Preparation and Structural Characterization of Complex Oxide Eutectic Precursors from Polymer–Salt Xerogels Obtained by Microwave-Assisted Drying


Abstract
:
1. Introduction
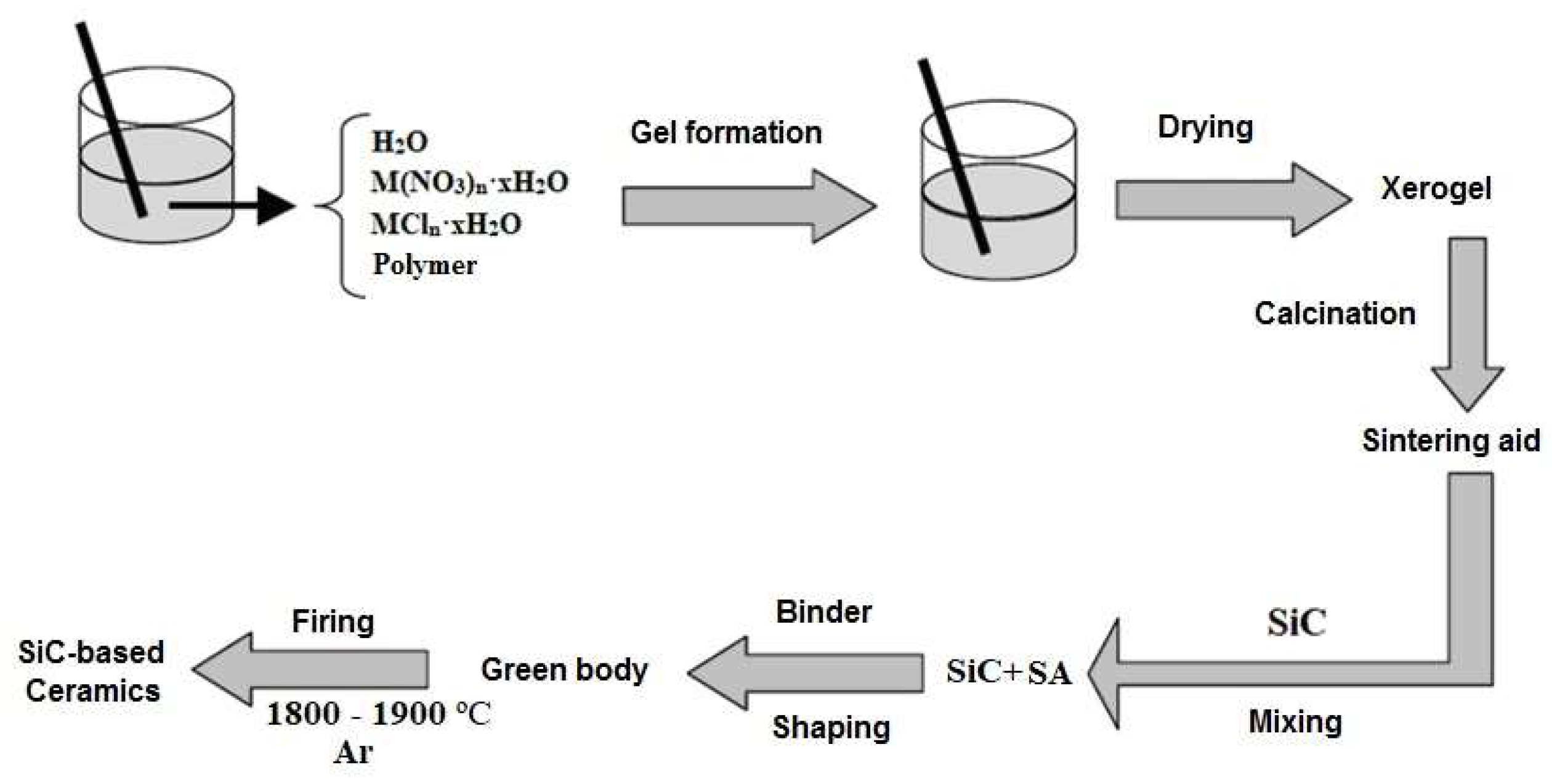
- Solvent evaporation and formation of gel structure;
- Gel drying into xerogel;
- Xerogel calcination to obtain the desired composition.
