2.1. Materials
The starting materials for synthesis of (tartrate-salen)Mn(III) polymer complexes including 1,2-diaminocyclohexane (mixture of isomers) (Sigma-Aldrich Inc., St. Louis, MO, USA);
L-(+)- and
D-(-)-tartaric acids (Sigma-Aldrich Inc., St. Louis, MO, USA); sodium
L-(+)- and
D-(-)-tartrate dihydrates (Sigma-Aldrich Inc., St. Louis, MO, USA); 2-
tert-butylphenol(Sigma-Aldrich Inc., St. Louis, MO, USA); paraformaldehyde (Sigma-Aldrich Inc., St. Louis, MO, USA); tetrabutylammonium bromide (TBAB) (Sigma-Aldrich Inc., St. Louis, MO, USA); catalytic substrates such as styrene, α-methylstyrene (Sigma-Aldrich Inc., St. Louis, MO, USA),
trans-stilbene (Sigma-Aldrich Inc., St. Louis, MO, USA), and indene(Sigma-Aldrich Inc., St. Louis, MO, USA); materials that were used for modification of supports like 3-aminopropyltrimethoxysilane (3-APTMS), Pluronic P123 (average
Mn, 5800), tetraethyl orthosilicate (TEOS) (Sigma-Aldrich Inc., St. Louis, MO, USA), and 3-chloroperoxybenzoic acid (
mCPBA) (Sigma-Aldrich Inc., St. Louis, MO, USA), along with metal or non-metal salts; as well as HPLC-grade solvents were bought from Sigma-Aldrich Corporation without further purifications(Sigma-Aldrich Inc., St. Louis, MO, USA). The local suppliers provided organic solvents and silica gel of column and thin layer chromatography (all analytical reagents). At the same time, synthetic intermediates including (
R,R)-1,2-diammoniumcyclohexane mono-(+)-tartrate salt [
24], (
S,S)-1,2-diammoniumcyclohexane mono-(-)-tartrate salt [
24], 3-
tert-butyl-5-chloromethyl-2-hydroxybenzaldehyde [
24], Zn(II)/Al(III) LDH-[C
6H
5COO] (Zn/Al LDH, [Zn
2.09Al
