Graphitized Carbon Xerogels for Lithium-Ion Batteries




Abstract
:1. Introduction
2. Materials and Methods
2.1. Synthesis and Graphitization of the Carbon Xerogels Studied
2.2. Structural and Porous Characterization
2.3. Electrochemical Characterization
3. Results and Discussion
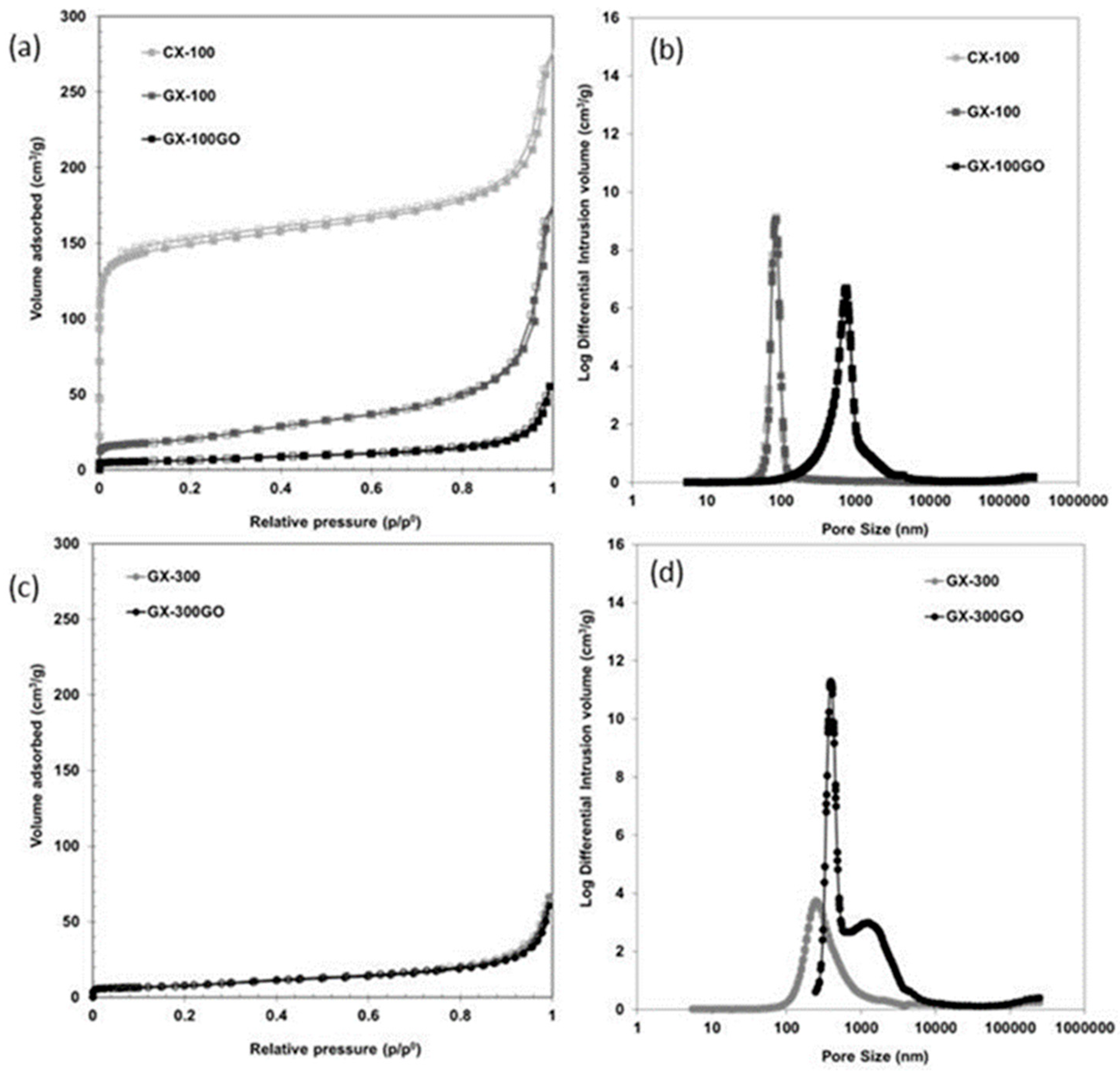
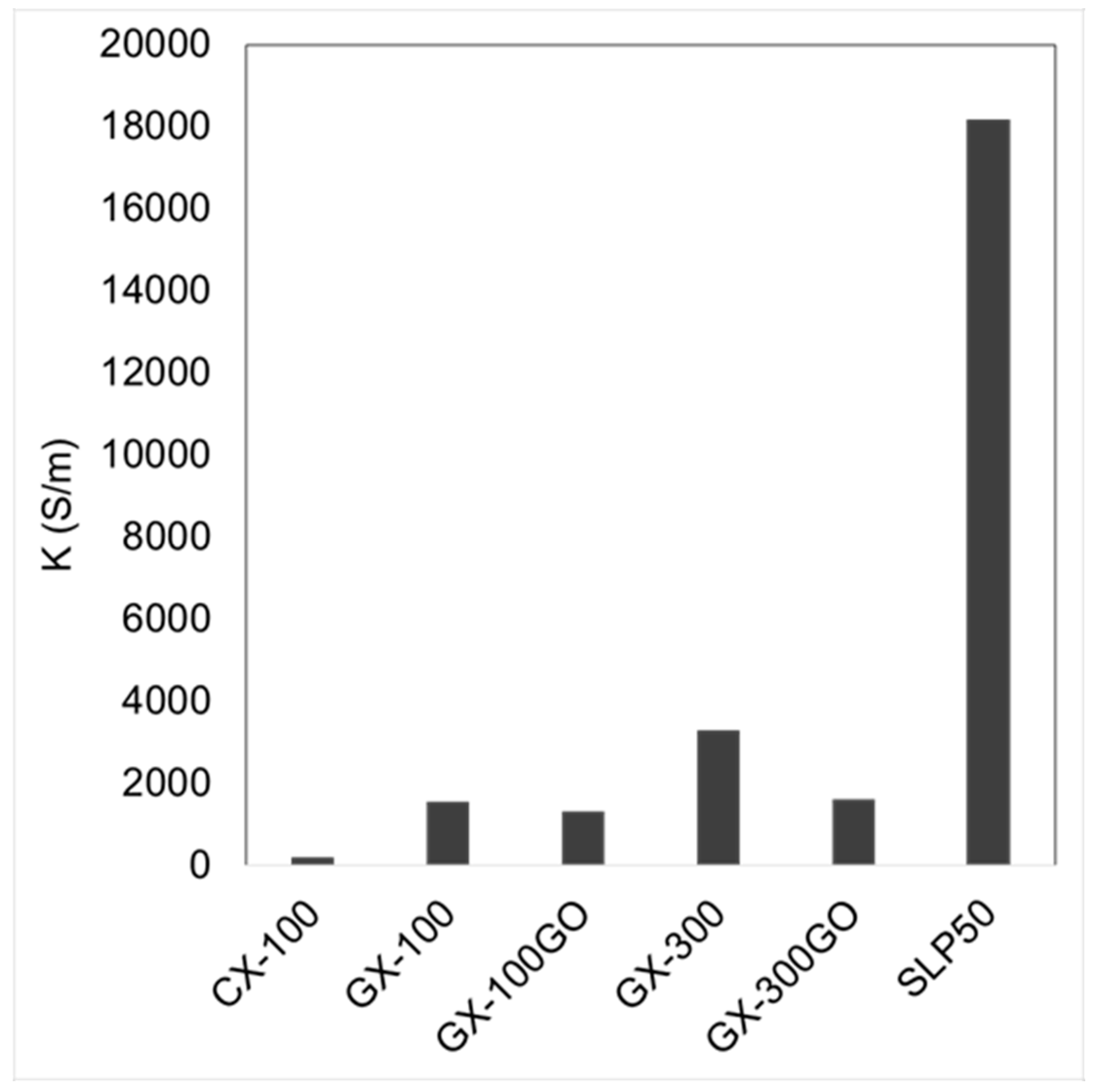
3.1. Structural and Chemical Characterization
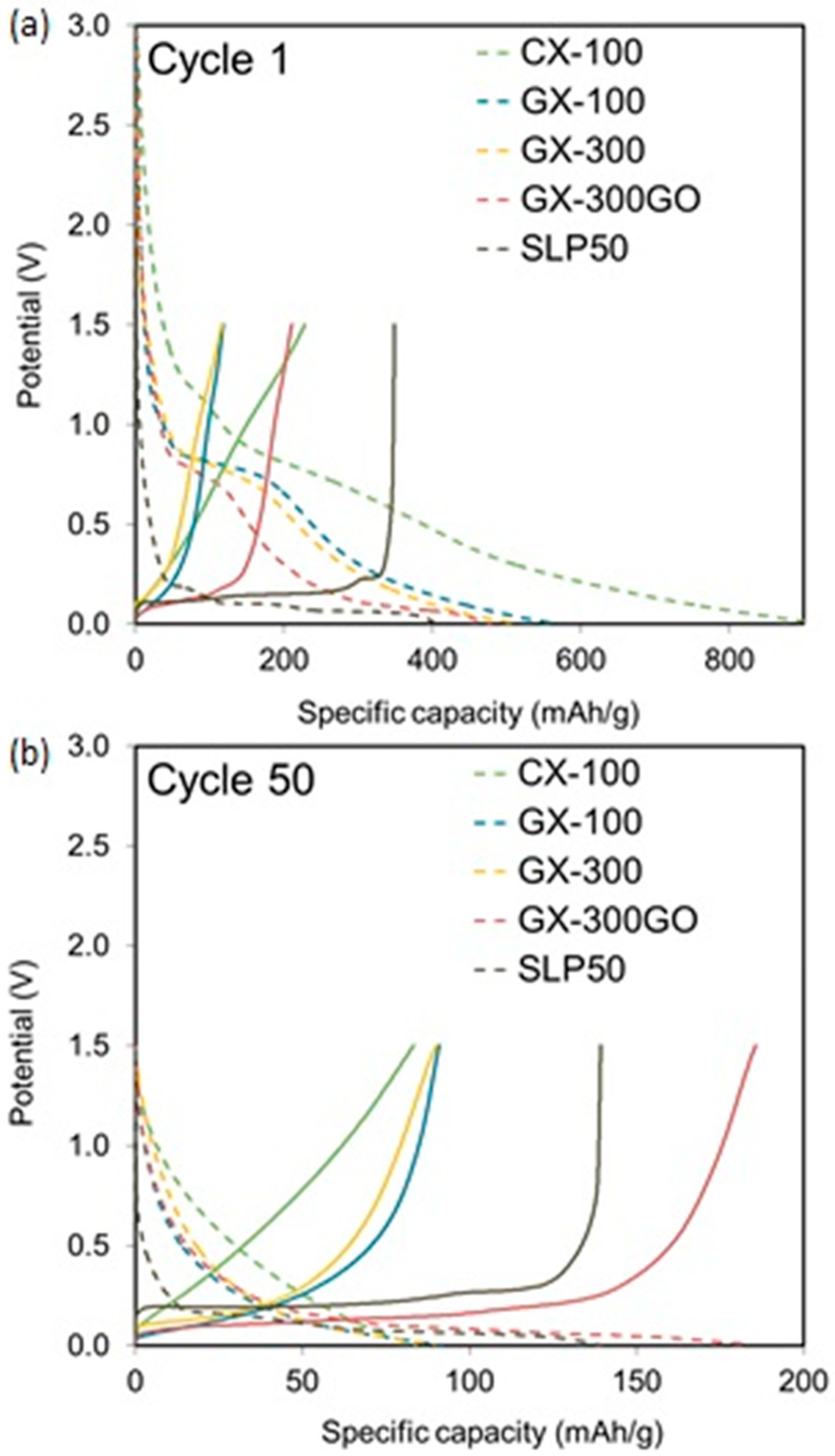
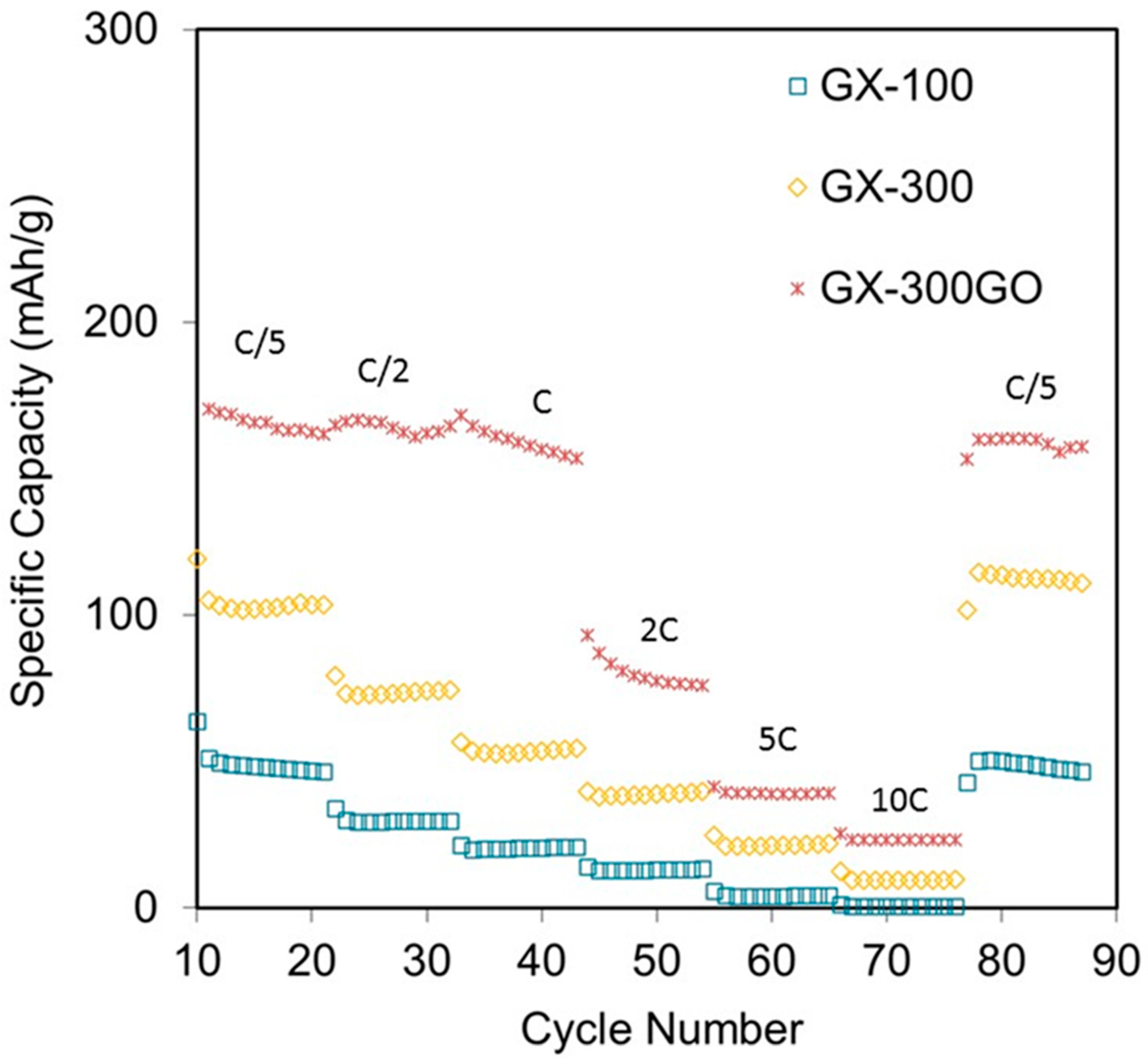
3.2. Electrochemical Characterization
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Cuesta, N.; Cameán, I.; Ramos, A.; García, A.B. Graphitized biogas–derived carbon nanofibers as anodes for lithium-ion batteries. Electrochim. Acta 2016, 222, 264–270. [Google Scholar] [CrossRef] [Green Version]
- Armaroli, N.; Balzani, V. Towards an electricity-powered world. Energy Environ. Sci. 2011, 4, 3193–3222. [Google Scholar] [CrossRef]
- Rey-Raap, N.; Piedboeuf, M.L.C.; Arenillas, A.; Menéndez, J.A.; Léonard, A.F.; Job, N. Aqueous and organic inks of bespoke carbon xerogels as models for studying the role of porosity in lithium-ion battery electrodes. Mater. Des. 2016, 109, 282–288. [Google Scholar] [CrossRef]
- Roberts, A.D.; Wang, S.; Li, X.; Zhang, H. Hierarchical porous nitrogen-rich carbon monoliths via ice-templating: High capacity and high-rate performance as lithium-ion battery anode materials. J. Mater. Chem. 2014, 43, 4341–4356. [Google Scholar] [CrossRef] [Green Version]
- Miranda, D.; Costa, C.M.; Lanceros-Mendez, S. Lithium ion rechargeable batteries: State of the art and future needs of microscopic theoretical models and simulations. J. Electroanal. Chem. 2015, 739, 97–110. [Google Scholar] [CrossRef]
- Wang, K.X.; Li, X.H.; Chen, J.S. Surface and interface engineering of electrode materials for lithium-ion batteries. Adv. Mater. 2015, 27, 527–545. [Google Scholar] [CrossRef]