2. Materials and Methods
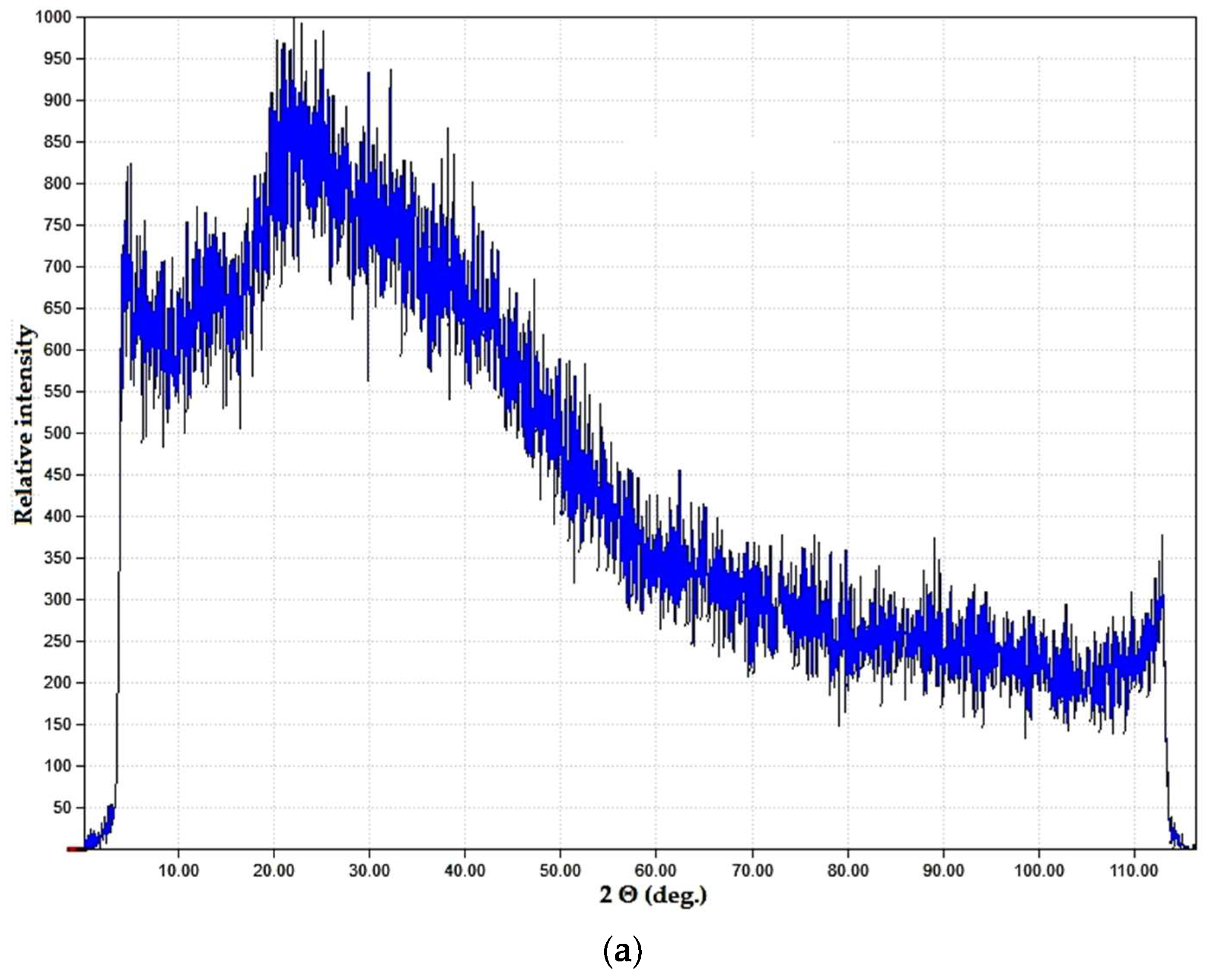
3. Results and Discussion
3.1. Sensitivity of Initial Components Towards Microwave Exposure
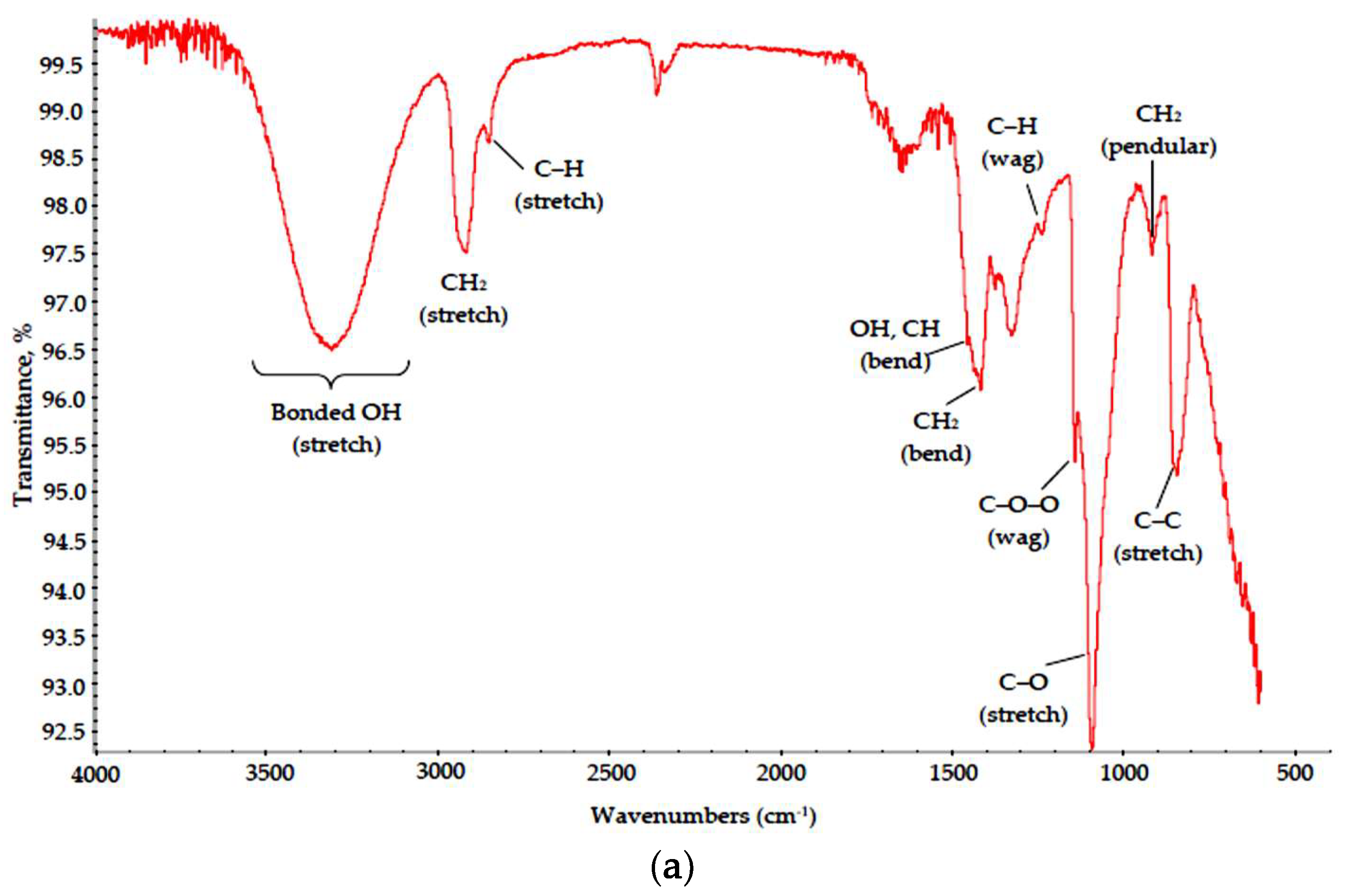
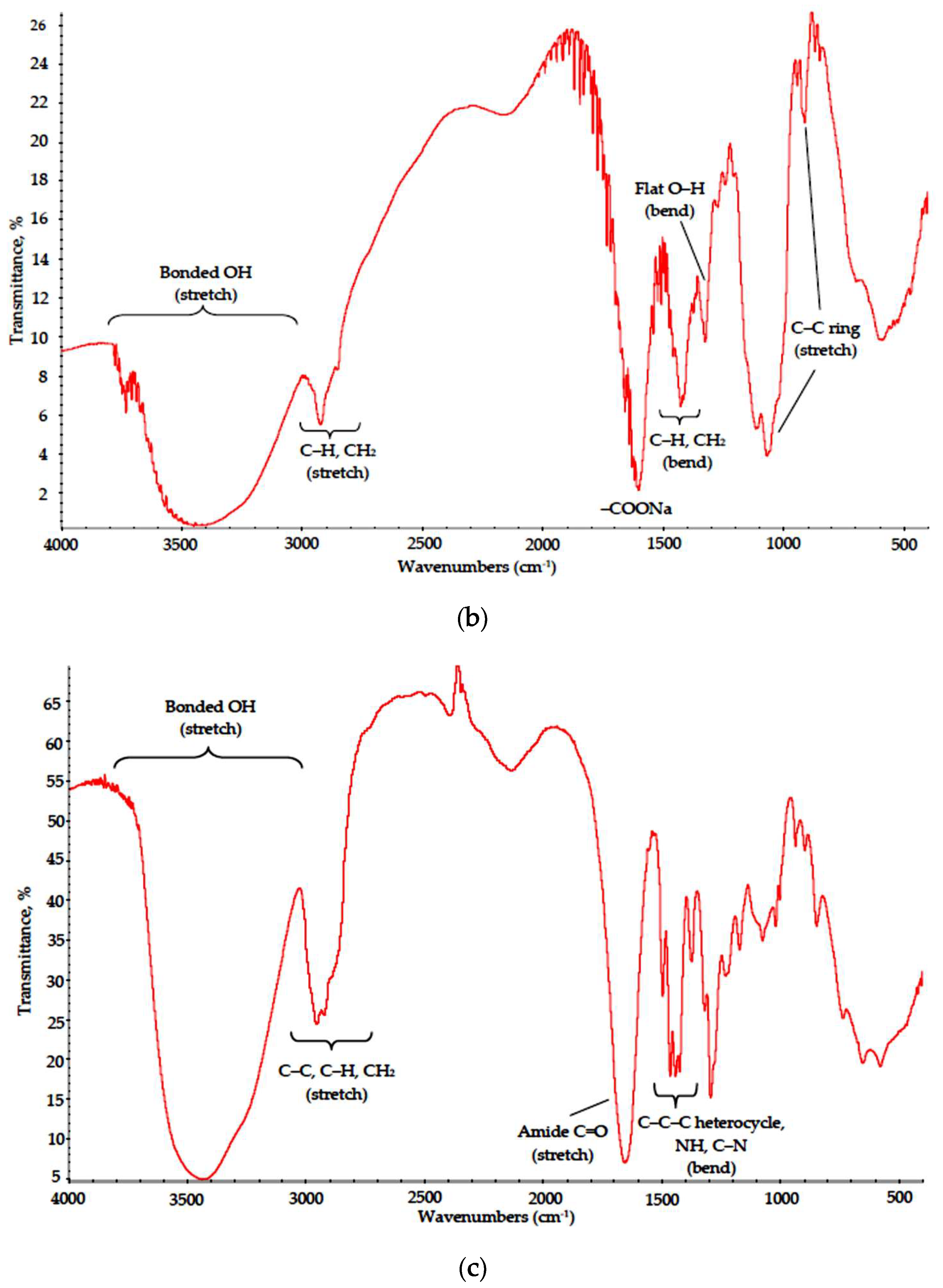
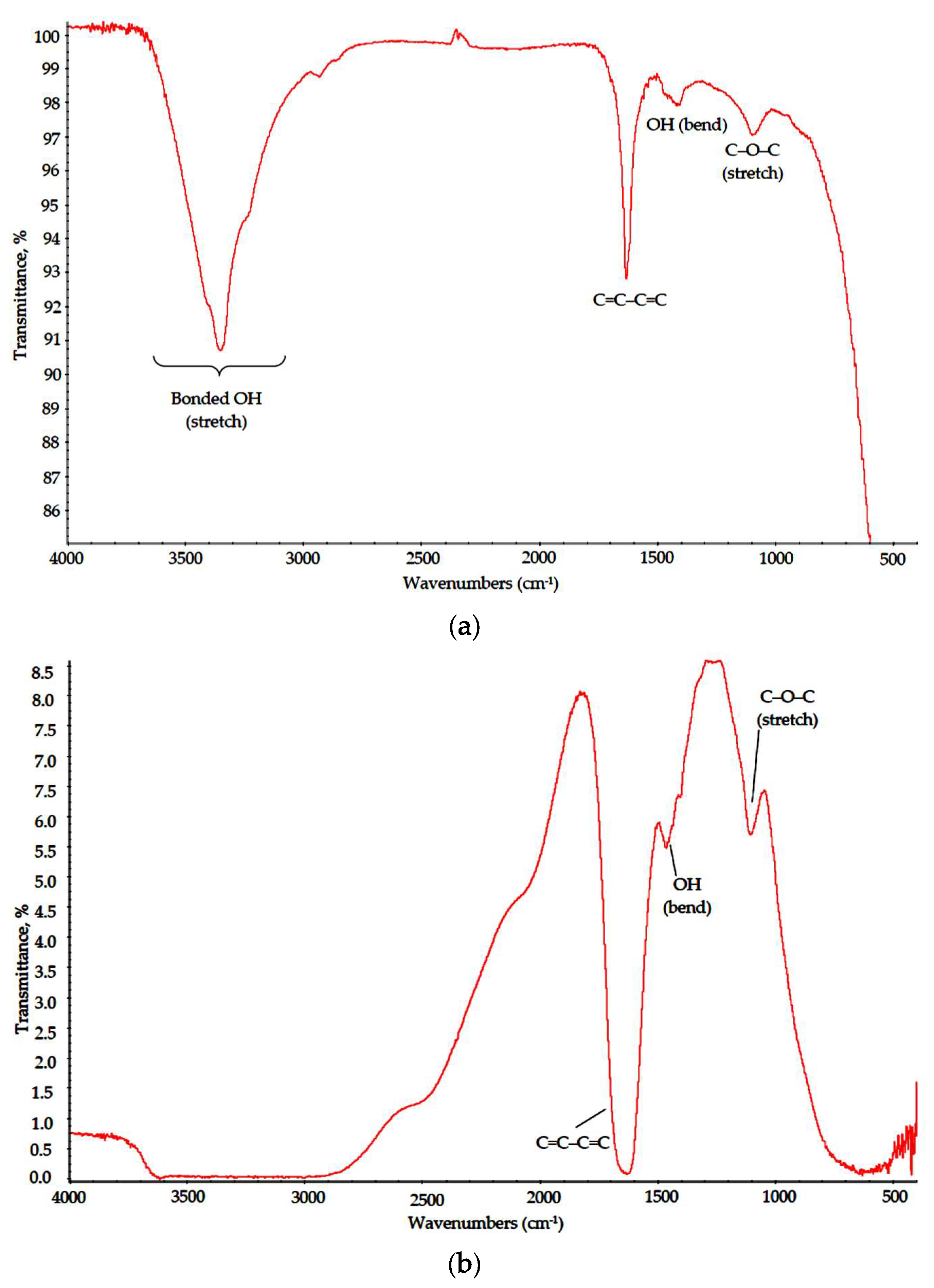
3.2. Polymer–Salt Systems
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Esposito, S. “Traditional” Sol-Gel Chemistry as a Powerful Tool for the Preparation of Supported Metal and Metal Oxide Catalysts. Materials 2019, 12, 668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Omidi, Z.; Ghasemi, A.; Bakhishi, S.R. Synthesis and Characterization of SiC Ultrafine Particles by Means of Sol-Gel and Carbothermal Reduction Methods. Ceram. Int. 2015, 41, 5779–5784. [Google Scholar] [CrossRef]
- Niederberger, M.; Pinna, N. Aqueous and Nonaqueous Sol-Gel Chemistry; Springer: London, UK, 2009; pp. 7–18. [Google Scholar]
- Sui, R.; Charpentier, P. Synthesis of metal oxide nanostructure by direct sol-gel chemistry in supercritical fluids. Chem. Rev. 2011, 112, 3057–3082. [Google Scholar] [CrossRef] [PubMed]
- Danks, A.E.; Hall, S.R.; Schnepp, Z. The Evolution of “Sol-Gel” Chemistry as a Technique for Material Synthesis. Mater. Horiz. 2016, 3, 91–112. [Google Scholar] [CrossRef] [Green Version]
