Synthesis and Evaluation of a Thermoresponsive Degradable Chitosan-Grafted PNIPAAm Hydrogel as a “Smart” Gene Delivery System

 ,
, 

Abstract

{kind=link}
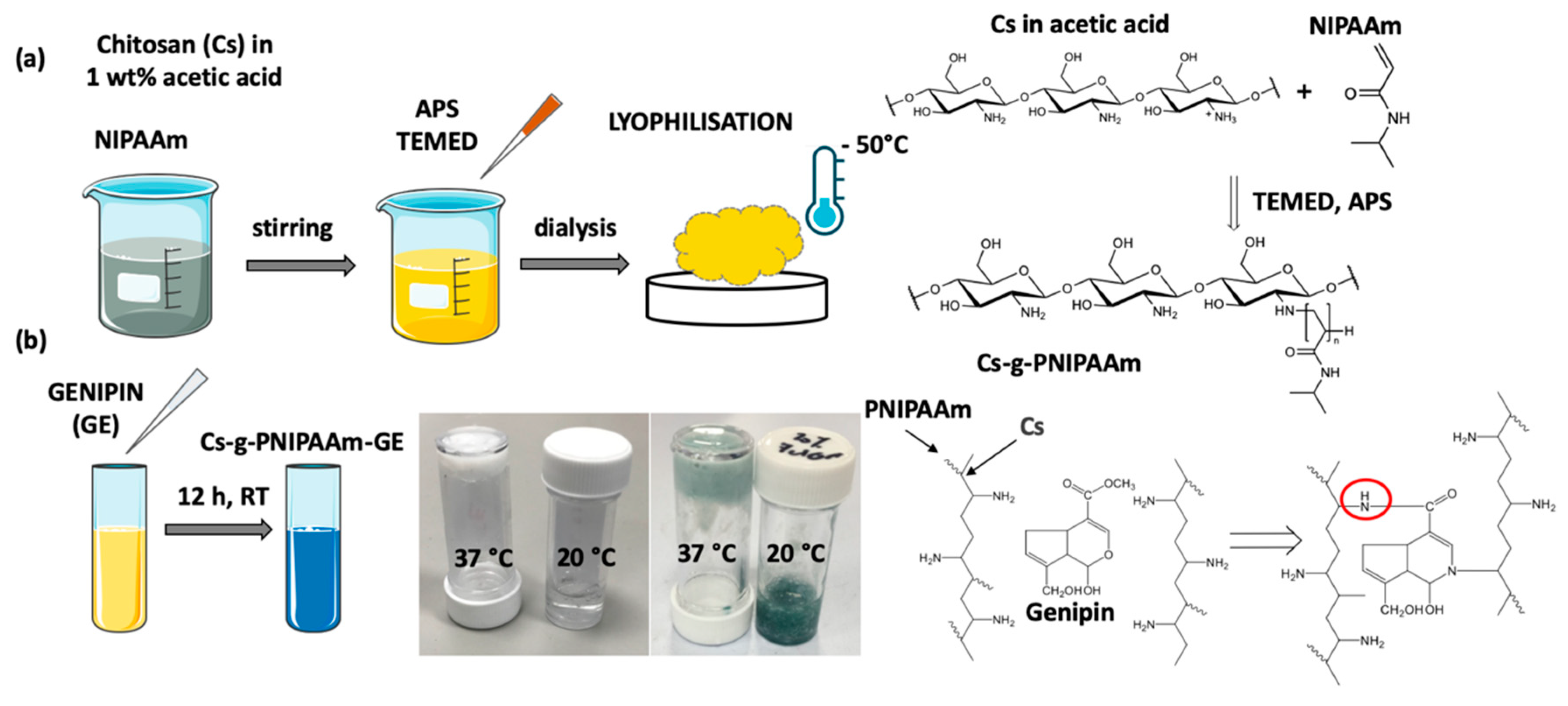{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of Cs-g-PNIPAAm
2.3. Synthesis of Nano-Complexes
2.4. Chemical Characterization of Cs-g-PNIPAAm Hydrogel
- (1)
- Nuclear magnetic resonance analysis
- (2)
- Fourier-transform infrared spectroscopy
- (3)
- Thermogravimetric analysis
- (4)
- Zeta potential analysis
2.5. Rheological Analysis
2.6. Scanning Electron Microscopy (SEM)
2.7. Swelling Ratio of the Cs-g-PNIPAAm Hydrogels
2.8. Injectability Analysis
2.9. Indirect Cytotoxicity Assessment
2.10. RALA/pEGFP-N1 NP Delivery from Cs-g-PNIPAAm Hydrogel
2.11. Statistical Analysis
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Hoare, T.R.; Kohane, D.S. Hydrogels in drug delivery: Progress and challenges. Polymer 2008, 49, 1993–2007. [Google Scholar] [CrossRef]
- Caló, E.; Khutoryanskiy, V.V. Biomedical applications of hydrogels: A review of patents and commercial products. Eur. Polym. J. 2015, 65, 252–267. [Google Scholar] [CrossRef]
- Li, J.; Mooney, D.J. Designing hydrogels for controlled drug delivery. Nat. Rev. Mater. 2016, 1, 16071. [Google Scholar] [CrossRef] [PubMed]
- Constantin, M.; Bucatariu, S.-M.; Doroftei, F.; Fundueanu, G. Smart composite materials based on chitosan microspheres embedded in thermosensitive hydrogel for controlled delivery of drugs. Carbohydr. Polym. 2017, 157, 493–502. [Google Scholar] [CrossRef]
- Bhattarai, N.; Gunn, J.; Zhang, M. Chitosan-based hydrogels for controlled, localized drug delivery. Adv. Drug Deliv. Rev. 2010, 62, 83–99. [Google Scholar] [CrossRef]
- Mao, S.; Sun, W.; Kissel, T. Chitosan-based formulations for delivery of DNA and siRNA. Adv. Drug Deliv. Rev. 2010, 62, 12–27. [Google Scholar] [CrossRef]
- Steinle, H.; Ionescu, T.-M.; Schenk, S.; Golombek, S.; Kunnakattu, S.-J.; Özbek, M.T.; Schlensak, C.; Wendel, H.P.; Avci-Adali, M. Incorporation of Synthetic mRNA in Injectable Chitosan-Alginate Hybrid Hydrogels for Local and Sustained Expression of Exogenous Proteins in Cells. Int. J. Mol. Sci. 2018, 19, 1313. [Google Scholar] [CrossRef]
- Han, H.D.; Mora, E.M.; Roh, J.W.; Nishimura, M.; Lee, S.J.; Stone, R.L.; Bar-Eli, M.; Lopez-Berestein, G.; Sood, A.K. Chitosan hydrogel for localized gene silencing. Cancer Boil. Ther. 2011, 11, 839–845. [Google Scholar] [CrossRef]
- Chalanqui, M.; Pentlavalli, S.; McCrudden, C.; Chambers, P.; Ziminska, M.; Dunne, N.; McCarthy, H. Influence of alginate backbone on efficacy of thermo-responsive alginate-g-P(NIPAAm) hydrogel as a vehicle for sustained and controlled gene delivery. Mater. Sci. Eng. C 2019, 95, 409–421. [Google Scholar] [CrossRef]
- Wang, K.; Kievit, F.M.; Florczyk, S.J.; Stephen, Z.R.; Zhang, M. 3D Porous Chitosan–Alginate Scaffolds as an In Vitro Model for Evaluating Nanoparticle-Mediated Tumor Targeting and Gene Delivery to Prostate Cancer. Biomacromolecules 2015, 16, 3362–3372. [Google Scholar] [CrossRef]
