
First Screen-Printed Sensor (Electrochemically Activated Screen-Printed Boron-Doped Diamond Electrode) for Quantitative Determination of Rifampicin by Adsorptive Stripping Voltammetry

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Apparatus
2.2. Reagents and Solutions
2.3. Preparation of Activated Screen-Printed Boron-Doped Diamond Electrode (aSPBDDE)
2.4. Rifampicin (RIF) Differential Pulse Adsorptive Stripping Voltammetric (DPAdSV) Analysis
2.5. RIF High-Performance Liquid Chromatography (HPLC)/PDA Analysis
3. Results and Discussion
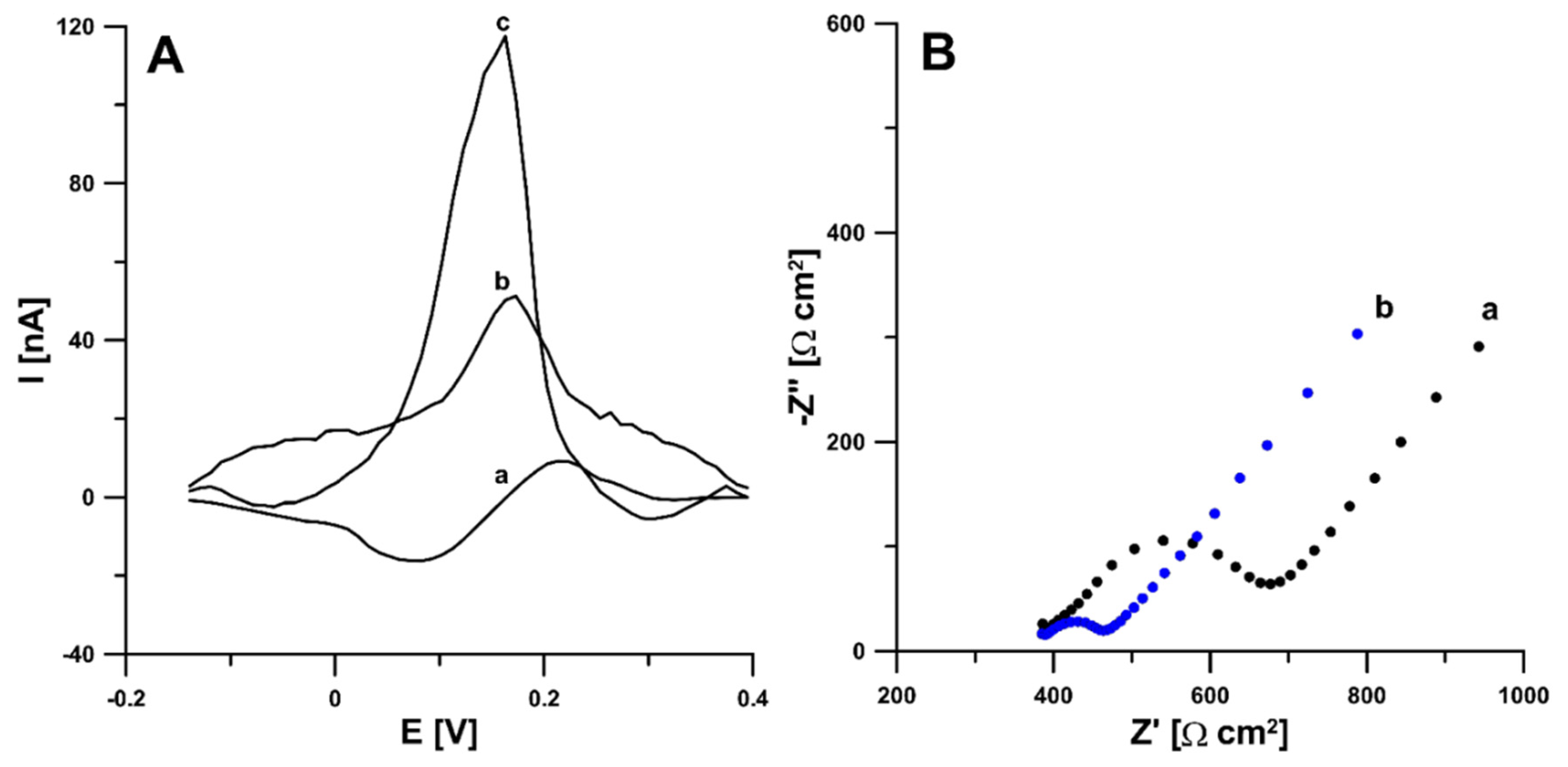
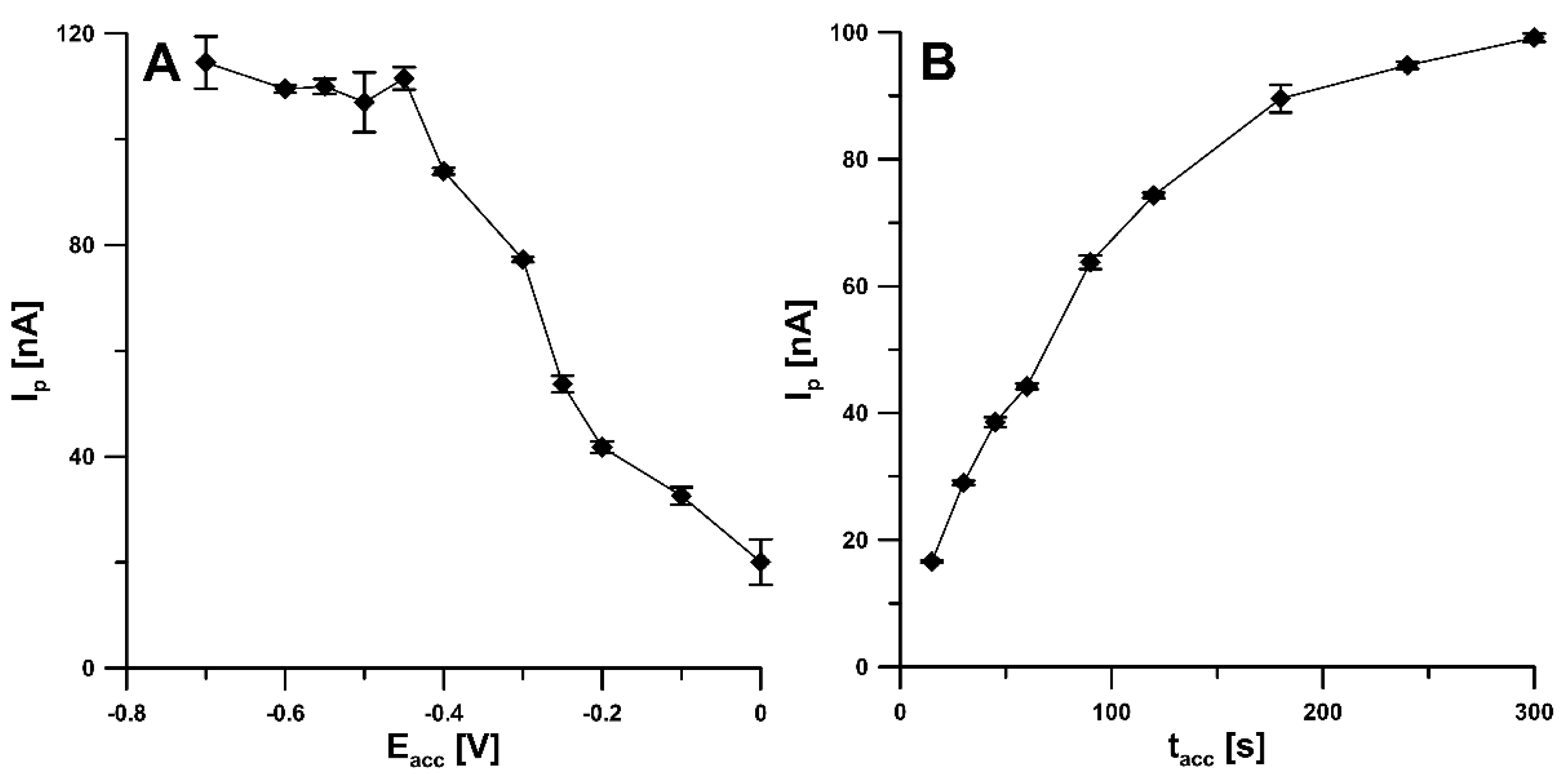
3.1. Characteristics of aSPBDDE Sensors
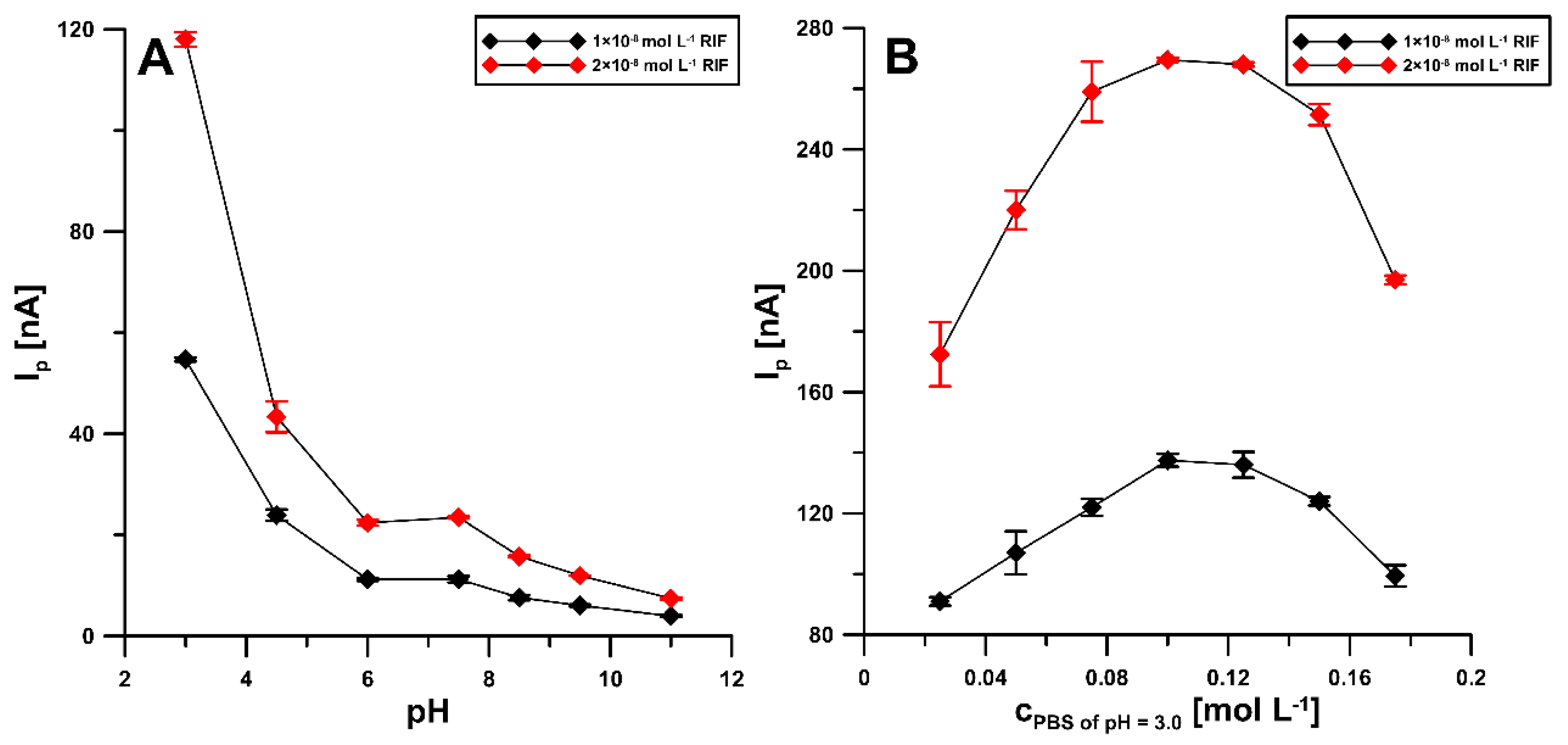
3.2. Influence of pH and Concentration of Supporting Electrolyte
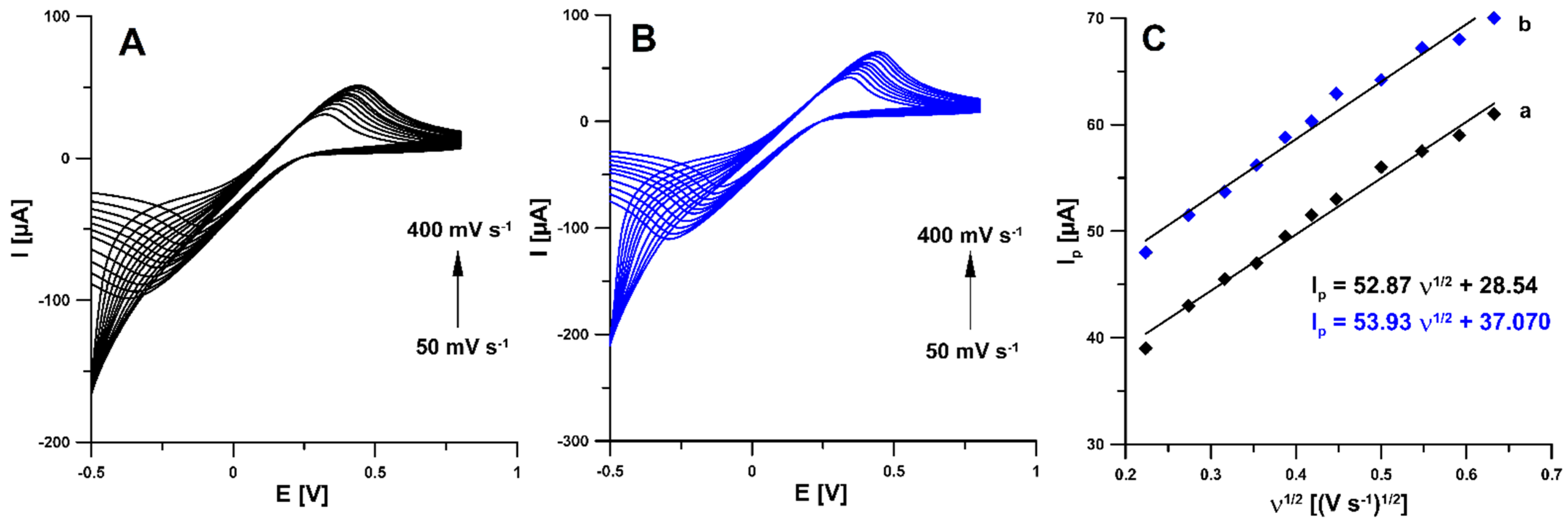
3.3. Cyclic Voltammetry (CV) Behaviour of RIF
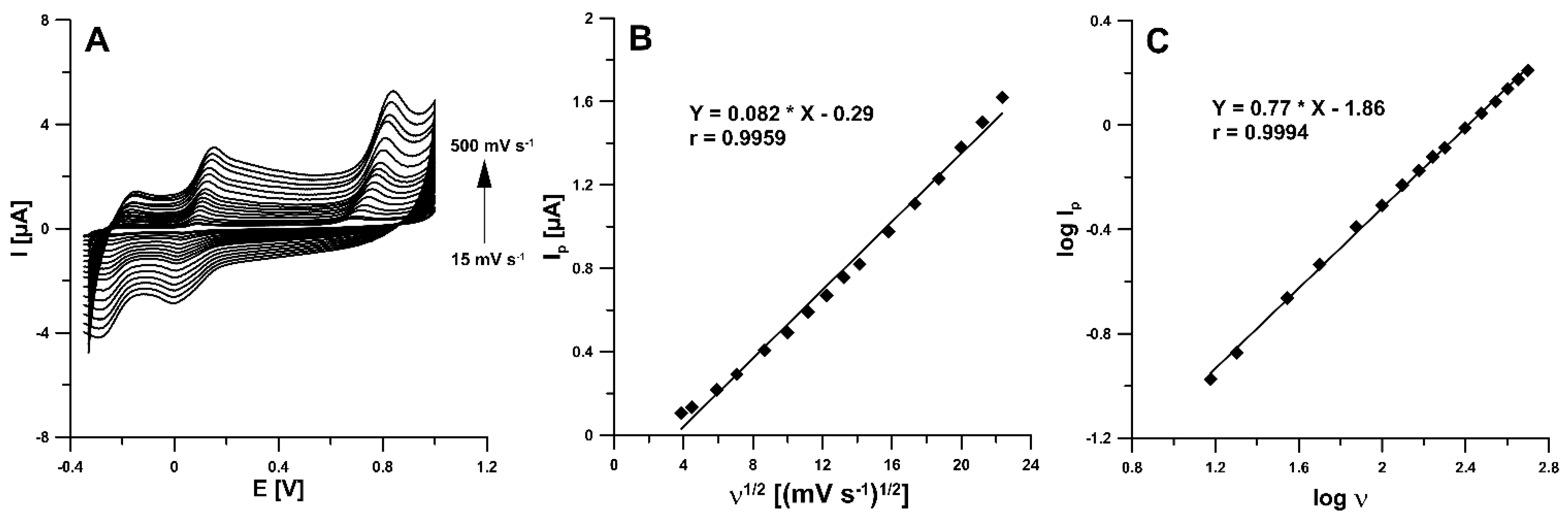
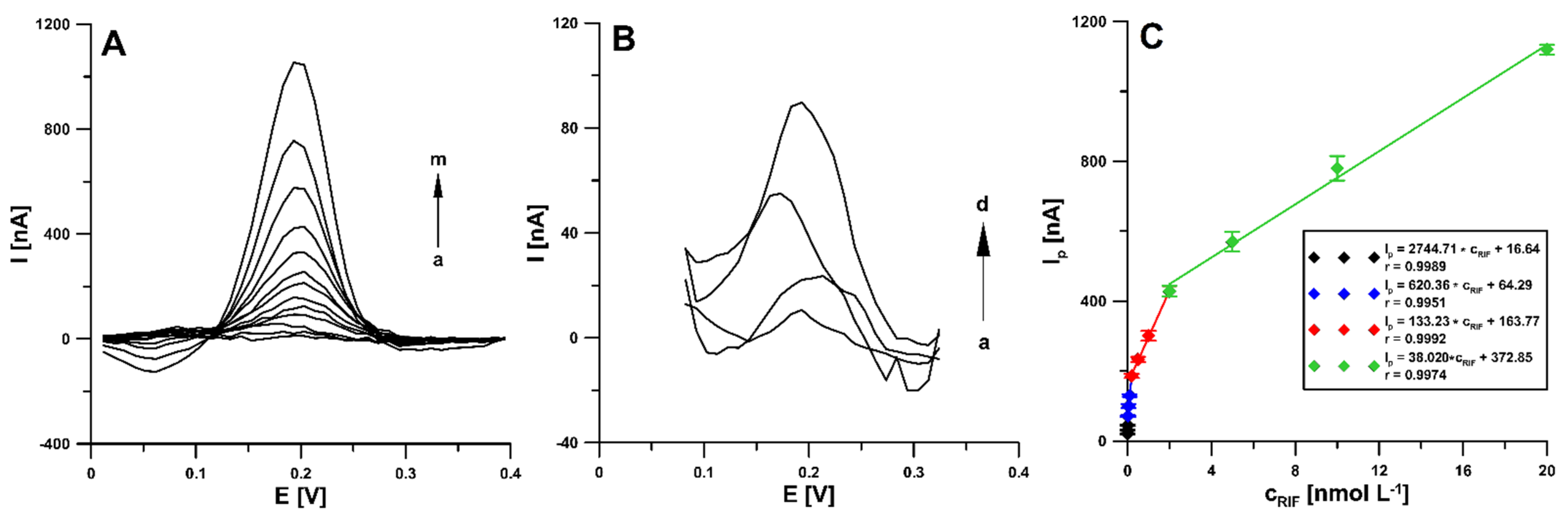
3.4. Optimization of DPAdSV Parameters
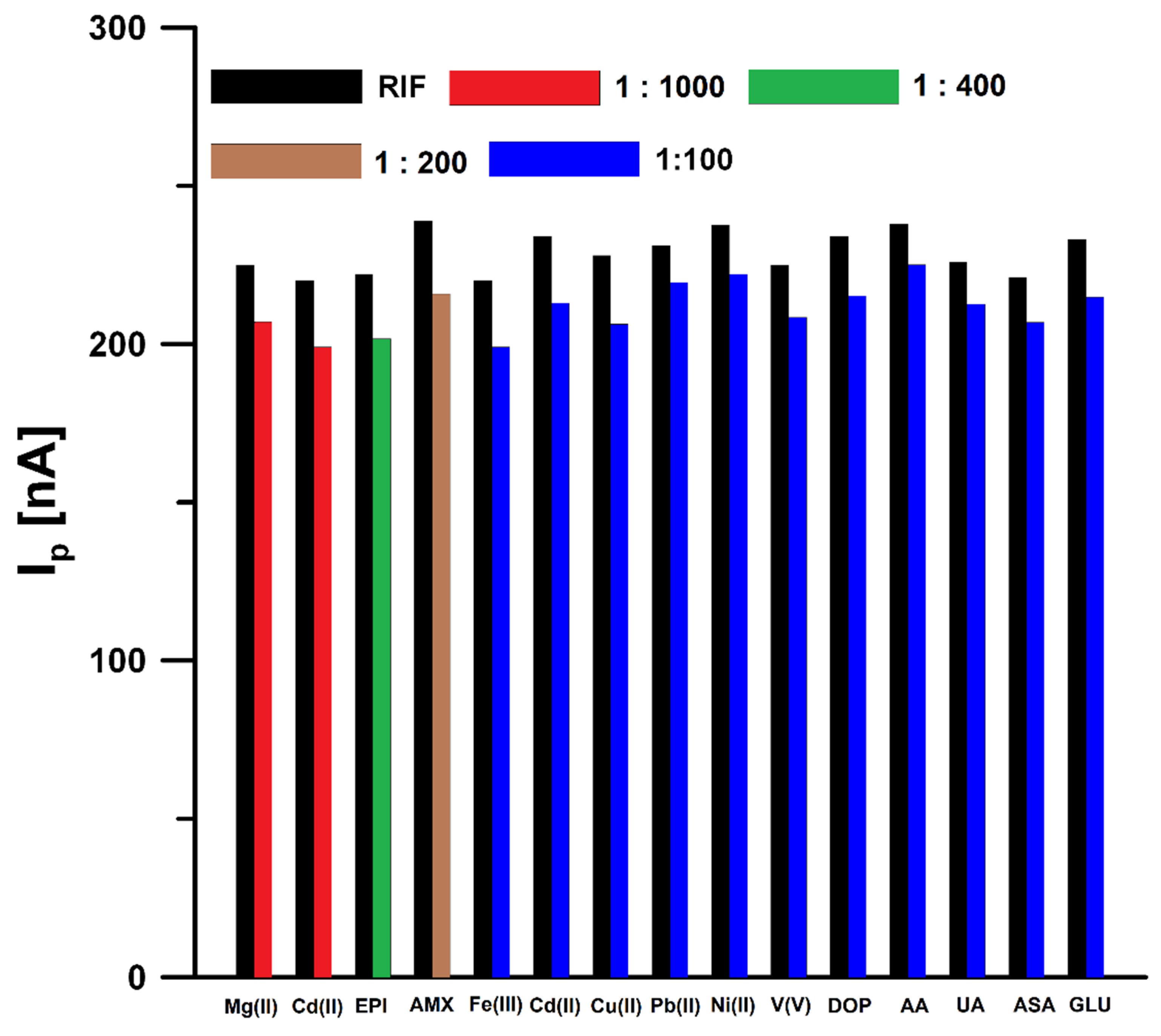
3.5. Interference Studies
3.6. Analytical Characteristic
3.7. Real Sample Analysis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tilinca, M.; Hancu, G.; Mircia, E.; Iriminescu, D.; Rusu, A.; Vlad, R.A.; Barabás, E. Simultaneous determination of isoniazid and rifampicin by UV spectrophotometry. Farmacia 2017, 65, 219–224. [Google Scholar]