- Nasi, R.; Esposito, S.; Freyria, F.S.; Armandi, M.; Gadhi, T.A.; Hernandez, S.; Rivolo, P.; Ditaranto, N.; Bonelli, B. Application of Reverse Micelle Sol-Gel Synthesis for Bulk Doping and Heteroatoms Surface Enrichment in Mo-Doped TiO2 Nanoparticles. Materials 2019, 12, 937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turova, N.Y.; Turevskaya, E.P.; Kessler, V.G.; Yanovskaya, M.L. The Chemistry of Metal Alkoxides; Kluwer Academic Publishers: New York, NY, USA, 2002; p. 568. [Google Scholar]
- Brinker, C.J.; Scherer, G.W. Sol-gel Science: The Physics and Chemistry of Sol-gel Processing; Academic Press: London, UK, 1990; pp. 97–234. [Google Scholar]
- Pechini, M.P. Method of Preparing Lead and Alkaline Earth Titanates and Niobates and Coating Method Using the Same to Form A Capacitor. US Patent 3330697A, 7 November 1967. [Google Scholar]
- Belyakov, A.V.; Kulikov, N.A. Production of Nanopowders of Yttrium-Aluminum Garnet by the Pechini Method. Refract. Ind. Ceram. 2011, 52, 61–62. [Google Scholar] [CrossRef]
- Russkikh, O.V.; Ivanov, D.V.; Isupova, L.A.; Chezganov, D.S.; Ostroushko, A.A. Synthesis, Morphology, and Activity of La1–xAgxMnO3 ± y Catalysts. Kinet. Catal. 2016, 57, 712–721. [Google Scholar] [CrossRef]