0.69(OH)
5.23]
1.00[C
6H
5COO]
0.62·H
2O) [
25], Mn5 [
26], and iodosylbenzene (PhIO) [
27] were synthesized according to literatures.
2.2. Characterization
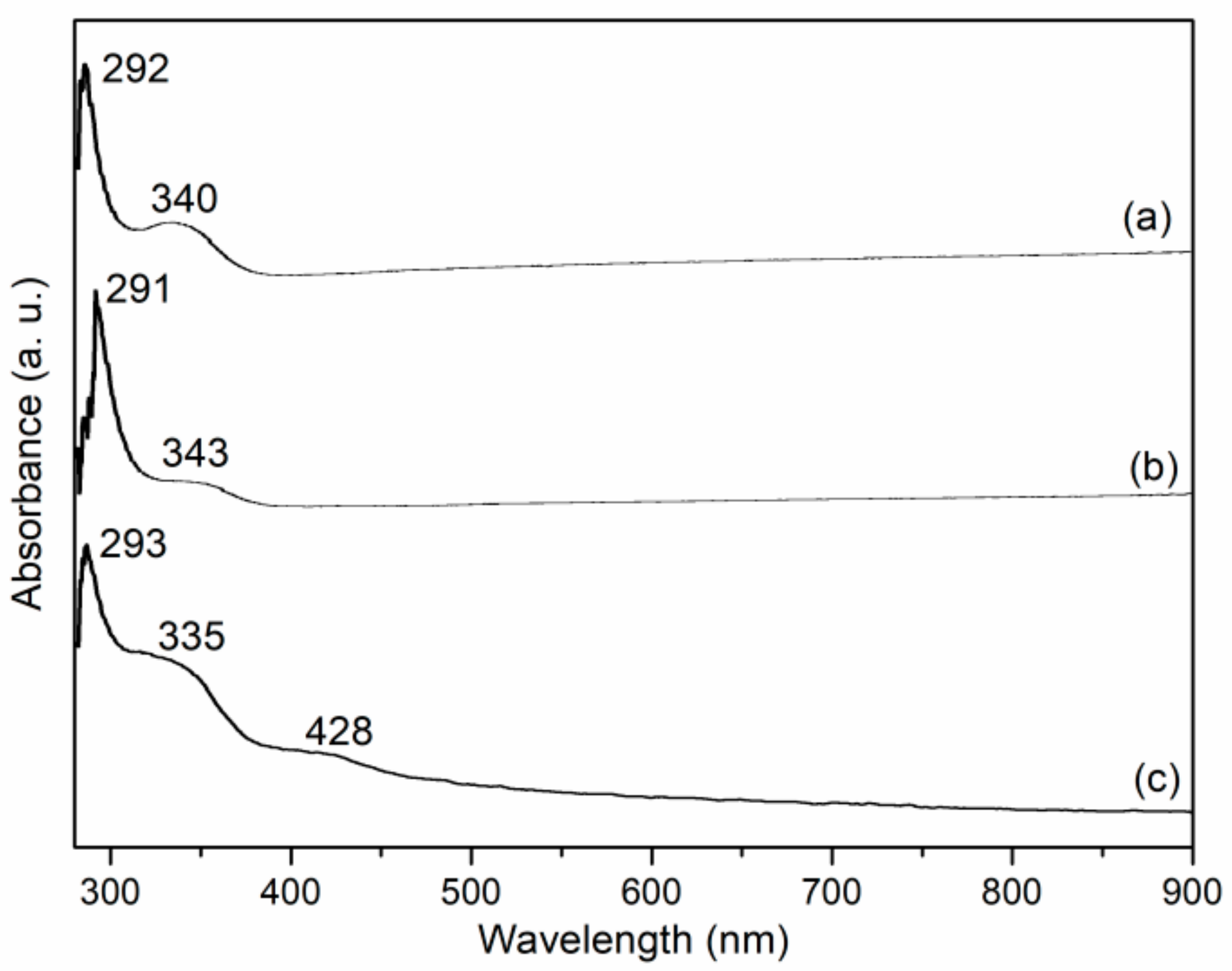
H1 NMR of organic intermediates were tested on Bruker ADVANCE III (400MHz) (Bruker Corporation, Bruker ADVANCE III, Billerica, MA, USA). UV-Vis spectra were collected on Shimadzu UV-1800 (sample of 10−3 mol L−1 Mn in CH2Cl2, 290–550 nm) (Shimadzu Corporation, UV-1800, Kyoto, Japan). FT-IR spectroscopy was measured in KBr pellets over Bruker Tensor 27 spectrometer (400–4000 cm−1) (Bruker Corporation, Bruker Tensor 27 spectrometer, Billerica, MA, USA). The elemental analyses (C, H, and N) were carried out on Elementar VarioEL III. ESI-HRMS (positive molecular ions) were probed on microOTOF-Q II (Bruker.Daltonics production) (Bruker Corporation, microOTOF-Q II, Billerica, MA, USA). The number (Mn)- and weight (Mw)-average molecular and polydispersity indices (PDI, Mw/Mn) of four polymeric salen ligands were detected on gel permeation chromatography (GPC, Waters 1515–2414, Styragel HT3 THF column, 40 °C, THF as eluent, 1 mL min−1 flow rate, calibrated by polystyrene standard) (Waters Corporation, Waters 1515–2414, Milford, MA, USA). Metal contents of synthetic catalysts were detected by inductively coupled plasma atomic emission spectrometry (ICP-AES) on ICPE-9000 (Shimadzu Corporation, ICPE-9000, Kyoto, Japan). Optional rotations of chiral ligands were obtained on Perkin-Elmer 341 (λ = 587 nm, 25 °C, values formatted as absolute rotation , sample of 0.01 g mL−1 in CH2Cl2) (PerkinElmer, Inc., Perkin-Elmer 341, Waltham, MA, USA).