- Wei, L.; Wang, J.; Yan, Z.-Y. Development of a sensitive and rapid method for rifampicin impurity analysis using supercritical fluid chromatography. J. Pharm. Biomed. Anal. 2015, 114, 341–347. [Google Scholar]
- Kumar, S.; Bouic, P.J.; Rosenkranz, B. A validated stable HPLC method for the simultaneous determination of rifampicin and 25-O-desacetyl rifampicin—evaluation of in vitro metabolism. Acta Chromatogr. 2019, 31, 92–98. [Google Scholar] [CrossRef]
- Chatterjee, K.; Kuo, C.W.; Chen, A.; Chen, P. Detection of residual rifampicin in urine via fluorescence quenching of gold nanoclusters on paper. J. Nanobiotechnol. 2015, 13, 46. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, A.; Waterhouse, D.; Ardrey, A.; Ward, S.A. Quantification of rifampicin in human plasma and cerebrospinal fluid by a highly sensitive and rapid liquid chromatographic–tandem mass spectrometric method. J. Pharm. Biomed. Anal. 2012, 70, 523–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oswald, S.; Peters, J.; Venner, M.; Siegmund, W. LC–MS/MS method for the simultaneous determination of clarithromycin, rifampicin and their main metabolites in horse plasma, epithelial lining fluid and broncho-alveolar cells. J. Pharm. Biomed. Anal. 2011, 55, 194–201. [Google Scholar] [CrossRef]