- Ostroushko, A.A.; Adamova, L.V.; Koveza, E.V. Sorption and Thermodynamic Characteristics of Compositions of Water-Soluble Nonionic Polymers and Nanocluster Polyoxomolybdates. Russian J. Phys. Chem. A 2018, 92, 2242–2246. [Google Scholar] [CrossRef]
- Andrianov, N.T.; Abdel’-Gavad, S.R.; Zenkova, N.V. Synthesis and Sintering of Cordierite Sol-Gel Powders Based on Different Magnesium Salts. Glass Ceram. 2006, 63, 415–418. [Google Scholar] [CrossRef]
- Belyakov, A.V.; Faikov, P.P.; Tsvigunov, A.N.; Andrianov, N.T.; Ivleva, Y.V. Synthesis of Aluminomagnesian Spinel with Excess Magnesium Oxide Under Varying Flow Rates of Cation Mass Transfer. Glass Ceram. 2006, 63, 46–51. [Google Scholar] [CrossRef]
- Anokhin, A.S.; Strel’nikova, S.S.; Andrianov, N.T.; Makarov, N.A.; Zhirov, D.A.; Solntsev, K.A. Sol-Gel Synthesis and Properties of Doped Lanthanum Chromite Nanopowders. Inorg. Mater. 2013, 49, 935–938. [Google Scholar] [CrossRef]
- Weerakkody, C.; Guild, C.; Achola, L.A.; Palo, J.; Suib, S.L. Effects of Microwave and Ultrasound Exposure to Microsphere Particles Made out of Different Classes of Inorganic and Organic Materials. J. Ind. Eng. Chem. 2018, 65, 26–30. [Google Scholar] [CrossRef]