- Hu, M.; Pang, X.; Zhou, Z. Recent progress in high-voltage lithium ion batteries. J. Power Sources 2013, 237, 229–242. [Google Scholar] [CrossRef]
- Scrosati, B.; Garche, J. Lithium batteries: Status, prospects and future. J. Power Sources 2010, 195, 2419–2430. [Google Scholar] [CrossRef]
- Yoon, S.; Lee, S.; Kim, S.; Park, K.W.; Cho, D.; Jeong, Y. Carbon nanotube film anodes for flexible lithium ion batteries. J. Power Sources 2015, 279, 495–501. [Google Scholar] [CrossRef]
- Collins, J.; Gourdin, G.; Foster, M.; Qu, D. Carbon surface functionalities and SEI formation during Li intercalation. Carbon 2015, 92, 193–244. [Google Scholar] [CrossRef]
- Lee, K.T.; Lytle, J.C.; Ergang, N.S.; Oh, S.M.; Stein, A. Synthesis and Rate Performance of Monolithic Macroporous Carbon Electrodes for Lithium-Ion Secondary Batteries. Adv. Funct. Mater. 2005, 15, 547–556. [Google Scholar] [CrossRef]
- Scrosati, B. Recent advances in lithium ion battery materials. Electrochim. Acta 2000, 45, 2461–2466. [Google Scholar] [CrossRef]
- Kakunuri, M.; Vennamalla, S.; Sharma, C.S. Synthesis of carbon xerogel nanoparticles by inverse emulsion polymerization of resorcinol–formaldehyde and their use as anode materials for lithium-ion battery. RSC Adv. 2015, 5, 4747–4753. [Google Scholar] [CrossRef]
- Goriparti, S.; Miele, E.; de Angelis, F.; di Fabrizio, E.; Zaccaria, R.P.; Capiglia, C. Review on recent progress of nanostructured anode materials for Li-ion batteries. J. Power Sources 2014, 257, 421–443. [Google Scholar] [CrossRef] [Green Version]
- Lahiri, I.; Choi, W. Carbon Nanostructures in Lithium Ion Batteries: Past, Present, and Future. Crit. Rev. Solid State Mater. Sci. 2013, 38, 128–166. [Google Scholar] [CrossRef]