- Lee, J.H. Injectable hydrogels delivering therapeutic agents for disease treatment and tissue engineering. Biomater. Res. 2018, 22, 27. [Google Scholar] [CrossRef] [PubMed]
- DiMatteo, R.; Darling, N.J.; Segura, T. In situ forming injectable hydrogels for drug delivery and wound repair. Adv. Drug Deliv. Rev. 2018, 127, 167–184. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, L.; Guo, B.; Ma, P.X. Cytocompatible injectable carboxymethyl chitosan/N-isopropylacrylamide hydrogels for localized drug delivery. Carbohydr. Polym. 2014, 103, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Pentlavalli, S.; Chambers, P.; Sathy, B.N.; O’Doherty, M.; Chalanqui, M.; Kelly, D.J.; Haut-Donahue, T.; McCarthy, H.O.; Dunne, N. Simple Radical Polymerization of Poly(Alginate-Graft-N-Isopropylacrylamide) Injectable Thermoresponsive Hydrogel with the Potential for Localized and Sustained Delivery of Stem Cells and Bioactive Molecules. Macromol. Biosci. 2017, 17, 1700118. [Google Scholar] [CrossRef]
- Teotia, A.K.; Sami, H.; Kumar, A. Thermo-Responsive Polymers; Elsevier BV: Amsterdam, The Netherlands, 2015; pp. 3–43. [Google Scholar]
- Mao, Z.; Ma, L.; Yan, J.; Yan, M.; Gaoa, C.; Shen, J. The gene transfection efficiency of thermoresponsive N,N,N-trimethyl chitosan chloride-g-poly(N-isopropylacrylamide) copolymer. Biomaterials 2007, 28, 4488–4500. [Google Scholar] [CrossRef]
- Cheng, N.; Gopinath, A.; Wang, L.; Iagnemma, K.; Hosoi, A.E. Thermally Tunable, Self-Healing Composites for Soft Robotic Applications. Macromol. Mater. Eng. 2014, 299, 1279–1284. [Google Scholar] [CrossRef]
- Chuang, C.-Y.; Don, T.-M.; Chiu, W.-Y. Synthesis of chitosan-based thermo- and pH-responsive porous nanoparticles by temperature-dependent self-assembly method and their application in drug release. J. Polym. Sci. Part A: Polym. Chem. 2009, 47, 5126–5136. [Google Scholar] [CrossRef]
- Chen, J.-P.; Cheng, T.-H. Thermo-Responsive Chitosan-graft-poly(N-isopropylacrylamide) Injectable Hydrogel for Cultivation of Chondrocytes and Meniscus Cells. Macromol. Biosci. 2006, 6, 1026–1039. [Google Scholar] [CrossRef]
- Lanzalaco, S.; Armelin, E. Poly(N-isopropylacrylamide) and Copolymers: A Review on Recent Progresses in Biomedical Applications. Gels 2017, 3, 36. [Google Scholar] [CrossRef]
- Alpar, H.O.; Eyles, J.E.; Williamson, E.D.; Somavarapu, S. Intranasal vaccination against plague, tetanus and diphtheria. Adv. Drug Deliv. Rev. 2001, 51, 173–201. [Google Scholar] [CrossRef]
- Kraber, S. How to Get Started with Design-Expert Software; Stat-Ease Inc.: Minneapolis, MN, USA, 2013. [Google Scholar]
- Mi, F.-L.; Shyu, S.-S.; Chen, C.-T.; Schoung, J.-Y. Porous chitosan microsphere for controlling the antigen release of Newcastle disease vaccine: Preparation of antigen-adsorbed microsphere and in vitro release. Biomaterials 1999, 20, 1603–1612. [Google Scholar] [CrossRef]