Porosity parameters including BET surface area, pore volume, pore radius, and pore size distribution of catalysts were determined on Micromeritics ASAP 2020 (testing N2 adsorption isotherms, 77.35 K) (Micromeritics Instrument Corporation, Micromeritics ASAP 2020, Norcross, GA, USA). Surface area was obtained by using multi-point Brunauer-Emmett-Teller (BET) method (P/P0 = 0.06–0.3). Total pore volume was calculated from adsorbed N2 (P/P0 = 0.97); pore volume and pore radius were determined by using Barrett-Joyner-Halenda (BJH) method. Bulk densities of the synthesized catalysts were detected on SOTAX TD2 density detector (CAMAG Corporation, SOTAX TD2, Muttenz, Switzerland). X-ray photoelectron spectroscopy (XPS) were carried out on Kratos Axis Ultra DLD (Kratos Analytical Ltd., Kratos Axis Ultra DLD, Manchester, UK), irradiation source was monochromatic Al Kα X-ray (1486.6 eV). The X-ray diffraction (XRD) of powdered catalysts was tested on Shimadzu XRD-6000 (Cu-Ka1, λ = 1.54059 Å) (Shimadzu Corporation, XRD-6000, Kyoto, Japan), and diffractions were collected (2θ = 4–55°, 0.02° intervals). Scanning electron microscopy (SEM) was carried out on JSM-6700F (JEOL) (JEOL, Ltd., JSM-6700F, Tokyo, Japan). Particle size and zeta potential of the synthesized catalysts were measured on Zetasizer Nano ZS90, Malvern (in CH2Cl2, 298 K) (Malvern Panalytical Ltd., Zetasizer Nano ZS90, Malvern, UK).
Thin layer chromatography (TLC) was performed on glass plates coated with GF254 silica gel (coloration in phosphomolybdic acid (PMA)/ethanol, 5 wt.%). Conversions of substrates and enantiomeric excesses of epoxide products were both detected by chiral HPLC (Daicel Chiralcel OD-H column, 150 mm × 4.6 mm; 5 μm particle size; mobile phase: n-hexane/2-propanol, 97/3, v/v; flow rate: 1.0 mL min−1; column temperature: 300 K; pressure: 5.0–7.0 MPa; sample concentration: 1.0 mg mL−1 in n-hexane; 10 μL injected; Waters chromatograph, system controller: Waters 1525, binary hplc pump; UV-Vis detector: Waters 2998, photodiode array detector; UV detection: 242 nm, λ = 210–400 nm).
2.3. Synthesis of Chiral Dimeric Salicylaldehyde (Compound 3, Scheme 1)
As shown in
Scheme 1, 3-
tert-butyl-5-chloromethyl-2-hydroxybenzaldehyde (Compound 1,
Scheme 1; 3.25 g, 14.4 mmol), sodium
L-(+)-tartrate dihydrate (compound 2a,
Scheme 1; 1.65 g, 7.2 mmol), and dry triethylamine (30 mL) were mixed into a round-bottomed flask (250 mL). With continuous stirring, the resulting orange solution was heated at 110 °C for 3 h. It can be seen that small crystals (NaCl) were gradually precipitated from the purple solution when the reaction was continued. Then, the solvent was removed under reduced pressure, after the total solution was cooled to room temperature. The CH
2Cl
2 (100 mL) was subsequently added to dilute residue. The CH
2Cl
2 layer obtained was washed with water (3 × 50 mL) and brine (3 × 50 mL), then dried over anhydrous Na
2SO
4, and filtered. After the removal of solvent by using rotary evaporation, residue was further purified by using column chromatography (SiO
2, 200–300 mesh; petroleum ether/ethyl acetate, 6/1,
v/
v, adding a few drops of Et
3N) to give chiral salicylaldehyde dimer (compound 3a,
Scheme 1; yellow sticky solid, 1.49 g, 39% yield).