- Chen, S.; Wang, Y.; Ahn, H.; Wang, G. Microwave hydrothermal synthesis of high performance tin–graphene nanocomposites for lithium ion batteries. J. Power Sources 2012, 216, 22–27. [Google Scholar] [CrossRef]
- Ogumi, Z.; Inaba, M. Carbon anodes. In Advances in Lithium Batteries; van Schalkwijk, W.A., Scrosati, B., Eds.; Kluwer Academic Publishers: Berlin, Germany, 2002; pp. 79–103. [Google Scholar]
- Alegre, C.; Sebastián, D.; Gálvez, M.E.; Baquedano, E.; Moliner, R.; Aricó, A.S.; Baglio, V.; Lázaro, M.J. N-Doped Carbon Xerogels as Pt Support for the Electro-Reduction of Oxygen. Materials 2017, 10, 1092. [Google Scholar] [CrossRef]
- Alegre, C.; Gálvez, M.E.; Moliner, R.; Baglio, V.; Stassi, A.; Aricó, A.S.; Lazaro, M.J. Platinum Ruthenium Catalysts Supported on Carbon Xerogel for Methanol Electro-Oxidation: Influence of the catalyst Synthesis Method. ChemCatChem 2013, 5, 3370–3780. [Google Scholar] [CrossRef]
- Alegre, C.; Gálvez, M.E.; Moliner, R.; Baglio, V.; Aricó, A.S.; Lázaro, M.J. Towards an optimal synthesis route for the preparation of highly mesoporous carbón xerogel-supported Pt catalysts for the oxygen reduction reaction. Appl. Catal. B Environ. 2014, 147, 947–957. [Google Scholar] [CrossRef] [Green Version]
- Rey-Raap, N.; Menéndez, J.A.; Arenillas, A. Optimization of the process variables in the microwave-induced synthesis of carbon xerogels. J. Sol Gel Sci. Technol. 2014, 69, 488–497. [Google Scholar] [CrossRef] [Green Version]
- Alonso-Buenaposada, I.D.; Rey-Raap, N.; Calvo, E.G.; Menéndez, J.A.; Arenillas, A. Effect of methanol content in commercial formaldehyde solutions on the porosity of RF carbon xerogels. J. Non Cryst. Solids 2015, 426, 13–18. [Google Scholar] [CrossRef] [Green Version]
- Rey-Raap, N.; Menéndez, J.A.; Arenillas, A. Simultaneous adjustment of the main chemical variables to fine-tune the porosity of carbon xerogels. Carbon 2014, 78, 490–499. [Google Scholar] [CrossRef] [Green Version]