- Richer, A.; Göbbels, M. Phase Equilibria and Crystal Chemistry in the System CaO – Al2O3 – Y2O3. J. Phase Equilibria Diffus. 2010, 31, 157–163. [Google Scholar] [CrossRef]
- Index (Inorganic) to the Powder-Diffraction File; ASTM: New-York, NY, USA, 1985.
- SDBSWeb: National Institute of Advanced Industrial Science and Technology. Available online: https://sdbs.db.aist.go.jp (accessed on 12 December 2019).
- Semenovich, G.M.; Khramova, T.S. IR and NMR Spectroscopy of Polymers. A Handbook on Physico-Chemistry of Polymers; Lipatov, Y.S., Ed.; Naukova Dumka: Kiev, USSR, 1985; Volume 3, pp. 486–487. (In Russian) [Google Scholar]
- Rakhimov, A.I.; Rakhimova, N.A.; Petrosyan, E.V. A Study on Surface Structure of Carboxymethylcellulose with Octafluorpentyl Moiety. Fluorine Notes 2014, 95, 5–6. Available online: http://www.notes.fluorine1.ru/public/2014/4_2014/letters/rusletter2.html (accessed on 12 December 2019). (In Russian).
- Melnikova, O.A.; Samkova, I.A.; Melnikov, M.Y.; Petrov, A.Y.; Eltsov, O.S. IR-Spectroscopic Studies of Chemical Structure of Polymeric Complexes of Medicinal Substances on the Basis of Pollyvinilpirrolidon. Adv. Curr. Nat. Sci. 2016, 8, 42–49. Available online: https://natural-sciences.ru/ru/article/view?id=36076 (accessed on 12 December 2019). (In Russian).
