, P.S. Patient-specific Analysis of Periodontal and Peri-implant Microbiomes. J. Dent. Res. 2013, 92, 168S–175S. [Google Scholar] [CrossRef] [Green Version]
- Dabdoub, S.M.; Ganesan, S.M.; Kumar, P.S. Comparative metagenomics reveals taxonomically idiosyncratic yet functionally congruent communities in periodontitis. Sci. Rep. 2016, 6, 38993. [Google Scholar] [CrossRef]
- Koyanagi, T.; Sakamoto, M.; Takeuchi, Y.; Ohkuma, M.; Izumi, Y. Analysis of microbiota associated with peri-implantitis using 16S rRNA gene clone library. J. Oral Microbiol. 2010, 2, 5104. [Google Scholar] [CrossRef]
- Shchipkova, A.Y.; Nagaraja, H.N.; Kumar, P.S. Subgingival Microbial Profiles of Smokers with Periodontitis. J. Dent. Res. 2010, 89, 1247–1253. [Google Scholar] [CrossRef] [Green Version]
- Human Microbiome Project Consortium. A framework for human microbiome research. Nature 2012, 13, 215–221. [Google Scholar]
- Zhou, Y.; Gao, H.; Mihindukulasuriya, K.A.; La Rosa, P.S.; Wylie, K.M.; Vishnivetskaya, T.; Podar, M.; Warner, B.I.; Tarr, P.; Nelson, D.E.; et al. Biogeography of the ecosystems of the healthy human body. Genome Biol. 2013, 14, R1. [Google Scholar] [CrossRef]
- Huse, S.M.; Ye, Y.; Zhou, Y.; Fodor, A.A. A Core Human Microbiome as Viewed through 16S rRNA Sequence Clusters. PLoS ONE 2012, 7, e34242. [Google Scholar] [CrossRef]
- Nasidze, I.; Li, J.; Quinque, D.; Tang, K.; Stoneking, M. Global diversity in the human salivary microbiome. Genome Res. 2009, 19, 636–643. [Google Scholar] [CrossRef] [Green Version]
- Mason, M.R.; Nagaraja, H.N.; Camerlengo, T.; Joshi, V.; Kumar, P.S. Deep Sequencing Identifies Ethnicity-Specific Bacterial Signatures in the Oral Microbiome. PLoS ONE 2013, 8, e77287. [Google Scholar] [CrossRef]
- Rylev, M.; Kilian, M. Prevalence and distribution of principal periodontal pathogens worldwide. J. Clin. Periodontol. 2008, 35, 346–361. [Google Scholar] [CrossRef]
- Haffajee, A.D.; Bogren, A.; Hasturk, H.; Feres, M.; Lopez, N.J.; Socransky, S.S. Subgingival microbiota of chronic periodontitis subjects from different geographic locations. J. Clin. Periodontol. 2004, 31, 996–1002. [Google Scholar] [CrossRef]
- Kim, T.; Kang, N.; Lee, S.B.; Eickholz, P.; Pretzl, B.; Kim, C.K. Differences in subgingival microflora of Korean and German periodontal patients. Arch. Oral Biol. 2009, 54, 223–229. [Google Scholar] [CrossRef]
- Flores, G.E.; Caporaso, J.G.; Henley, J.B.; Rideout, J.R.; Domogala, D.; Chase, J.; Leff, J.W.; Vázquez-Baeza, Y.; González, A.; Knight, R.; et al. Temporal variability is a personalized feature of the human microbiome. Genome Biol. 2014, 15, 531. [Google Scholar] [CrossRef]
- Xu, X.; He, J.; Xue, J.; Wang, Y.; Li, K.; Zhang, K.; Guo, Q.; Liu, X.; Zhou, Y.; Cheng, L.; et al. Oral cavity contains distinct niches with dynamic microbial communities. Environ. Microbiol. 2015, 17, 699–710. [Google Scholar] [CrossRef]
- Zheng, W.; Tsompana, M.; Ruscitto, A.; Sharma, A.; Genco, R.; Sun, Y.; Buck, M.J. An accurate and efficient experimental approach for characterization of the complex oral microbiota. Microbiome 2015, 3, 48. [Google Scholar] [CrossRef]
- Lagier, J.C.; Dubourg, G.; Million, M.; Cadoret, F.; Bilen, M.; Fenollar, F.; Levasseur, A.; Rolain, J.M.; Fournier, P.E.; Raoult, D. Culturing the human microbiota and culturomics. Nat. Rev. Genet. 2018, 16, 540–550. [Google Scholar] [CrossRef] [Green Version]
