
Recovery of Platinum Group Metals from Spent Automotive Catalysts Using Lithium Salts and Hydrochloric Acid


Abstract
:1. Introduction
2. Materials and Methods
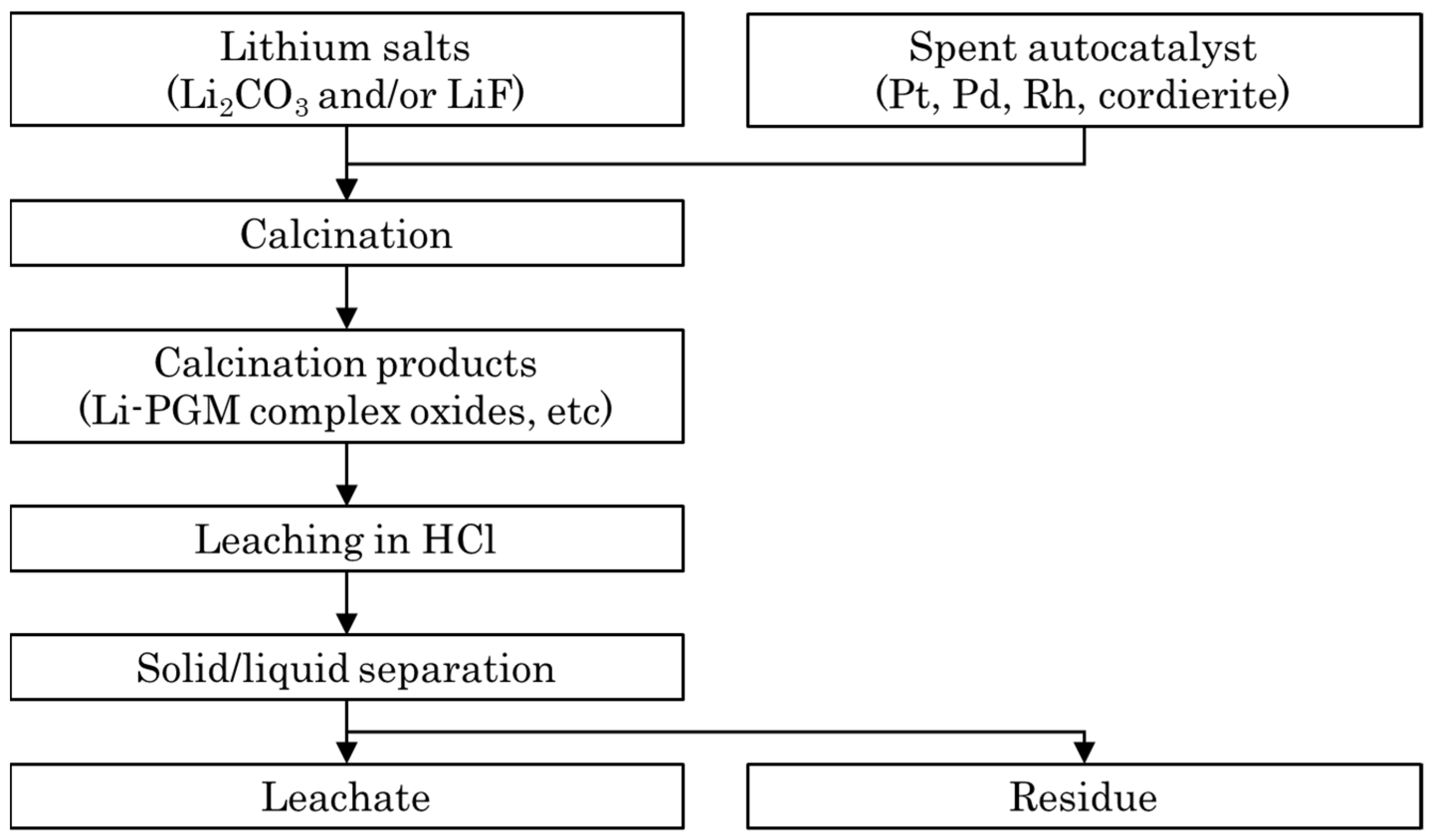
2.1. Sample Preparation
2.1.1. Starting Materials
2.1.2. Sample Preparation
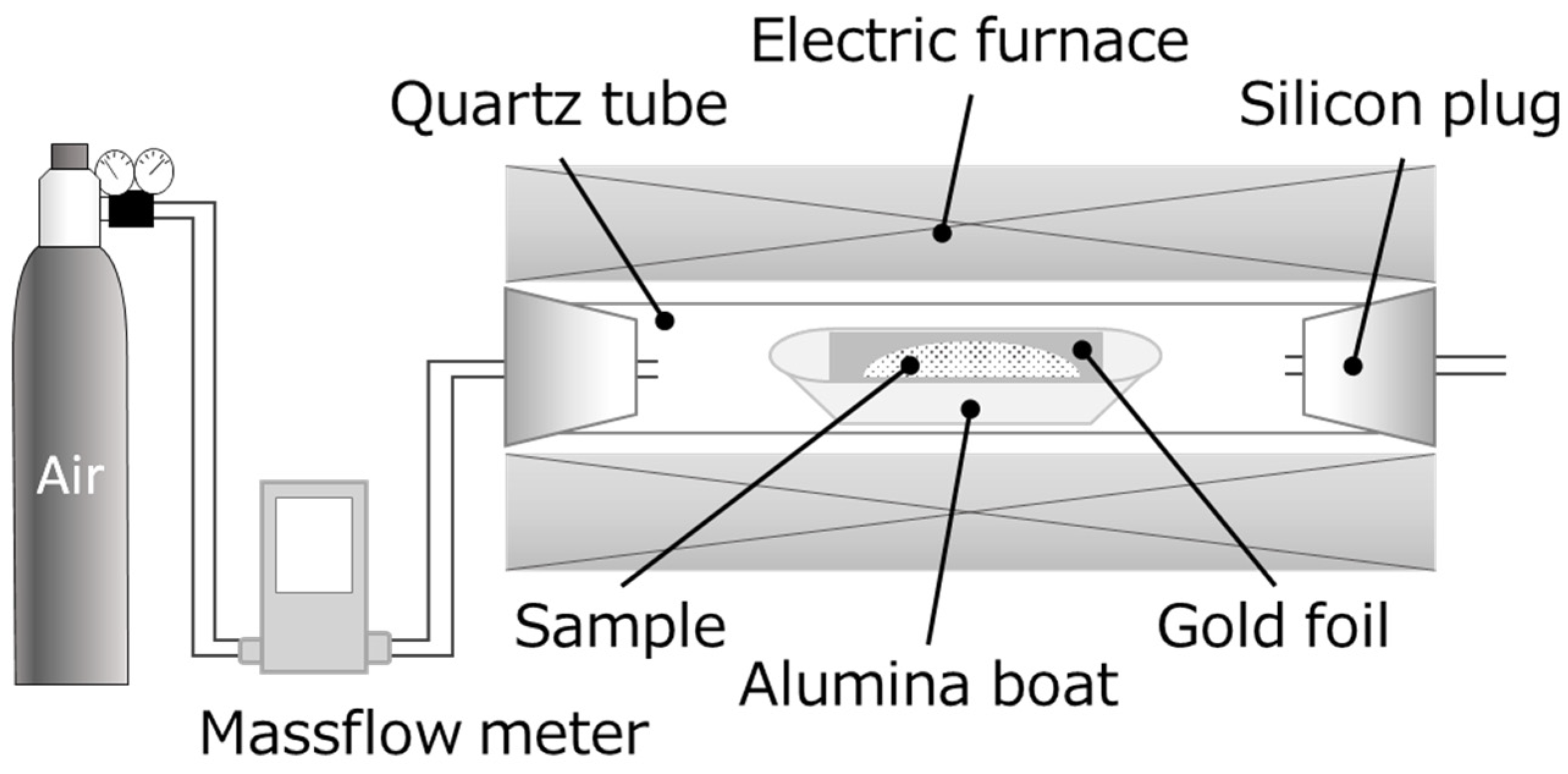