1H NMR (CDCl
3, 400 MHz) δ
H (ppm): 1.27 (9H, s), 3.95 (4H, s), 4.55 (2H, s), 7.18–7.23 (2H, m), 7.50–7.61 (2H, m), 9.85 (2H, s). FT-IR (tested in KBr pellets) σ (cm
−1): 3432 (m), 3398–3280 (br, s), 2939 (s), 2856 (s), 1719 (vs). ESI-HRMS (positive, m/z): 553.6002 (Calcd. for [M+Na]
+ 553.5519).
= −65 (
c = 0.01 g mL
−1, CH
2Cl
2). Ideal formula of 3a is C
28H
34O
10. Anal. Calcd.: C, 63.4; H, 6.4. Found: C, 62.8; H, 7.0. Synthesis of 3b was identical to 3a except for replacement of sodium
L-(+)-tartrate dihydrate with sodium
D-(-)-tartrate dihydrate (
Section S1, Supplementary data).
2.4. Synthesis of Chiral Ligands (Compound L, Scheme 1)
(
R,R)-1,2-diammoniumcyclohexane mono-(+)-tartrate salt (0.45 g, 1.7 mmol), anhydrous K
2CO
3 (0.47 g, 3.4 mmol), and distilled water (15 mL) were mixed into a round-bottomed flask (250 mL) at room temperature. Then, dry ethanol (6 mL) was introduced under magnetic stirring. The resulting cloudy solution was heated at 75 °C for 2 h under stirring, and then cooled to room temperature. Next, this solution was extracted by CH
2Cl
2 (4 × 5 mL) to obtain free diamine (Compound 4a,
Scheme 1). The CH
2Cl
2 phase was then slowly added to a pre-prepared ethanol solution of compound 3a (0.9 g, 1.7 mmol, in 20 mL,
Scheme 1) at room temperature under stirring. The orange solution was refluxed at 80 °C for 3 h, whose solvent was evaporated under reduced pressure. The crude product thus obtained was dissolved in CH
2Cl
2 (30 mL), the organic layer was washed with distilled water (50 mL), brine (50 mL), dried over anhydrous Na
2SO
4, and then filtered. Solvent was removed by using rotary evaporation, and Compound L1 was obtained as yellow sticky solid (0.96 g).
1H NMR (400 MHz, CDCl
3) δ
H (ppm): 1.41 (9H, s), 1.40–1.56 (8H, m), 2.33 (2H, s), 3.93 (4H, s), 4.53 (2H, s), 7.40–7.44 (2H, m), 7.50–7.55 (2H, m), 9.88 (2H, s). FT-IR (KBr) σ (cm
−1): 3442 (br, s), 2958 and 2867 (m), 2930 (m), 1772 (w), 1649 (s), 1559 (w), 1267 (w).
= −116 (
c = 0.01 g mL
−1, CH
2Cl
2).