- Hari, P.R.; Chandy, T.; Sharma, C.P. Chitosan/calcium?alginate beads for oral delivery of insulin. J. Appl. Polym. Sci. 1996, 59, 1795–1801. [Google Scholar] [CrossRef]
- Yadavalli, T.; Ramasamy, S.; Chandrasekaran, G.; Michael, I.; Therese, H.A.; Chennakesavulu, R. Dual responsive PNIPAM–chitosan targeted magnetic nanopolymers for targeted drug delivery. J. Magn. Magn. Mater. 2015, 380, 315–320. [Google Scholar] [CrossRef]
- Cao, Y.; Zhang, C.; Shen, W.; Cheng, Z.; Yu, L.L.; Ping, Q. Poly(N-isopropylacrylamide)–chitosan as thermosensitive in situ gel-forming system for ocular drug delivery. J. Control. Release 2007, 120, 186–194. [Google Scholar] [CrossRef]
- Oupicky, D.; Reschel, T.; Koňák, Č.; Oupická, L. Temperature-Controlled Behavior of Self-Assembly Gene Delivery Vectors Based on Complexes of DNA with Poly(l-lysine)-graft-poly(N-isopropylacrylamide). Macromolecules 2003, 36, 6863–6872. [Google Scholar] [CrossRef]
- Kang, Z.; Meng, Q.; Liu, K. Peptide-based gene delivery vectors. J. Mater. Chem. B 2019, 7, 1824–1841. [Google Scholar] [CrossRef]
- McCarthy, H.O.; McCaffrey, J.; McCrudden, C.M.; Zholobenko, A.; Ali, A.A.; McBride, J.W.; Massey, A.S.; Pentlavalli, S.; Chen, K.-H.; Cole, G.; et al. Development and characterization of self-assembling nanoparticles using a bio-inspired amphipathic peptide for gene delivery. J. Control. Release 2014, 189, 141–149. [Google Scholar] [CrossRef]
- Feng, X.D.; Guo, X.Q.; Qiu, K.Y. Study of the initiation mechanism of the vinyl polymerization with the system persulfate/N,N,N’,N’-tetramethylethylenediamine. Die Makromolekulare Chemie 1988, 189, 77–83. [Google Scholar] [CrossRef]
- Muzzarelli, R. Genipin-crosslinked chitosan hydrogels as biomedical and pharmaceutical aids. Carbohydr. Polym. 2009, 77, 1–9. [Google Scholar] [CrossRef]
- Ziminska, M.; Dunne, N.; Hamilton, A.R. Porous Materials with Tunable Structure and Mechanical Properties via Templated Layer-by-Layer Assembly. ACS Appl. Mater. Interfaces 2016, 8, 21968–21973. [Google Scholar] [CrossRef]
- Kuśnieruk, S.; Wojnarowicz, J.; Chodara, A.; Chudoba, T.; Gierlotka, S.; Lojkowski, W. Influence of hydrothermal synthesis parameters on the properties of hydroxyapatite nanoparticles. Beilstein J. Nanotechnol. 2016, 7, 1586–1601. [Google Scholar] [CrossRef] [PubMed]
- Philip, C.; Michelle, O.; Sreekanth, P.; Marine, C.; Helen, M.; Nicholas, D.; Binulal, S.; Hannah, P.; Daniel, K.; Tammy, H.D. Delivery of self-assembling osteogenic nanoparticles via a thermo-responsive nanofibre reinforced hydrogel. Front. Bioeng. Biotechnol. 2016, 4. [Google Scholar] [CrossRef]
- ISO 10993-5:2009 Biological Evaluation of Medical Devices — Part 5: Tests for in vitro Cytotoxicity. [Cited 1 Nov 2019]. Available online: https://www.iso.org/standard/36406.html (accessed on 12 October 2019).