- Ostroushko, A.A.; Mogil’nikov, Y.V.; Popov, K.A. Polymer-Salt Compositions Containing Anionic d-Metal Species. Russ. J. Inorg. Chem. 1998, 43, 840–845. [Google Scholar]
- Wicks, B. Direct Observations of the Internal Structure of Carbon Fibers. J. Mater. Sci. 1971, 6, 173–175. [Google Scholar] [CrossRef]
- Si, Y.; Samulski, E.T. Synthesis of Water Soluble Graphene. Nanoletters 2008, 8, 1679–1682. [Google Scholar] [CrossRef] [PubMed]
- Frank, H.P. The lactam-amino acid equilibria for ethylpyrrolidone and polyvinylpyrrolidone. J. Polym. Sci. 1954, 12, 565–575. [Google Scholar] [CrossRef]




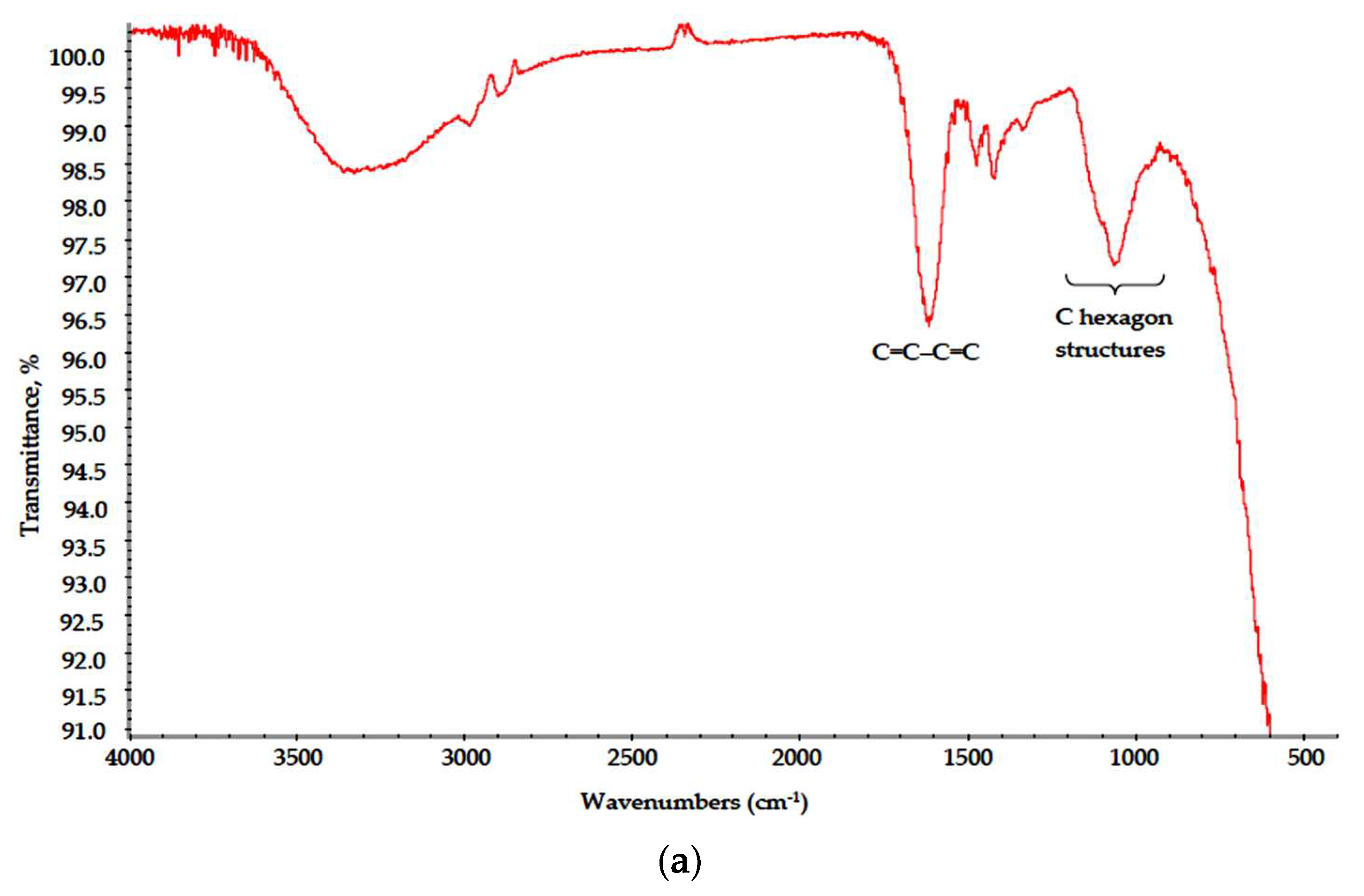

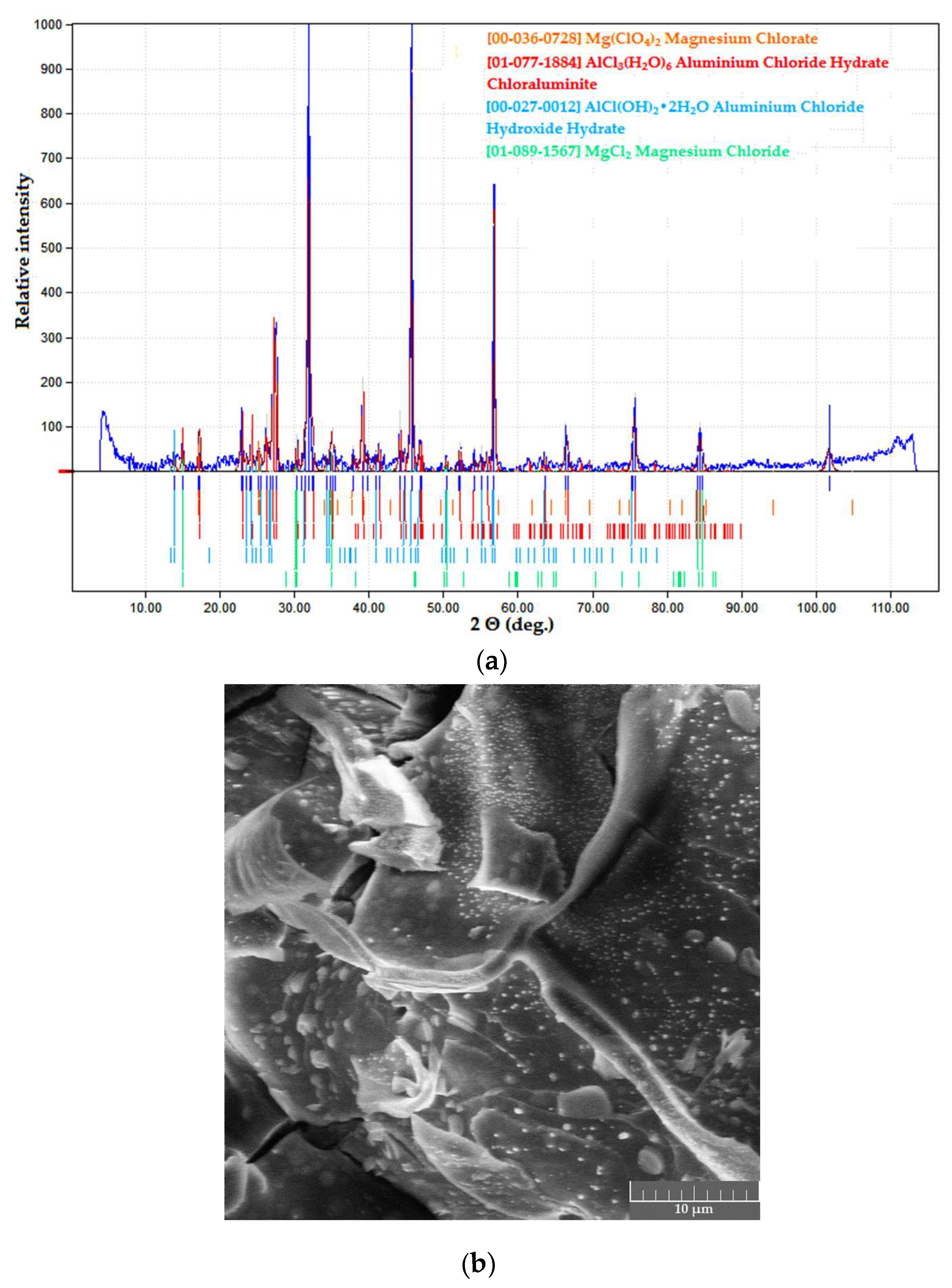
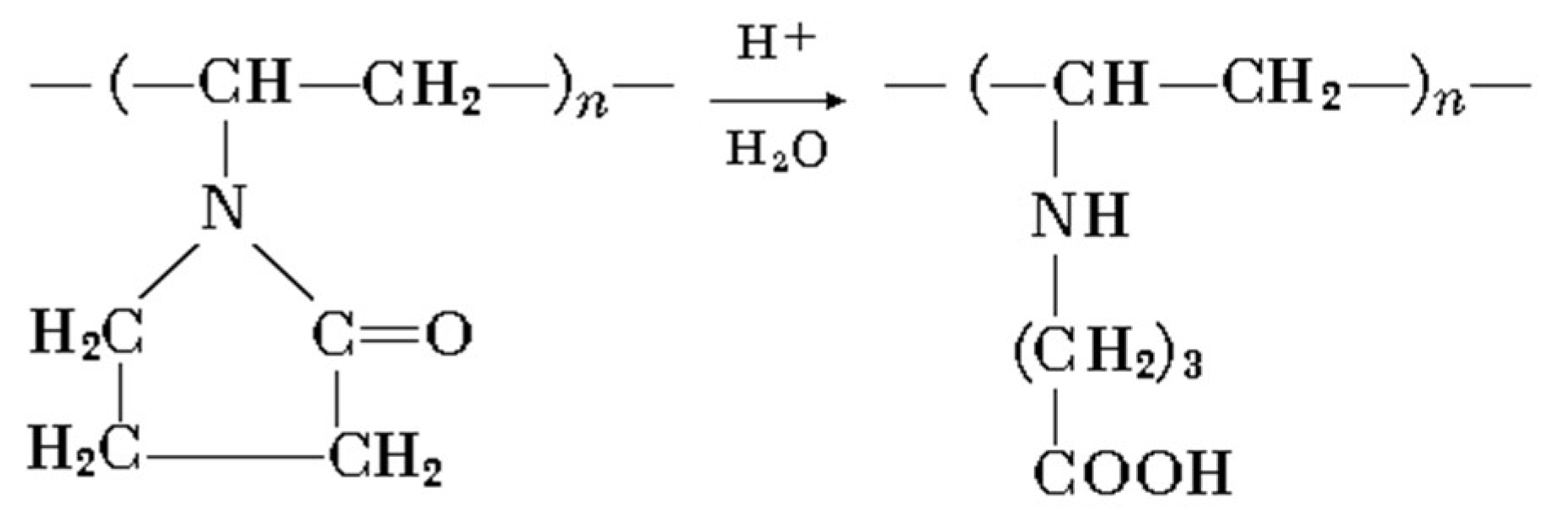

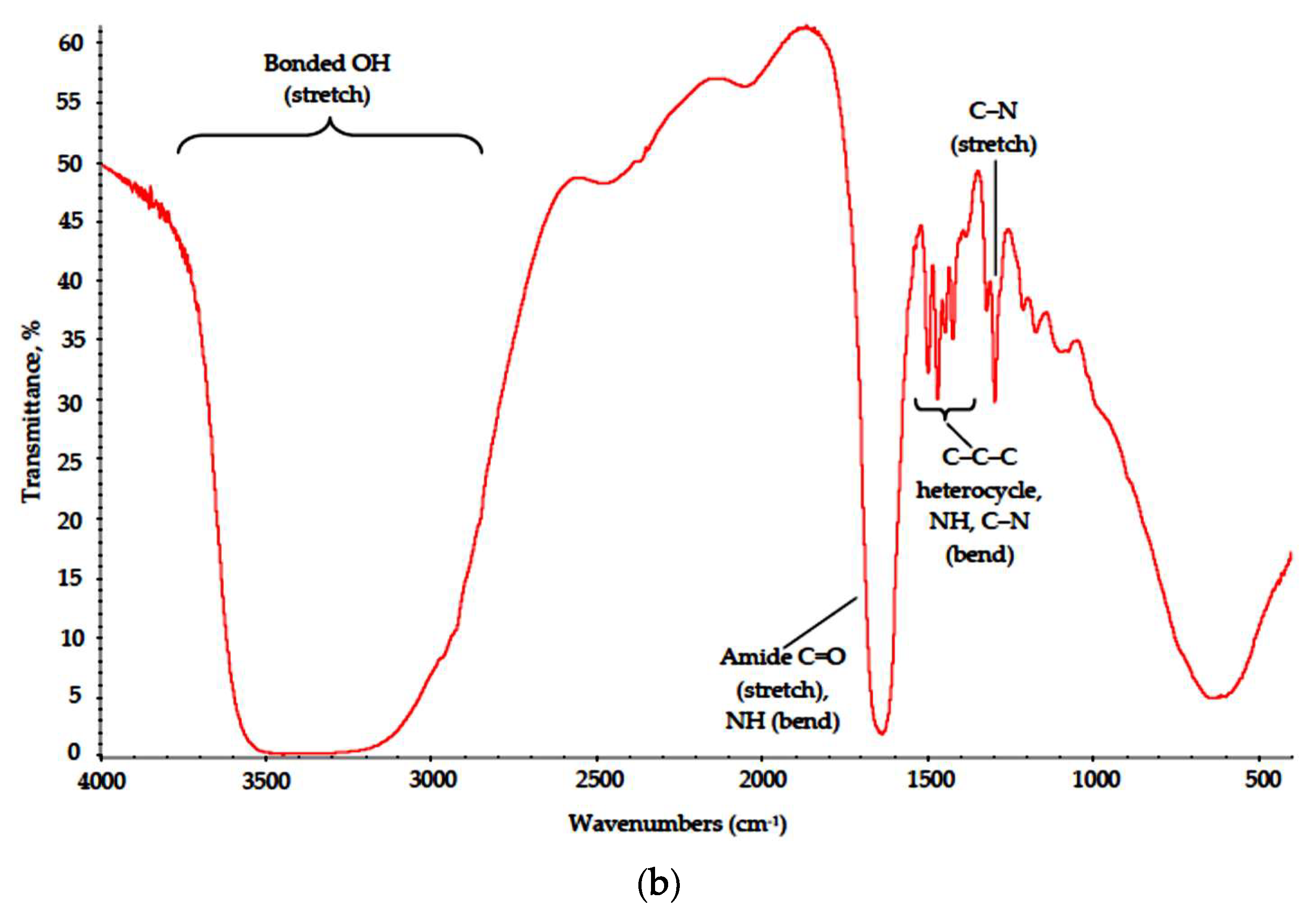