2.2. Calcination Experiments
2.3. Leaching Experiments
2.3.1. Hydrochloric Acid
2.3.2. Aqua Regia
2.4. Characterization
2.4.1. Thermogravimetric–Differential Thermal Analysis
2.4.2. Elemental Analysis
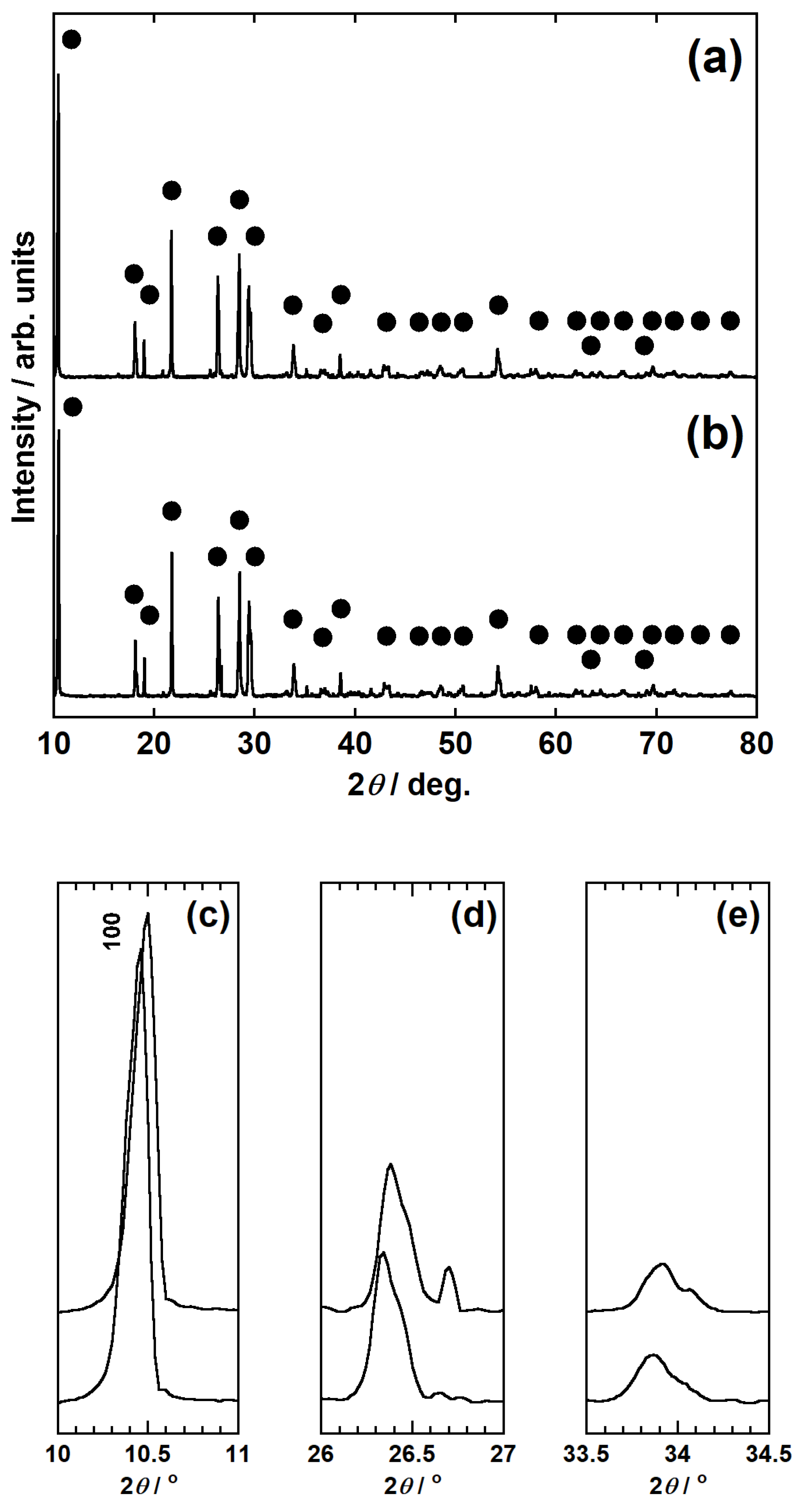
2.4.3. XRD Analysis
3. Results and Discussion
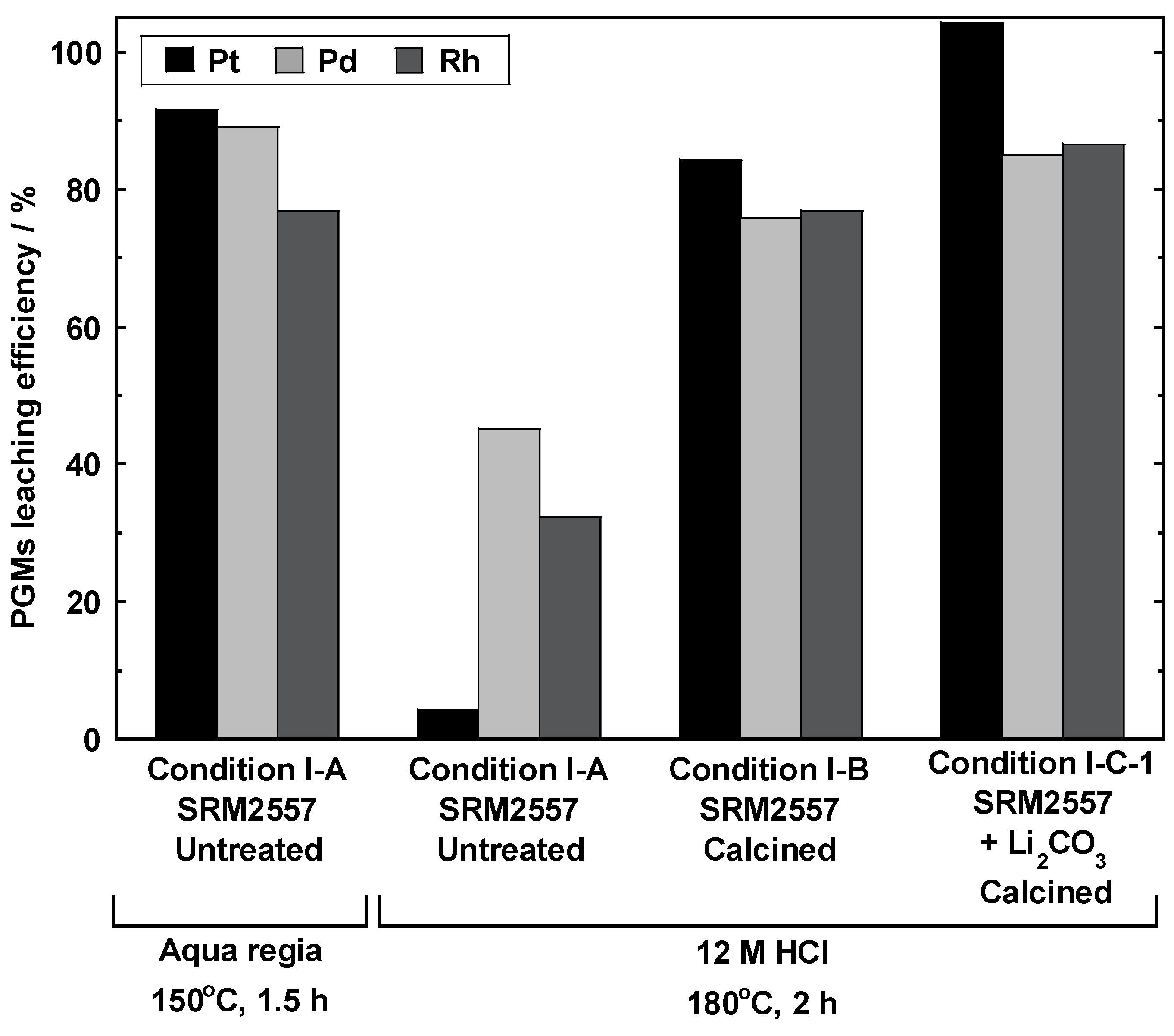