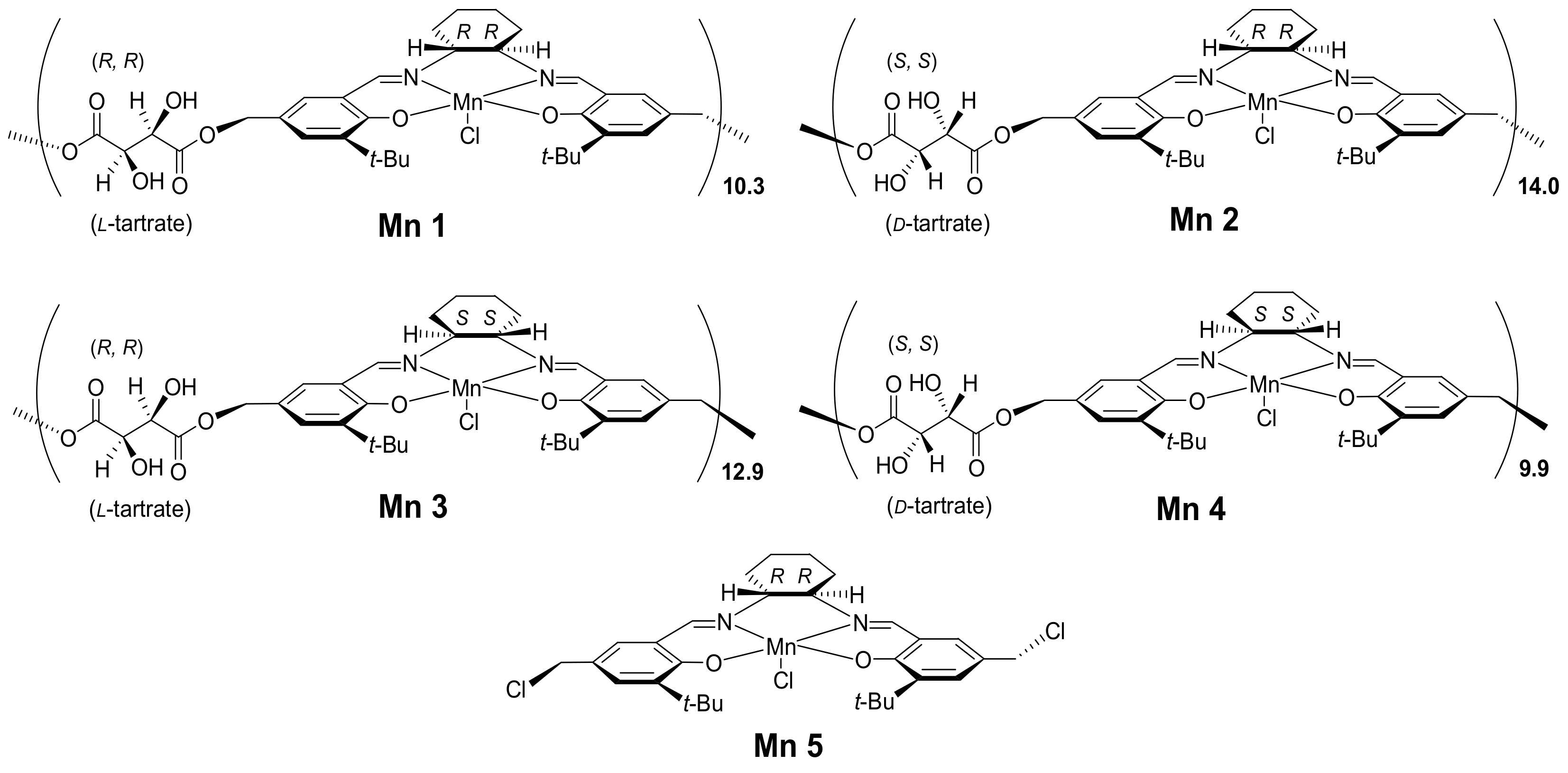
Mn = 6262,
Mw = 13150, PDI (
Mw/
Mn) = 2.1. Based on
Mn, number of tartrate-salen monomers was 10.3, then the ideal formula of L1 was deduced as (C
4H
4O
6·C
30H
40O
2N
2)
10.3. Anal. Calcd.: C, 67.1; H, 7.2; N, 4.6. Found: C, 66.8; H, 6.7; N, 5.5. Synthesis of Compounds L2, L3, and L4 were identical to L1 except for substitution of chiral tartrate and diamine counterparts (
Section S2, Supplementary data).