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Oxide | CaO | Al2O3 | Y2O3 | MgO | Melting Point, °C | Eutectic Phases | |
|---|---|---|---|---|---|---|---|
| Eutectic System | |||||||
| CaO–Al2O3–Y2O3 (CAY) | 32.0 | 37.0 | 31.0 | — | 1675 | 2CaO·Y2O3·Al2O3, CaO·Al2O3, 3Y2O3·5Al2O3 | |
| MgO–Al2O3–Y2O3 (MAY) | — | 43.0 | 50.9 | 6.1 | 1775 | Al2O3, MgO·Al2O3, 3Y2O3·5Al2O3 | |
| Name | Unit Cell Formula | Grade | Regulatory Document (in Russian) |
|---|---|---|---|
| PVA | 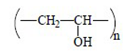 | 16/1 | GOST 10799-78 |
| Na-CMC |  | pharmaceutical | TU 2231-034-79249837-2006 |
| PVP | 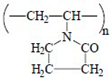 | K-90 | FSP 42-0345-4367-03 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vartanyan, M.; Voytovich, I.; Gorbunova, I.; Makarov, N. Preparation and Structural Characterization of Complex Oxide Eutectic Precursors from Polymer–Salt Xerogels Obtained by Microwave-Assisted Drying. Materials 2020, 13, 1808. https://doi.org/10.3390/ma13081808
Vartanyan M, Voytovich I, Gorbunova I, Makarov N. Preparation and Structural Characterization of Complex Oxide Eutectic Precursors from Polymer–Salt Xerogels Obtained by Microwave-Assisted Drying. Materials. 2020; 13(8):1808. https://doi.org/10.3390/ma13081808
Chicago/Turabian StyleVartanyan, Maria, Ilya Voytovich, Irina Gorbunova, and Nikolay Makarov. 2020. "Preparation and Structural Characterization of Complex Oxide Eutectic Precursors from Polymer–Salt Xerogels Obtained by Microwave-Assisted Drying" Materials 13, no. 8: 1808. https://doi.org/10.3390/ma13081808