- Rakuša, Ž.T.; Roškar, R.; Andrejc, A.K.; Lušin, T.T.; Faganeli, N.; Grabnar, I.; Mrhar, A.; Kristl, A.; Trontelj, J. Fast and simple LC-MS/MS method for rifampicin quantification in human plasma. Int. J. Anal. Chem. 2019, 2019, 4848236. [Google Scholar]
- van Ewijk-Beneken Kolmer, E.W.J.; Teulen, M.J.A.; van der Hombergh, E.C.A.; van Erp, N.E.; te Brake, L.H.M.; Aarnoutse, R.E. Determination of protein-unbound, active rifampicin in serum by ultrafiltration and ultra performance liquid chromatography with UV detection. A method suitable for standard and high doses of rifampicin. J. Chromatogr. B 2017, 1063, 42–49. [Google Scholar] [CrossRef]
- Szipszky, C.; Van Aartsen, D.; Criddle, S.; Rao, P.; Zentner, I.; Justine, M.; Mduma, E.; Mpagama, S.; Al-Shaer, M.H.; Peloquin, C.; et al. Determination of rifampin concentrations by urine colorimetry and mobile phone readout for personalized dosing in tuberculosis treatment. J. Pediatric Infect. Dis. Soc. 2021, 10, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Alonso-Lomillo, M.A.; Dominguez-Renedo, O.; Arcos-Martinez, M.J. Optimization of a cyclodextrin-based sensor for rifampicin monitoring. Electrochim. Acta 2005, 50, 1807–1811. [Google Scholar] [CrossRef]
- Tyszczuk, K.; Korolczuk, M. New protocol for determination of rifampicine by adsorptive stripping voltammetry. Electroanalysis 2009, 21, 101–106. [Google Scholar] [CrossRef]