- Van Der Horst, J.; Buijs, M.J.; Laine, M.L.; Wismeijer, D.; Loos, B.G.; Crielaard, W.; Zaura, E. Sterile paper points as a bacterial DNA-contamination source in microbiome profiles of clinical samples. J. Dent. 2013, 41, 1297–1301. [Google Scholar] [CrossRef]
- Perez-Chaparro, P.; Duarte, P.; Pannuti, C.; Figueiredo, L.; Mestnik, M.; Gonçalves, C.; Faveri, M.; Feres, M. Evaluation of human and microbial DNA content in subgingival plaque samples collected by paper points or curette. J. Microbiol. Methods 2015, 111, 19–20. [Google Scholar] [CrossRef]
- Padial-Molina, M.; López-Martínez, J.; O’Valle, F.; Galindo-Moreno, P. Microbial Profiles and Detection Techniques in Peri-Implant Diseases: A Systematic Review. J. Oral Maxillofac. Res. 2016, 7, e10. [Google Scholar] [CrossRef]
- Sedghizadeh, P.P.; Yooseph, S.; Fadrosh, D.W.; Zeigler-Allen, L.; Thiagarajan, M.; Salek, H.; Farahnik, F.; Williamson, S.J. Metagenomic investigation of microbes and viruses in patients with jaw osteonecrosis associated with bisphosphonate therapy. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. 2012, 114, 764–770. [Google Scholar] [CrossRef] [Green Version]
- Young, G.; Turner, S.; Davies, J.K.; Sundqvist, G.; Figdor, D. Bacterial DNA Persists for Extended Periods after Cell Death. J. Endod. 2007, 33, 1417–1420. [Google Scholar] [CrossRef]
- Mason, C.E.; Afshinnekoo, E.; Tighe, S.; Wu, S.; Levy, S. International Standards for Genomes, Transcriptomes, and Metagenomes. J. Biomol. Tech. JBT 2017, 28, 8–18. [Google Scholar] [CrossRef] [Green Version]
- Lan, Y.; Rosen, G.; Hershberg, R. Marker genes that are less conserved in their sequences are useful for predicting genome-wide similarity levels between closely related prokaryotic strains. Microbiome 2016, 4, 18. [Google Scholar] [CrossRef] [Green Version]
- Mao, D.P.; Zhou, Q.; Chen, C.Y.; Quan, Z.X. Coverage evaluation of universal bacterial primers using the metagenomic datasets. BMC Microbiol. 2012, 12, 66. [Google Scholar] [CrossRef]
- Charalampakis, G.; Belibasakis, G.N. Microbiome of peri-implant infections: Lessons from conventional, molecular and metagenomic analyses. Virulence 2015, 6, 183–187. [Google Scholar] [CrossRef] [Green Version]
- Ruppé, E.; Greub, G.; Schrenzel, J. Messages from the first International Conference on Clinical Metagenomics (ICCMg). Microbes Infect. 2017, 19, 223–228. [Google Scholar] [CrossRef] [Green Version]
- Lagier, J.C.; Armougom, F.; Million, M.; Hugon, P.; Pagnier, I.; Robert, C.; Bittar, F.; Fournous, G.; Gimenez, G.; Maraninchi, M.; et al. Microbial culturomics: Paradigm shift in the human gut microbiome study. Clin. Microbiol. Infect. 2012, 18, 1185–1193. [Google Scholar] [CrossRef]
- Dubourg, G.; Lagier, J.C.; Robert, C.; Armougom, F.; Hugon, P.; Metidji, S.; Dione, N.; Dangui, N.P.M.; Pfleiderer, A.; Abrahao, J.; et al. Culturomics and pyrosequencing evidence of the reduction in gut microbiota diversity in patients with broad-spectrum antibiotics. Int. J. Antimicrob. Agents 2014, 44, 117–124. [Google Scholar] [CrossRef]
- Lagier, J.-C.; Hugon, P.; Khelaifia, S.; Fournier, P.-E.; La Scola, B.; Raoult, D. The Rebirth of Culture in Microbiology through the Example of Culturomics to Study Human Gut Microbiota. Clin. Microbiol. Rev. 2015, 28, 237–264. [Google Scholar] [CrossRef]