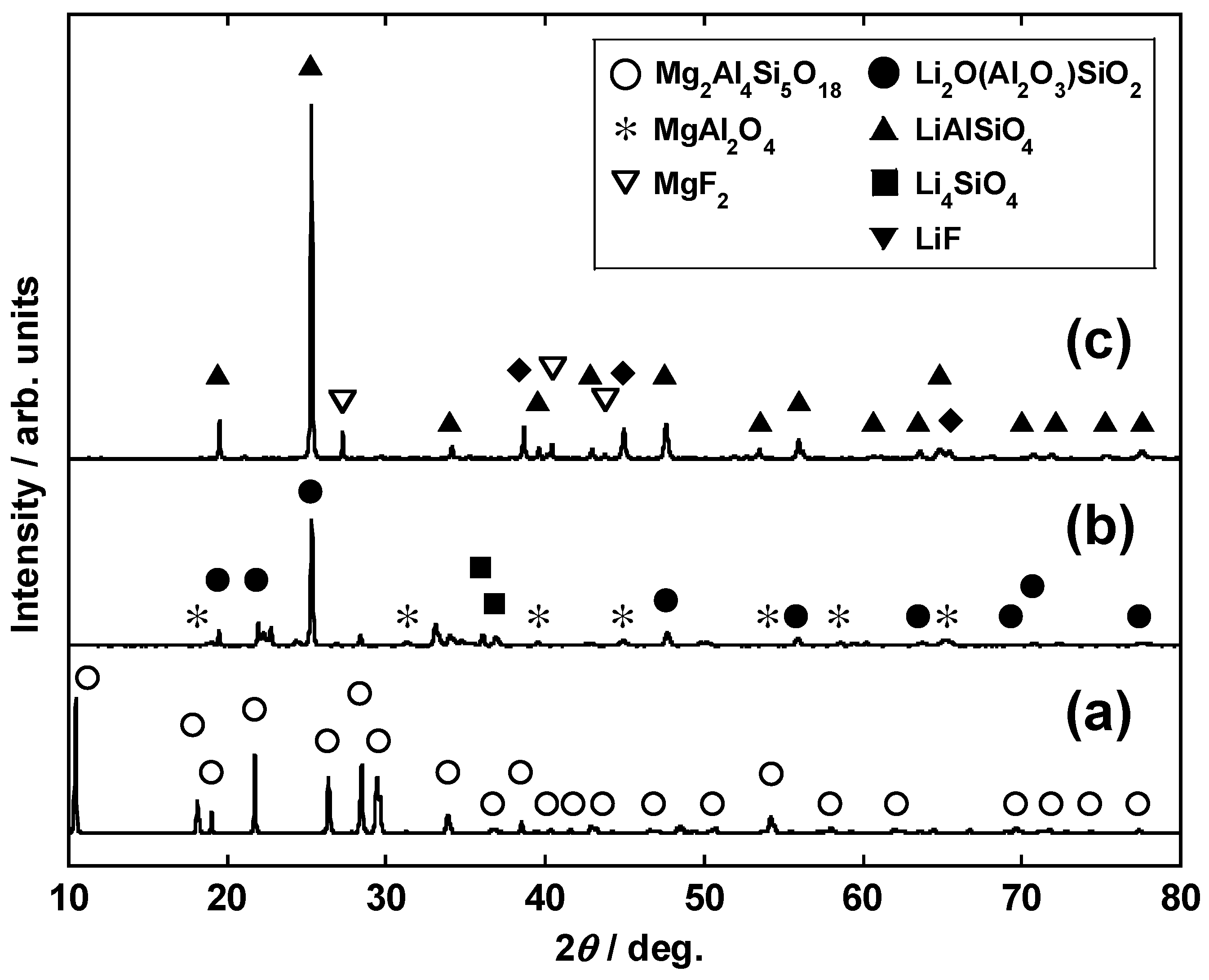
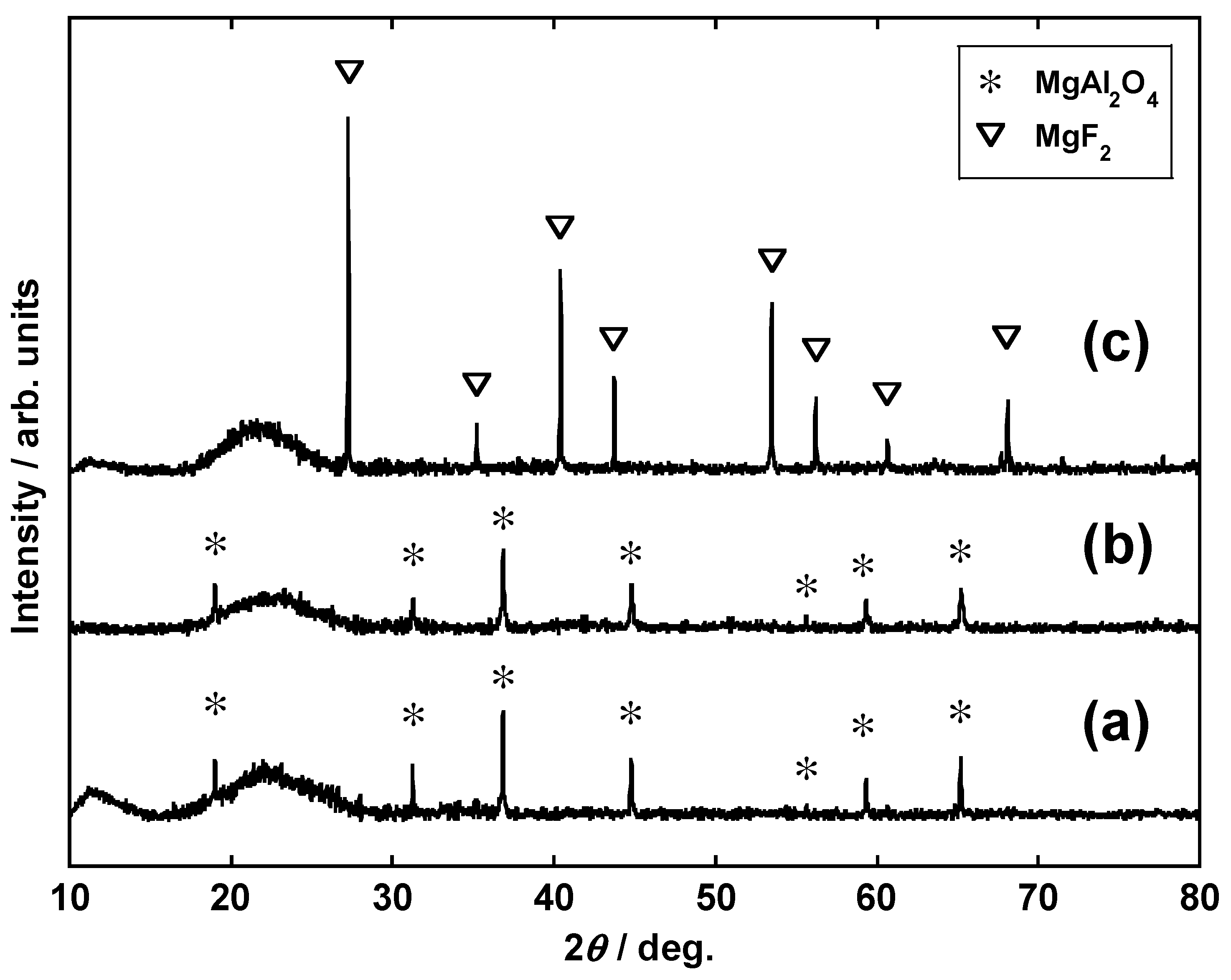
3.1. Calcination and Influence of Li2CO3 Addition