- Fathi, M.; Alami-Milani, M.; Geranmayeh, M.H.; Barar, J.; Erfan-Niya, H.; Omidi, Y. Dual thermo-and pH-sensitive injectable hydrogels of chitosan/(poly(N-isopropylacrylamide-co-itaconic acid)) for doxorubicin delivery in breast cancer. Int. J. Boil. Macromol. 2019, 128, 957–964. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.-C.; Chen, C.-H.; Wang, C.-Z.; Wang, Y.-H.; Chang, J.-K.; Wang, G.-J.; Ho, M.-L.; Wang, C.-K. Preparation of porous bioceramics using reverse thermo-responsive hydrogels in combination with rhBMP-2 carriers: In vitro and in vivo evaluation. J. Mech. Behav. Biomed. Mater. 2013, 27, 64–76. [Google Scholar] [CrossRef]
- Wang, Y.; Pitto-Barry, A.; Habtemariam, A.; Canelon, I.R.; Sadler, P.J.; Barry, N.P.E. Nanoparticles of chitosan conjugated to organo-ruthenium complexes. Inorg. Chem. Front. 2016, 3, 1058–1064. [Google Scholar] [CrossRef]
- Li, G.; Zhuang, Y.; Mu, Q.; Wang, M.; Fang, Y. Preparation, characterization and aggregation behavior of amphiphilic chitosan derivative having poly (l-lactic acid) side chains. Carbohydr. Polym. 2008, 72, 60–66. [Google Scholar] [CrossRef]
- Alves, N.M.; Mano, J.F. Chitosan derivatives obtained by chemical modifications for biomedical and environmental applications. Int. J. Boil. Macromol. 2008, 43, 401–414. [Google Scholar] [CrossRef]
- Sosnik, A.; Imperiale, J.C.; Vázquez-González, B.; Raskin, M.M.; Muñoz-Muñoz, F.; Burillo, G.; Cedillo, G.; Bucio, E. Mucoadhesive thermo-responsive chitosan-g-poly( N-isopropylacrylamide) polymeric micelles via a one-pot gamma-radiation-assisted pathway. Colloids Surfaces B: Biointerfaces 2015, 136, 900–907. [Google Scholar] [CrossRef]
- Hamman, J. Chitosan Based Polyelectrolyte Complexes as Potential Carrier Materials in Drug Delivery Systems. Mar. Drugs 2010, 8, 1305–1322. [Google Scholar] [CrossRef]
- Ritthidej, G.C. Nasal Delivery of Peptides and Proteins with Chitosan and Related Mucoadhesive Polymers; Elsevier BV: Amsterdam, The Netherlands, 2011; pp. 47–68. [Google Scholar]
- Rwei, S.-P.; Tuan, H.N.A.; Chiang, W.-Y.; Way, T.-F.; Hsu, Y.-J. Synthesis and Drug Delivery Application of Thermo- and pH-Sensitive Hydrogels: Poly(β-CD-co-N-Isopropylacrylamide-co-IAM). Materials 2016, 9, 1003. [Google Scholar] [CrossRef]
- Mu, Q.; Fang, Y. Preparation of thermosensitive chitosan with poly(N-isopropylacrylamide) side at hydroxyl group via O-maleoyl-N-phthaloyl-chitosan (MPCS). Carbohydr. Polym. 2008, 72, 308–314. [Google Scholar] [CrossRef]
- Queiroz, M.; Melo, K.R.T.; Sabry, D.A.; Sassaki, G.L.; Rocha, H.A.O. Does the Use of Chitosan Contribute to Oxalate Kidney Stone Formation? Mar. Drugs 2014, 13, 141–158. [Google Scholar] [CrossRef] [PubMed]
- Gunathilake, T.M.S.U.; Ching, Y.C.; Chuah, C.H. Enhancement of Curcumin Bioavailability Using Nanocellulose Reinforced Chitosan Hydrogel. Polymers 2017, 9, 64. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Koh, J. Physiochemical, Optical and Biological Activity of Chitosan-Chromone Derivative for Biomedical Applications. Int. J. Mol. Sci. 2012, 13, 6102–6116. [Google Scholar] [CrossRef] [PubMed]