- Rawool, C.R.; Srivastava, A.K. A dual template imprinted polymer modified electrochemical sensor based on Cu metal organic framework/mesoporous carbon for highly sensitive and selective recognition of rifampicin and isoniazid. Sens. Actuators B Chem. 2019, 288, 493–506. [Google Scholar] [CrossRef]
- Zou, J.; Huang, L.-L.; Jiang, X.-Y.; Jiao, F.-P.; Yu, J.-G. Electrochemical behaviors and determination of rifampicin on graphene nanoplatelets modified glassy carbon electrode in sulfuric acid solution. Desalination Water Treat. 2018, 120, 272–281. [Google Scholar] [CrossRef]
- Watanabe, T.; Shimizu, T.K.; Tateyama, Y.; Kim, Y.; Kawai, M.; Einaga, Y. Giant electric double-layer capacitance of heavily boron-doped diamond electrode. Diam. Relat. Mater. 2010, 19, 772–777. [Google Scholar] [CrossRef]
- Divyapriya, G.; Nidheesh, P.V. Electrochemically generated sulfate radicals by boron doped diamond and its environmental applications. Curr. Opin. Solid State Mater. Sci. 2021, 25, 100921. [Google Scholar] [CrossRef]
- Zhi, J.; Luo, D. Fabrication and electrochemical behavior of vertically aligned boron-doped diamond nanorod forest electrodes. Electrochem. Commun. 2009, 11, 1093–1096. [Google Scholar]
- Shi, D.; Liu, L.; Zhai, Z.; Chen, B.; Lu, Z.; Zhang, C.; Yuan, Z.; Zhou, M.; Yang, B.; Huang, N.; et al. Effect of oxygen terminated surface of boron-doped diamondmthin-film electrode on seawater salinity sensing. J. Mater. Sci. Technol. 2021, 86, 1–10. [Google Scholar] [CrossRef]