- Rey-Raap, N.; Calvo, E.G.; Arenillas, A.; Menéndez, J.Á. High surface area carbon xerogels: Microwave vs. conventional activation with KOH. Chim. Oggi Chem. Today 2012, 30, 30–32. [Google Scholar]
- Sebastián, D.; Alegre, C.; Gálvez, M.E.; Moliner, R.; Lázaro, M.J.; Aricó, A.S.; Baglio, V. Towards new generation fuel cell electrocatalysts base don xerogel-nanofiber carbón composites. J. Mater. Chem. A 2014, 2, 13713–13722. [Google Scholar] [CrossRef]
- Xia, X.H.; Zhang, X.F.; Yi, S.Q.; Liu, H.B.; Chen, Y.X.; Chen, H.; Yang, L.; He, Y.D. Preparation of high specific surface area composite carbon cryogels from self-assembly of graphene oxide and resorcinol monomers for supercapacitors. J. Solid State Electrochem. 2016, 20, 1793–1802. [Google Scholar] [CrossRef]
- Meng, F.; Zhang, X.; Xu, B.; Yue, S.; Guo, H.; Luo, Y. Alkali-treated graphene oxide as a solid base catalyst: Synthesis and electrochemical capacitance of graphene/carbon composite aerogels. J. Mater. Chem. 2011, 21, 18537–18539. [Google Scholar] [CrossRef]
- Marković, Z.M.; Babić, B.M.; Dramićanin, M.D.; Antunović, I.D.H.; Pavlović, V.B.; Peruško, D.B.; Marković, B.M.T. Preparation of highly conductive carbon cryogel based on pristine graphene. Synth. Met. 2012, 162, 743–747. [Google Scholar] [CrossRef]
- Liu, L.; Yang, J.; Meng, Q. Graphene cross-linked phenol–formaldehyde hybrid organic and carbon xerogel during ambient pressure drying. J. Sol Gel Sci. Technol. 2013, 66, 1–5. [Google Scholar] [CrossRef]
- Ling, Z.; Wang, G.; Dong, Q.; Qian, B.; Zhang, M.; Li, C.; Qiu, J. An ionic liquid template approach to graphene–carbon xerogel composites for supercapacitors with enhanced performance. J. Mater. Chem. A 2014, 2, 14329–14333. [Google Scholar] [CrossRef]