- Mellati, A.; Dai, S.; Bi, J.; Jin, B.; Zhang, H. A biodegradable thermosensitive hydrogel with tuneable properties for mimicking three-dimensional microenvironments of stem cells. RSC Adv. 2014, 4, 63951–63961. [Google Scholar] [CrossRef]
- Ma, X.; Li, R.; Ren, J.; Lv, X.C.; Zhao, X.H.; Ji, Q.; Xia, Y.Z. Restorable, high-strength poly(N-isopropylacrylamide) hydrogels constructed through chitosan-based dual macro-cross-linkers with rapid response to temperature jumps. RSC Adv. 2017, 7, 47767–47774. [Google Scholar] [CrossRef]
- Bao, H.; Li, L.; Leong, W.C.; Gan, L.H. Thermo-Responsive Association of Chitosan-graft-Poly(N-isopropylacrylamide) in Aqueous Solutions. J. Phys. Chem. B 2010, 114, 10666–10673. [Google Scholar] [CrossRef]
- Das, D.; Pham, T.T.H.; Noh, I. Characterizations of hyaluronate-based terpolymeric hydrogel synthesized via free radical polymerization mechanism for biomedical applications. Colloids Surf. B 2018, 170, 64–75. [Google Scholar] [CrossRef]
- Wang, L.; Li, B.; Xu, F.; Xu, Z.; Wei, D.; Feng, Y.; Wang, Y.; Jia, D.; Zhou, Y. UV-crosslinkable and thermo-responsive chitosan hybrid hydrogel for NIR-triggered localized on-demand drug delivery. Carbohydr. Polym. 2017, 174, 904–914. [Google Scholar] [CrossRef]
- Strachota, B.; Zhigunov, A.; Konefał, R.; Puffr, R.; Matějka, L.; Spěváček, J.; Dybal, J. Poly(N-isopropylacrylamide)-clay based hydrogels controlled by the initiating conditions: Evolution of structure and gel formation. Soft Matter 2015, 11, 9291–9306. [Google Scholar] [CrossRef]
- Neta, P.; Maruthamuthu, P.; Carton, P.M.; Fessenden, R.W. Formation and reactivity of the amino radical. J. Phys. Chem. 1978, 82, 1875–1878. [Google Scholar] [CrossRef]
- Shirangi, M.; Toraño, J.S.; Sellergren, B.; Hennink, W.E.; Somsen, G.W.; Van Nostrum, C.F. Methyleneation of Peptides by N,N,N,N-Tetramethylethylenediamine (TEMED) under Conditions Used for Free Radical Polymerization: A Mechanistic Study. Bioconjugate Chem. 2014, 26, 90–100. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.-W.; Liu, X.; Miller, A.L.; Cheng, Y.-S.; Yeh, M.-L.; Lu, L. Strengthening injectable thermo-sensitive NIPAAm-g-chitosan hydrogels using chemical cross-linking of disulfide bonds as scaffolds for tissue engineering. Carbohydr. Polym. 2018, 192, 308–316. [Google Scholar] [CrossRef] [PubMed]
- Heuts, J.P.A.; Russell, G.T.; Smith, G.B.; Van Herk, A.M. The Importance of Chain-Length Dependent Kinetics in Free-Radical Polymerization: A Preliminary Guide. Macromol. Symp. 2007, 248, 12–22. [Google Scholar] [CrossRef]
- Uliyanchenko, E.; Van Der Wal, S.; Schoenmakers, P.J. Challenges in polymer analysis by liquid chromatography. Polym. Chem. 2012, 3, 2313. [Google Scholar] [CrossRef]
- Brar, A.S.; Goyal, A.K.; Hooda, S. Two-dimensional NMR studies of acrylate copolymers. Pure Appl. Chem. 2009, 81, 389–415. [Google Scholar] [CrossRef]
- Beshah, K. Sequential assignments of polymers by 2D NMR techniques: Application to stereochemical configuration assignment of poly(acrylic acid). Die Makromol Chemie. 1993, 194, 3311–3321. [Google Scholar] [CrossRef]