- Kapałka, A.; Fóti, G.; Comninellis, C. Investigations of electrochemical oxygen transfer reaction on boron-doped diamond electrodes. Electrochim. Acta 2007, 53, 1954–1961. [Google Scholar] [CrossRef]
- McLaughlin, M.H.S.; Pakpour-Tabrizi, A.C.; Jackman, R.B. A detailed EIS study of boron doped diamond electrodes with gold nanoparticles for high sensitivity mercury detection. Sci. Rep. 2021, 11, 9505. [Google Scholar] [CrossRef] [PubMed]
- Goutal, S.; Auvity, S.; Legrand, T.; Hauquier, F.; Cisternino, S.; Chapy, H.; Saba, W.; Tournier, N. Validation of a simple HPLC-UV method for rifampicin determination in plasma: Application to the study of rifampicin arteriovenous concentration gradient. J. Pharmaceut. Biomed. 2016, 123, 173–178. [Google Scholar] [CrossRef] [PubMed]
- González-Sánchez, M.I.; Gómez-Monedero, B.; Agrisuelas, J.; Iniesta, J.; Valero, E. Highly activated screen-printed carbon electrodes by electrochemical treatment with hydrogen peroxide. Electrochem. Commun. 2018, 91, 36–40. [Google Scholar] [CrossRef]
- González-Sánchez, M.I.; Gómez-Monedero, B.; Agrisuelas, J.; Iniesta, J.; Valero, E. Electrochemical performance of activated screen printed carbon electrodes for hydrogen peroxide and phenol derivatives sensing. J. Electroanal. Chem. 2019, 839, 75–82. [Google Scholar] [CrossRef] [Green Version]