2.5. Synthesis of (Tartrate-Salen)Mn(III) Polymer Complexes (Compound Mn, Scheme 1)
Compound L1 (
Scheme 1, 0.81 g), Mn(OAc)
2·4H
2O (0.62 g, 2.55 mmol), and anhydrous ethanol (10 mL) were mixed into a round-bottomed flask (100 mL), which was then refluxed at 75 °C for 3 h under N
2 protection. After protection was removed, LiCl·H
2O (0.46 g, 7.65 mmol) was added. The solution that obtained was further stirred at 75 °C for 1 h in air, then solvent was evaporated under reduced pressure, and the resulting brown powders were collected and thoroughly washed with distilled water (3 × 30 mL), then Compound Mn1 was obtained as brown powders (0.76 g). FT-IR (in KBr pellets) σ (cm
−1): 3433 (br, s), 2957 and 2868 (m), 2930 (m), 1741 (w), 1618 (s), 1544 (w), 1268 (w), 567 (w).
= +129° (
c = 0.01 g mL
−1, CH
2Cl
2). According to L1, the ideal formula of Compound Mn1 was summarized as (C
4H
4O
6·C
30H
38O
2N
2MnCl·2H
2O)
10.3. Anal. Calcd.: C, 55.7; H, 6.2; N, 3.8. Found: C, 56.3; H, 6.1; N, 4.5. Mn
3+ content was 1.10 mmol g
−1 by ICP-AES. The syntheses of other polymer complexes (Compounds Mn2, Mn3, Mn4) were identical to Mn1 (
Section S3, Supplementary data).
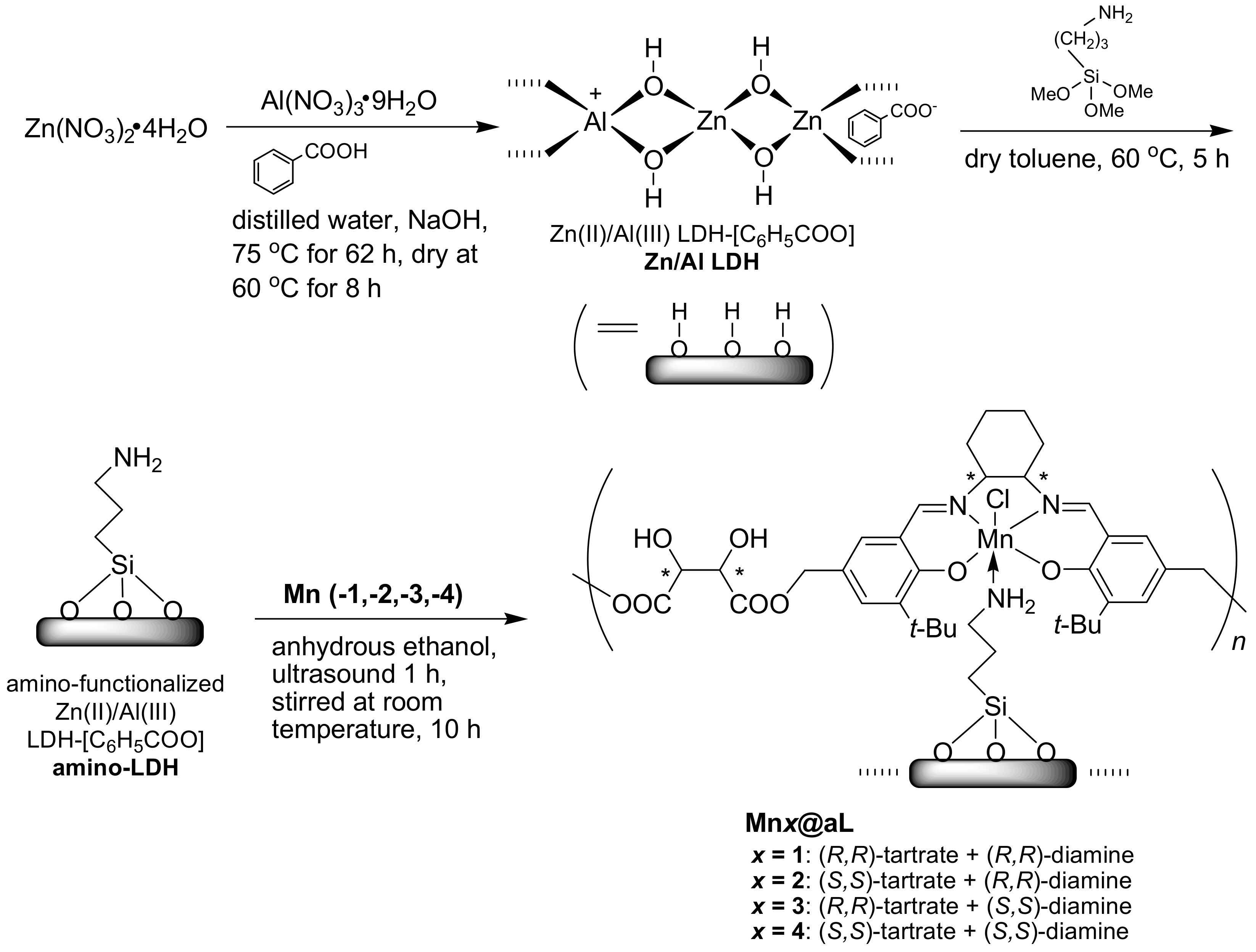
2.7. Synthesis of Amino-LDH-Supported (Tartrate-Salen)Mn(III) Polymers (Mnx@aL, x = 1, 2, 3, 4)
Compound Mn1 (
Scheme 1, 0.20 g), amino-LDH (0.40 g), and anhydrous ethanol (20 mL) were mixed into a round-bottomed flask (250 mL), which was then dispersed under ultrasound for 1 h, and stirred for 10 h at room temperature. After total solvent was removed by rotary-evaporation, the residue was collected and washed with anhydrous ethanol (2 × 5 mL). Mn1@aL was obtained as yellowish-brown powders (0.42 g). FT-IR (KBr) σ (cm