- Chen, T.T.; Song, W.L.; Fan, L.Z. Engineering graphene aerogels with porous carbon of large surface area for flexible all-solid-state supercapacitors. Electrochim. Acta 2015, 165, 92–97. [Google Scholar] [CrossRef]
- Canal-Rodríguez, M.; Menéndez, J.A.; Montes-Morán, M.A.; Martín-Gullón, I.; Parra, J.B.; Arenillas, A. The role of conductive additives on the performance of hybrid carbon xerogels as electrodes in aqueous supercapacitors. Electrochim. Acta 2019, 295, 693–702. [Google Scholar] [CrossRef]
- Canal-Rodríguez, M.; Arenillas, A.; Menéndez, J.A.; Beneroso, D.; Rey-Raap, N. Carbon xerogels graphitized by microwave heating as anode materials in lithium-ion batteries. Carbon 2018, 137, 384–394. [Google Scholar] [CrossRef]
- Canal-Rodríguez, M.; Menéndez, J.A.; Arenillas, A. Performance of carbon xerogel-graphene hybrids as electrodes in aqueous supercapacitors. Electrochim. Acta 2018, 276, 28–36. [Google Scholar] [CrossRef]
- Canal-Rodríguez, M.; Arenillas, A.; Rey-Raap, N.; Ramos-Fernández, G.; Martín-Gullón, I.; Menéndez, J.A. Graphene-doped carbon xerogel combining high electrical conductivity and surface area for optimized aqueous supercapacitors. Carbon 2017, 118, 291–298. [Google Scholar] [CrossRef] [Green Version]
- Fromm, O.; Heckmann, A.; Rodehorst, U.C.; Frerichs, J.; Becker, D.; Winter, M.; Placke, T. Carbons from biomass precursors as anode materials for lithium ion batteries: New insights into carbonization and graphitization behavior and into their correlation to electrochemical performance. Carbon 2018, 128, 147–163. [Google Scholar] [CrossRef]
- Fuertes, A.B.; Alvarez, S. Graphitic mesoporous carbons synthesised through mesostructured silica templates. Carbon 2004, 42, 3049–3055. [Google Scholar] [CrossRef]