- Gosser, D.K. Cyclic Voltammetry: Simulation and Analysis of Reaction Mechanism; VCH: New York, NY, USA, 1993. [Google Scholar]
- Amidi, S.; Hosseinzadeh Ardakani, Y.; Amiri-Aref, M.; Ranjbari, E.; Sepehri, Z.; Bagheri, H. Sensitive electrochemical determination of rifampicin using gold nanoparticles/poly-melamine nanocomposite. RSC Adv. 2017, 7, 40111–40118. [Google Scholar] [CrossRef] [Green Version]
- Hammam, E.; Beltagi, A.M.; Ghoneim, M.M. Voltammetric assay of rifampicin and isoniazid drugs, separately and combined in bulk, pharmaceutical formulations and human serum at a carbon paste electrode. Microchem. J. 2004, 77, 53–62. [Google Scholar] [CrossRef]
- Grabarczyk, M.; Koper, A. How to determine uranium faster and cheaper by adsorptive stripping voltammetry in water samples containing surface active compounds. Electroanalysis 2011, 23, 1442–1446. [Google Scholar] [CrossRef]
- Mocak, J.; Bond, A.M.; Mitchell, S.; Scollary, G. A statistical overview of standard (IUPAC and ACS) and new procedures for determining the limits of detection and quantification: Application to voltammetric and stripping techniques. Pure Appl. Chem. 1997, 69, 297–328. [Google Scholar] [CrossRef]
- Zhang, N.; Brites Helu, M.; Zhang, K.; Fang, X.; Yin, H.; Chen, J.; Ma, S.; Fang, A.; Wang, C. Multiwalled Carbon Nanotubes-CeO2 Nanorods: A “Nanonetwork” Modified Electrode for Detecting Trace Rifampicin. Nanomaterials 2020, 10, 391. [Google Scholar] [CrossRef] [Green Version]