- Mirau, P.A. 2D NMR STUDIES OF SYNTHETIC POLYMERS. Bull. Magn. Reson. 1992, 13, 109–137. [Google Scholar]
- Vasavi, Y. Heteronuclear Multible Bond Correlation Spectroscopy-An Overview. Artic. Int. J. Pharm. Tech. Res. 2011, 3, 1410–1422. [Google Scholar]
- Huang, H.; Qi, X.; Chen, Y.; Wu, Z. Thermo-sensitive hydrogels for delivering biotherapeutic molecules: A review. Saudi Pharm. J. 2019, 27, 990–999. [Google Scholar] [CrossRef]
- Si, S.; Zhou, R.; Xing, Z.; Xu, H.; Cai, Y.; Zhang, Q. A study of hybrid organic/inorganic hydrogel films based on in situ-generated TiO2 nanoparticles and methacrylated gelatin. Fibers Polym. 2013, 14, 982–989. [Google Scholar] [CrossRef]
- Barbieri, M.; Cellini, F.; Cacciotti, I.; Peterson, S.D.; Porfiri, M. In situ temperature sensing with fluorescent chitosan-coated PNIPAAm/alginate beads. J. Mater. Sci. 2017, 52, 12506–12512. [Google Scholar] [CrossRef]
- Huang, K.-T.; Huang, C.-J. Novel Zwitterionic Nanocomposite Hydrogel as Effective Chronic Wound Healing Dressings; Springer Science and Business Media LLC: Berlin, Germany, 2015. [Google Scholar]
- Boucard, N.; David, L.; Rochas, C.; Montembault, A.; Viton, C.; Domard, A. Polyelectrolyte Microstructure in Chitosan Aqueous and Alcohol Solutions. Biomacromolecules 2007, 8, 1209–1217. [Google Scholar] [CrossRef] [PubMed]
- N, V.G.; Shivakumar, H. Investigation of Swelling Behavior and Mechanical Properties of a pH-Sensitive Superporous Hydrogel Composite. Iran. J. Pharm. Res.: IJPR 2012, 11, 481–493. [Google Scholar]
- Haq, M.A.; Su, Y.; Wang, D. Mechanical properties of PNIPAM based hydrogels: A review. Mater. Sci. Eng. C 2017, 70, 842–855. [Google Scholar] [CrossRef]
- Xu, R.; Ma, S.; Wu, Y.; Lee, H.; Zhou, F.; Liu, W. Adaptive control in lubrication, adhesion, and hemostasis by Chitosan-Catechol-pNIPAM. Biomater. Sci. 2019, 7, 3599–3608. [Google Scholar] [CrossRef]
- Zhang, X.; Cheng, L.; Feng, L.; Peng, Y.; Zhou, Z.; Yin, G.-Y.; Li, W.; Zhang, A. Thermoresponsive dendronized chitosan-based hydrogels as injectable stem cell carriers. Polym. Chem. 2019, 10, 2305–2315. [Google Scholar] [CrossRef]
- Yan, C.; Pochan, D.J. Rheological properties of peptide-based hydrogels for biomedical and other applications. Chem. Soc. Rev. 2010, 39, 3528–3540. [Google Scholar] [CrossRef]
- Ren, Z.; Wang, Y.; Ma, S.; Duan, S.; Yang, X.; Gao, P.; Zhang, X.; Cai, Q. Effective Bone Regeneration Using Thermosensitive Poly(N-Isopropylacrylamide) Grafted Gelatin as Injectable Carrier for Bone Mesenchymal Stem Cells. ACS Appl. Mater. Interfaces 2015, 7, 19006–19015. [Google Scholar] [CrossRef]
- Yan, C.; Altunbas, A.; Yucel, T.; Nagarkar, R.P.; Schneider, J.P.; Pochan, D.J. Injectable solid hydrogel: Mechanism of shear-thinning and immediate recovery of injectable β-hairpin peptide hydrogels. Soft Matter 2010, 6, 5143–5156. [Google Scholar] [CrossRef]