- Yoon, S.B.; Chai, G.S.; Kang, S.K.; Yu, J.S.; Gierszal, K.P.; Jaroniec, M. Graphitized Pitch-Based Carbons with Ordered Nanopores Synthesized by Using Colloidal Crystals as Templates. J. Am. Chem. Soc. 2005, 127, 4188–4189. [Google Scholar] [CrossRef]
- Frackowiak, E.; Lota, G.; Machnikowski, J.; Vix-Guterl, C.; Béguin, F. Optimisation of supercapacitors using carbons with controlled nanotexture and nitrogen content. Electrochim. Acta 2006, 51, 2209–2214. [Google Scholar] [CrossRef]
- Calvo, E.G.; Lufrano, F.; Staiti, P.; Brigandì, A.; Arenillas, A.; Menéndez, J.A. Optimizing the electrochemical performance of aqueous symmetric supercapacitors based on an activated carbon xerogel. J. Power Sources 2013, 241, 776–782. [Google Scholar] [CrossRef]
- Calvo, E.G.; Ferrera-Lorenzo, N.; Menéndez, J.A.; Arenillas, A. Microwave synthesis of micro-mesoporous activated carbon xerogels for high performance supercapacitors. Microporous Mesoporous Mater. 2013, 168, 206–212. [Google Scholar] [CrossRef]
- Zubizarreta, L.; Arenillas, A.; Menéndez, J.A.; Pis, J.J.; Pirard, J.P.; Job, N. Microwave drying as an effective method to obtain porous carbon xerogels. J. Non Cryst. Solids 2008, 354, 4024–4026. [Google Scholar] [CrossRef]
- Rodriguez-Pastor, I.; Ramos-Fernandez, G.; Varela-Rizo, H.; Terrones, M.; Martin-Gullon, I. Towards the understanding of the graphene oxide structure: How to control the formation of humic- and fulvic-like oxidized debris. Carbon 2015, 84, 299–309. [Google Scholar] [CrossRef] [Green Version]
- Feret, F.R. Determination of the crystallinity of calcined and graphitic cokes by X-ray diffraction. Analyst 1998, 123, 595–600. [Google Scholar] [CrossRef]
- Rey-Raap, N.; Calvo, E.G.; Bermúdez, J.M.; Cameán, I.; García, A.B.; Menéndez, J.A.; Arenillas, A. An electrical conductivity translator for carbons. Meas. J. Int. Meas. Confed. 2014, 56, 215–218. [Google Scholar] [CrossRef] [Green Version]