- Asadpour-Zeynali, K.; Mollarasouli, F. Novel electrochemical biosensor based on PVP capped CoFe2O4@CdSe core-shell nanoparticles modified electrode for ultra-trace level determination of rifampicin by square wave adsorptive stripping voltammetry. Biosens. Biolelectron. 2017, 92, 509–516. [Google Scholar] [CrossRef]
- Girousi, S.T.; Gherghi, I.C.; Karava, M.K. DNA-modified carbon paste electrode applied to the study of interaction between rifampicin (RIF) and DNA in solution and at the electrode surface. J. Pharm. Biomed. Anal. 2004, 36, 851–858. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | Linear Range (µM) | LOD (µM) | Application | Ref. |
|---|---|---|---|---|
| Spectrophotometry | 6.08–60.80 | 4.25 | Pharmaceutical formulations | [1] |
| Fluorescence quenching | 0.61–1000.0 | 0.085 | Human urine | [4] |
| HPLC | 0–2.0 | 5.86 | Herbal extracts, liver microsomes | [3] |
| LC-MS/MS | 0.030–7.78 | 0.30 | Human plasma | [5] |
| UPLC | 0.0790–31.60 | - | Human plasma | [8] |
| Amperometry | - | 1.69 | Pharmaceutical formulations, urine | [10] |
| DPAdSV | 0.0000020–0.020 | 0.00000022 | Bovine urine, river water | This work |
| RIF Concentration (µM) ± SD (n = 3) | ||||||
|---|---|---|---|---|---|---|
| Sample | Added | Found DPAdSV | RIF Concentration Found in Electrochemical Cell | Found HPLC/PDA | Recovery * (%) | Relative Error ** (%) |