−1): 3711 and 3674 (both w), 3648 and 3589 (both m), 3443 (br, s), 2960 and 2869 (both w), 2925 (w), 1770 (w), 1697 (m), 1626 (m), 1558 (m), 1261 (w), 1134 (s), 876 (w), 566 (w), and 420 (w). The ideal formula of Mn1@aL was [Zn
2.09Al
0.69(OH)
5.23]
1.00[C
6H
5COO]
0.62[C
3H
8NO
3Si]
0.21[C
4H
4O
6·C
30H
38O
2N
2MnCl]
0.06. Anal. Calcd.: C, 21.6; H, 3.2; N, 1.1. Found: C, 20.3; H, 2.6; N, 1.1. Mn
3+ content was 0.18 mmol g
−1 determined by ICP-AES. Mn
x@aL (
x = 2, 3, 4) were prepared according to the same process (
Section S4, Supplementary data).
2.8. Catalytic Activity
Alkene substrate (1 mmol), catalyst (1.5–6 mol% Mn according to alkene), oxidant (PhIO or m-CPBA, 1.2 mmol), NH4OAc (co-catalyst, 0.12 mmol), and CH2Cl2 (or other solvents, 5 mL) were mixed into a round-bottomed flask (100 mL) under ice bath (0 °C). The mixture was monitored by TLC with PMA coloration under vigorous stirring (petroleum ether/CH2Cl2, 2/1, v/v; Rf of styrene, α-methylstyrene, trans-stilbene, and indene: 0.89, 0.86, 0.72, 0.75; Rf of corresponding epoxides: 0.26, 0.23, 0.36, 0.32). After 6 h, the mixture was concentrated to dryness under reduced pressure, residue was extracted by using n-hexane (3 × 5 mL), and the left solid catalysts were combined with consumables for recycling. Hexane layer was concentrated under reduced pressure, and crude product was purified by a short column chromatography (alkaline Al2O3; petroleum ether/CH2Cl2, 2/1, v/v, a few drops of Et3N), then both conversion and e.e. value were reported on chiral HPLC.

 ,
, 






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

 and
and
 and
and

 and
and

 and
and