- Skowroński, J.M.; Knofczyński, K. Catalytically graphitized glass-like carbon examined as anode for lithium-ion cell performing at high charge/discharge rates. J. Power Sources 2009, 194, 81–87. [Google Scholar] [CrossRef]
- Rey-Raap, N.; Arenillas, A.; Menéndez, J.A. Formaldehyde in the synthesis of resorcinol-formaldehyde carbon gels. In Formaldehyde: Synthesis, Applications and Potential Health Effects; Nova Science Publishers: New York, NY, USA, 2015; pp. 31–60. [Google Scholar]
- Canal-Rodríguez, M.; Menéndez, A.J.; Arenillas, A. Carbon Xerogels: The Bespoke Nanoporous Carbons. In Porosity-Process, Technologies and Applications; IntechOpen: London, UK, 2017; pp. 69–89. [Google Scholar]
- Rey-Raap, N.; Arenillas, A.; Menéndez, J.A. A visual validation of the combined effect of pH and dilution on the porosity of carbon xerogels. Microporous Mesoporous Mater. 2016, 223, 89–93. [Google Scholar] [CrossRef] [Green Version]
- Pei, S.; Cheng, H.-M. The reduction of graphene oxide. Carbon 2012, 50, 3210–3228. [Google Scholar] [CrossRef]
- McAllister, M.J.; Li, J.L.; Adamson, D.H.; Schniepp, H.C.; Abdala, A.A.; Liu, J.; Herrera-Alonso, M.; Milius, D.L.; Car, R.; Prud’homme, R.K.; et al. Single Sheet Functionalized Graphene by Oxidation and Thermal Expansion of Graphite. Chem. Mater. 2007, 19, 4396–4404. [Google Scholar] [CrossRef]
- Spahr, M.E.; Buqa, H.; Würsig, A.; Goers, D.; Hardwick, L.; Novák, P.; Krumeich, F.; Dentzer, J.; Vix-Guterl, C. Surface reactivity of graphite materials and their surface passivation during the first electrochemical lithium insertion. J. Power Sources 2006, 153, 300–311. [Google Scholar] [CrossRef]
- Piedboeuf, M.-L.; Léonard, A.; Reichenauer, G.; Balzer, C.; Job, N. How do the micropores of carbon xerogels influence their electrochemical behavior as anodes for lithium-ion batteries? Microporous Mesoporous Mater. 2019, 275, 278–287. [Google Scholar] [CrossRef]