- ISO 11040-4:2015 Prefilled Syringes—Part 4: Glass Barrels for Injectables and Sterilized Subassembled Syringes Ready for Filling. [cited 1 Nov 2019]. Available online: https://www.iso.org/standard/58079.html (accessed on 10 October 2019).
- Lencina, M.M.S.; Iatridi, Z.; Villar, M.A.; Tsitsilianis, C. Thermoresponsive hydrogels from alginate-based graft copolymers. Eur. Polym. J. 2014, 61, 33–44. [Google Scholar] [CrossRef]
- Liu, M.; Song, X.; Wen, Y.; Zhu, J.; Li, J. Injectable Thermoresponsive Hydrogel Formed by Alginate-g-Poly(N-isopropylacrylamide) That Releases Doxorubicin-Encapsulated Micelles as a Smart Drug Delivery System. ACS Appl. Mater. Interfaces 2017, 9, 35673–35682. [Google Scholar] [CrossRef]
- Lencina, M.M.S.; Rizzo, C.; Demitri, C.; Andreucetti, N.; Maffezzoli, A. Rheological analysis of thermo-responsive alginate/PNIPAAm graft copolymers synthesized by gamma radiation. Radiat. Phys. Chem. 2019, 156, 38–43. [Google Scholar] [CrossRef]
- Kang, J.; Schwendeman, S.P. Pore Closing and Opening in Biodegradable Polymers and Their Effect on the Controlled Release of Proteins. Mol. Pharm. 2007, 4, 104–118. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, B.M.; Schwendeman, S.P. Characterization of the initial burst release of a model peptide from poly(D,L-lactide-co-glycolide) microspheres. J. Control. Release 2002, 82, 289–307. [Google Scholar] [CrossRef]
- Ribeiro, C.A.; Martins, M.V.S.; Bressiani, A.H.; Bressiani, J.C.; Leyva, M.E.; De Queiroz, A.A.A. Electrochemical preparation and characterization of PNIPAM-HAp scaffolds for bone tissue engineering. Mater. Sci. Eng. C 2017, 81, 156–166. [Google Scholar] [CrossRef]
- A Cooperstein, M.; Canavan, H.E. Assessment of cytotoxicity of (N-isopropyl acrylamide) and poly(N-isopropyl acrylamide)-coated surfaces. Biointerphases 2013, 8, 19. [Google Scholar] [CrossRef]
- Fujimoto, K.L.; Ma, Z.; Nelson, D.M.; Hashizume, R.; Guan, J.; Tobita, K.; Wagner, W.R. Synthesis, characterization and therapeutic efficacy of a biodegradable, thermoresponsive hydrogel designed for application in chronic infarcted myocardium. Biomater. 2009, 30, 4357–4368. [Google Scholar] [CrossRef]
- Capella, V.; Rivero, R.E.; Liaudat, A.C.; Ibarra, L.E.; Roma, D.A.; Alustiza, F.; Mañas, F.; Barbero, C.A.; Bosch, P.; Rivarola, C.R.; et al. Cytotoxicity and bioadhesive properties of poly-N-isopropylacrylamide hydrogel. Heliyon 2019, 5, e01474. [Google Scholar] [CrossRef]
- Gandhi, A.; Paul, A.; Sen, S.O.; Sen, K.K. Studies on thermoresponsive polymers: Phase behaviour, drug delivery and biomedical applications. Asian J. Pharm. Sci. 2015, 10, 99–107. [Google Scholar] [CrossRef]
- Ohya, S.; Nakayama, Y.; Matsuda, T. In vivo evaluation of poly(N-isopropylacrylamide) (PNIPAM)-grafted gelatin as an in situ-formable scaffold. J. Artif. Organs 2005, 7, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Howling, G.I.; Dettmar, P.W.; Goddard, P.A.; Hampson, F.C.; Dornish, M.; Wood, E.J. The effect of chitin and chitosan on the proliferation of human skin fibroblasts and keratinocytes in vitro. Biomaterials 2001, 22, 2959–2966. [Google Scholar] [CrossRef]