- Masucci, L.; Quaranta, G.; Nagel, D.; Primus, S.; Romano, L.; Graffeo, R.; Ianiro, G.; Gasbarrini, A.; Cammarota, G.; Sanguinetti, M. Culturomics: Bacterial species isolated in 3 healthy donors for faecal microbiota transplantation in Clostridium difficile infection. Microbiol. Med. 2017, 32, 6510. [Google Scholar] [CrossRef]
- Patini, R.; Cattani, P.; Marchetti, S.; Isola, G.; Quaranta, G.; Gallenzi, P. Evaluation of Predation Capability of Periodontopathogens Bacteria by Bdellovibrio Bacteriovorus HD100. An in Vitro Study. Materials 2019, 12, 2008. [Google Scholar] [CrossRef]
- Facciolo, M.-T.; Riva, F.; Gallenzi, P.; Patini, R.; Gaglioti, D. A rare case of oral multisystem Langerhans cell histiocytosis. J. Clin. Exp. Dent. 2017, 9, e820–e824. [Google Scholar] [CrossRef] [Green Version]
- Pippi, R.; Santoro, M.; Patini, R. The central odontogenic fibroma: How difficult can be making a preliminary diagnosis. J. Clin. Exp. Dent. 2016, 8, e223–e225. [Google Scholar] [CrossRef]
- Coviello, V.; Zareh Dehkhargani, S.; Patini, R.; Cicconetti, A. Surgical ciliated cyst 12 years after Le Fort I maxillary advancement osteotomy: A case report and review of the literature. Oral Surg. 2017, 10, 165–170. [Google Scholar] [CrossRef]
- Pippi, R.; Santoro, M.; Patini, R. Fibrolipoma of the Oral Cavity: Treatment Choice in a Case with an Unusual Location. J. Clin. Diagn. Res. 2017, 11, ZJ07–ZJ08. [Google Scholar] [CrossRef]
- Patini, R.; Coviello, V.; Riminucci, M.; Corsi, A.; Cicconetti, A. Early-stage diffuse large B-cell lymphoma of the submental region: A case report and review of the literature. Oral Surg. 2017, 10, 56–60. [Google Scholar] [CrossRef]
- Pippi, R.; Pietrantoni, A.; Patini, R.; Santoro, M.; Santoro, M. Is telephone follow-up really effective in early diagnosis of inflammatory complications after tooth extraction? Med. Oral Patol. Oral Cir. Bucal 2018, 23, e707–e715. [Google Scholar] [CrossRef]
- Paduano, S.; Bucci, R.; Rongo, R.; Silva, R.; Michelotti, A. Prevalence of temporomandibular disorders and oral parafunctions in adolescents from public schools in Southern Italy. CRANIO 2018, 14, 1–6. [Google Scholar] [CrossRef]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martellacci, L.; Quaranta, G.; Patini, R.; Isola, G.; Gallenzi, P.; Masucci, L. A Literature Review of Metagenomics and Culturomics of the Peri-implant Microbiome: Current Evidence and Future Perspectives. Materials 2019, 12, 3010. https://doi.org/10.3390/ma12183010
Martellacci L, Quaranta G, Patini R, Isola G, Gallenzi P, Masucci L. A Literature Review of Metagenomics and Culturomics of the Peri-implant Microbiome: Current Evidence and Future Perspectives. Materials. 2019; 12(18):3010. https://doi.org/10.3390/ma12183010
Chicago/Turabian StyleMartellacci, Leonardo, Gianluca Quaranta, Romeo Patini, Gaetano Isola, Patrizia Gallenzi, and Luca Masucci. 2019. "A Literature Review of Metagenomics and Culturomics of the Peri-implant Microbiome: Current Evidence and Future Perspectives" Materials 12, no. 18: 3010. https://doi.org/10.3390/ma12183010
APA StyleMartellacci, L., Quaranta, G., Patini, R., Isola, G., Gallenzi, P., & Masucci, L. (2019). A Literature Review of Metagenomics and Culturomics of the Peri-implant Microbiome: Current Evidence and Future Perspectives. Materials, 12(18), 3010. https://doi.org/10.3390/ma12183010