- Guo, Z.; Wang, C.; Chen, M.; Li, M. Hard Carbon Derived from Coal Tar Pitch for Use as the Anode Material in Lithium Ion Batteries. Int. J. Electrochem. Sci. 2013, 8, 2702–2709. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Graphitic | Non-Graphitic | Graphitic Contribution | SBET | |||
|---|---|---|---|---|---|---|
| d002 (nm) | Lc (nm) | d002 (nm) | Lc (nm) | (%) | m2 g−1 | |
| CX-100 | – | – | – | – | – | 591 |
| GX-100 | 0.338 | 14.1 | 0.350 | 2.0 | 7.5 | 71 |
| GX-300 | 0.337 | 12.3 | 0.350 | 1.8 | 22.6 | 22 |
| GX-100GO | 0.338 | 12.9 | 0.352 | 1.4 | 32.9 | 29 |
| GX-300GO | 0.338 | 13.2 | 0.356 | 0.8 | 29.4 | 26 |
| SLP50 | 0.335 | 53 | – | – | 100 | 5 |
| Cirr (1stcycle) | R (50thcycle) | R (100thcycle) | Ec (1stcycle) | Ec (50thcycle) | |
|---|---|---|---|---|---|
| (%) | (%) | (%) | (%) | (%) | |
| CX-100 | 72.4 | 36.8 | 25.2 | 27.6 | 99.2 |
| GX-100 | 73.7 | 77.6 | 62.7 | 26.3 | 99.2 |
| GX-300 | 72.8 | 78.3 | 69.9 | 27.2 | 100.5 |
| GX-300GO | 51.4 | 87.7 | 77.9 | 48.6 | 100.5 |
| SLP50 | 11 | 59.6 | 43.8 | 89.0 | 99.9 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Canal-Rodríguez, M.; Arenillas, A.; Villanueva, S.F.; Montes-Morán, M.A.; Menénedez, J.A. Graphitized Carbon Xerogels for Lithium-Ion Batteries. Materials 2020, 13, 119. https://doi.org/10.3390/ma13010119
Canal-Rodríguez M, Arenillas A, Villanueva SF, Montes-Morán MA, Menénedez JA. Graphitized Carbon Xerogels for Lithium-Ion Batteries. Materials. 2020; 13(1):119. https://doi.org/10.3390/ma13010119
Chicago/Turabian StyleCanal-Rodríguez, Maria, Ana Arenillas, Sara F. Villanueva, Miguel A. Montes-Morán, and J. Angel Menénedez. 2020. "Graphitized Carbon Xerogels for Lithium-Ion Batteries" Materials 13, no. 1: 119. https://doi.org/10.3390/ma13010119