| Bystrzyca river | 0.001 0.05 | 0.000973 ± 0.0000240 0.0457 ± 0.00110 | 0.0000973 ± 0.00000240 0.00457 ± 0.000110 | <LOD 0.0471 ± 0.00250 | 97.3 91.4 | - 3.0 |
| Bovine urine | 1.0 50.0 | 0.920 ± 0.0070 49.30 ± 0.40 | 0.0000920 ± 0.00000070 0.00493 ± 0.000040 | <LOD 47.60 ± 0.018 | 92.0 98.6 | - 3.6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kozak, J.; Tyszczuk-Rotko, K.; Wójciak, M.; Sowa, I.; Rotko, M. First Screen-Printed Sensor (Electrochemically Activated Screen-Printed Boron-Doped Diamond Electrode) for Quantitative Determination of Rifampicin by Adsorptive Stripping Voltammetry. Materials 2021, 14, 4231. https://doi.org/10.3390/ma14154231
Kozak J, Tyszczuk-Rotko K, Wójciak M, Sowa I, Rotko M. First Screen-Printed Sensor (Electrochemically Activated Screen-Printed Boron-Doped Diamond Electrode) for Quantitative Determination of Rifampicin by Adsorptive Stripping Voltammetry. Materials. 2021; 14(15):4231. https://doi.org/10.3390/ma14154231
Chicago/Turabian StyleKozak, Jędrzej, Katarzyna Tyszczuk-Rotko, Magdalena Wójciak, Ireneusz Sowa, and Marek Rotko. 2021. "First Screen-Printed Sensor (Electrochemically Activated Screen-Printed Boron-Doped Diamond Electrode) for Quantitative Determination of Rifampicin by Adsorptive Stripping Voltammetry" Materials 14, no. 15: 4231. https://doi.org/10.3390/ma14154231