- Kondiah, P.J.; Choonara, Y.E.; Kondiah, P.P.; Marimuthu, T.; Kumar, P.; Du Toit, L.C.; Pillay, V. A Review of Injectable Polymeric Hydrogel Systems for Application in Bone Tissue Engineering. Molecules 2016, 21, 1580. [Google Scholar] [CrossRef] [PubMed]
- Steitz, J.; Britten, C.M.; Wolfel, T.; Tüting, T. Effective induction of anti-melanoma immunity following genetic vaccination with synthetic mRNA coding for the fusion protein EGFP.TRP2. Cancer Immunol. Immunother. 2005, 55, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Chu, X.; Li, N.; Fu, L.; Gu, H.; Zhang, N. Engineered DNA nanodrugs alleviate inflammation in inflammatory arthritis. Int. J. Pharm. 2020, 577, 119047. [Google Scholar] [CrossRef] [PubMed]
- Cole, G.; Ali, A.A.; McErlean, E.; Mulholland, E.J.; Short, A.; McCrudden, C.M.; McCaffrey, J.; Robson, T.; Kett, V.L.; Coulter, J.A.; et al. DNA vaccination via RALA nanoparticles in a microneedle delivery system induces a potent immune response against the endogenous prostate cancer stem cell antigen. Acta Biomater. 2019, 96, 480–490. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Lee, B.; Heo, J.H.; Lee, K.E.; Shankar, P.; Han, K.-H.; Lee, J.H. The Effect of ζ-Potential and Hydrodynamic Size on Nanoparticle Interactions in Hydrogels. Part. Part. Syst. Charact. 2018, 36, 1800292. [Google Scholar] [CrossRef]
- Ismail, F.A.; Napaporn, J.; Hughes, J.A.; Brazeau, G.A. In Situ Gel Formulations for Gene Delivery: Release and Myotoxicity Studies. Pharm. Dev. Technol. 2000, 5, 391–397. [Google Scholar] [CrossRef]
- Foroozandeh, P.; Aziz, A. Insight into Cellular Uptake and Intracellular Trafficking of Nanoparticles. Nanoscale Res. Lett. 2018, 13, 339. [Google Scholar] [CrossRef]









© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ziminska, M.; Wilson, J.J.; McErlean, E.; Dunne, N.; McCarthy, H.O. Synthesis and Evaluation of a Thermoresponsive Degradable Chitosan-Grafted PNIPAAm Hydrogel as a “Smart” Gene Delivery System. Materials 2020, 13, 2530. https://doi.org/10.3390/ma13112530
Ziminska M, Wilson JJ, McErlean E, Dunne N, McCarthy HO. Synthesis and Evaluation of a Thermoresponsive Degradable Chitosan-Grafted PNIPAAm Hydrogel as a “Smart” Gene Delivery System. Materials. 2020; 13(11):2530. https://doi.org/10.3390/ma13112530
Chicago/Turabian StyleZiminska, Monika, Jordan J. Wilson, Emma McErlean, Nicholas Dunne, and Helen O. McCarthy. 2020. "Synthesis and Evaluation of a Thermoresponsive Degradable Chitosan-Grafted PNIPAAm Hydrogel as a “Smart” Gene Delivery System" Materials 13, no. 11: 2530. https://doi.org/10.3390/ma13112530
APA StyleZiminska, M., Wilson, J. J., McErlean, E., Dunne, N., & McCarthy, H. O. (2020). Synthesis and Evaluation of a Thermoresponsive Degradable Chitosan-Grafted PNIPAAm Hydrogel as a “Smart” Gene Delivery System. Materials, 13(11), 2530. https://doi.org/10.3390/ma13112530