3.2. Impact of Li2CO3 Concentration in Li Salt Mixture (Conditions I-C and II)
3.3. Effects of Various Parameters on PGM Leaching
3.3.1. HCl Concentration and the Type of Li Salt (Conditions I-C-1 and I-C-5)
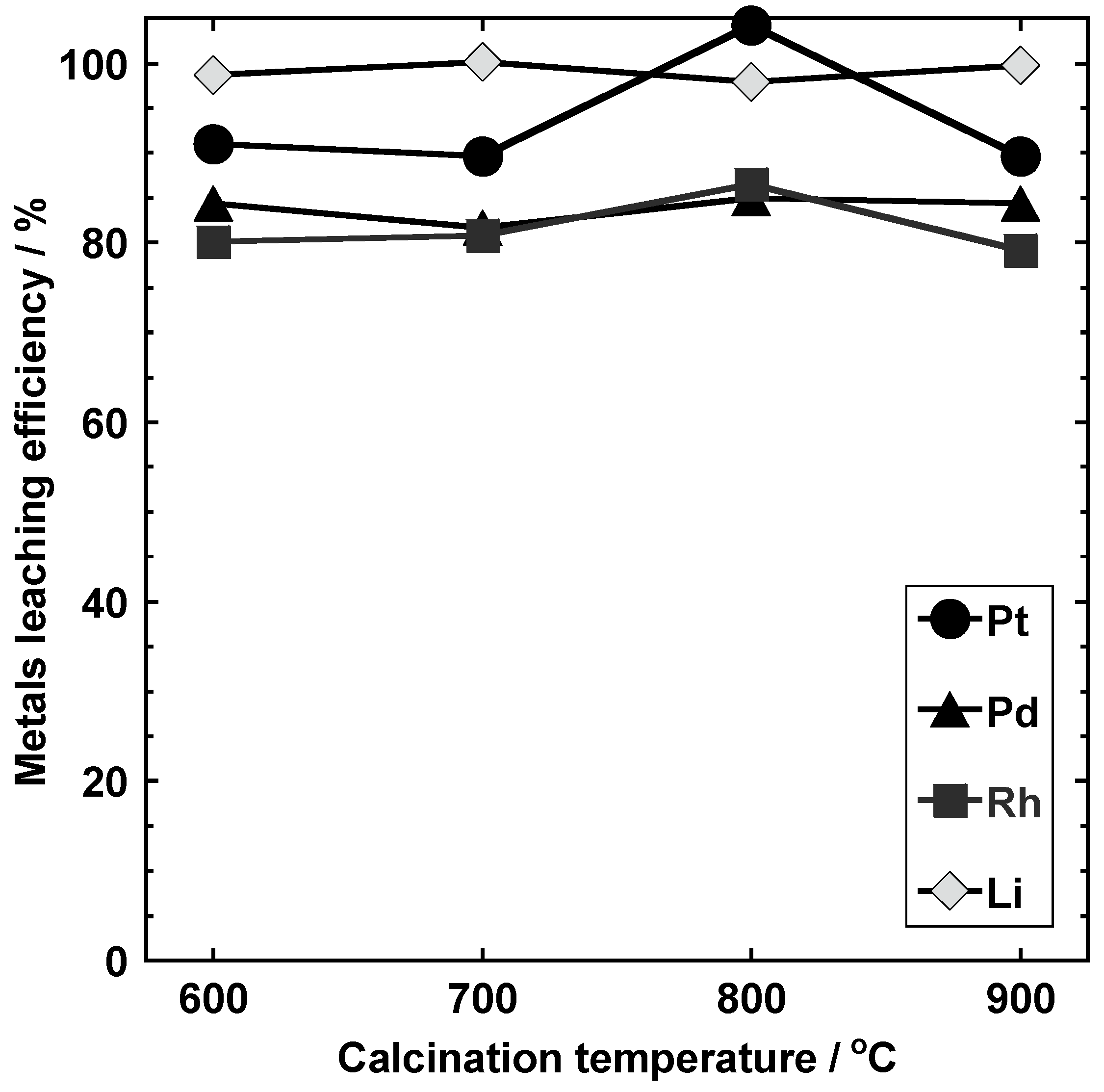
3.3.2. Calcination Temperature (Conditions I-D-1, I-D-2, I-D-3, and I-D-4)
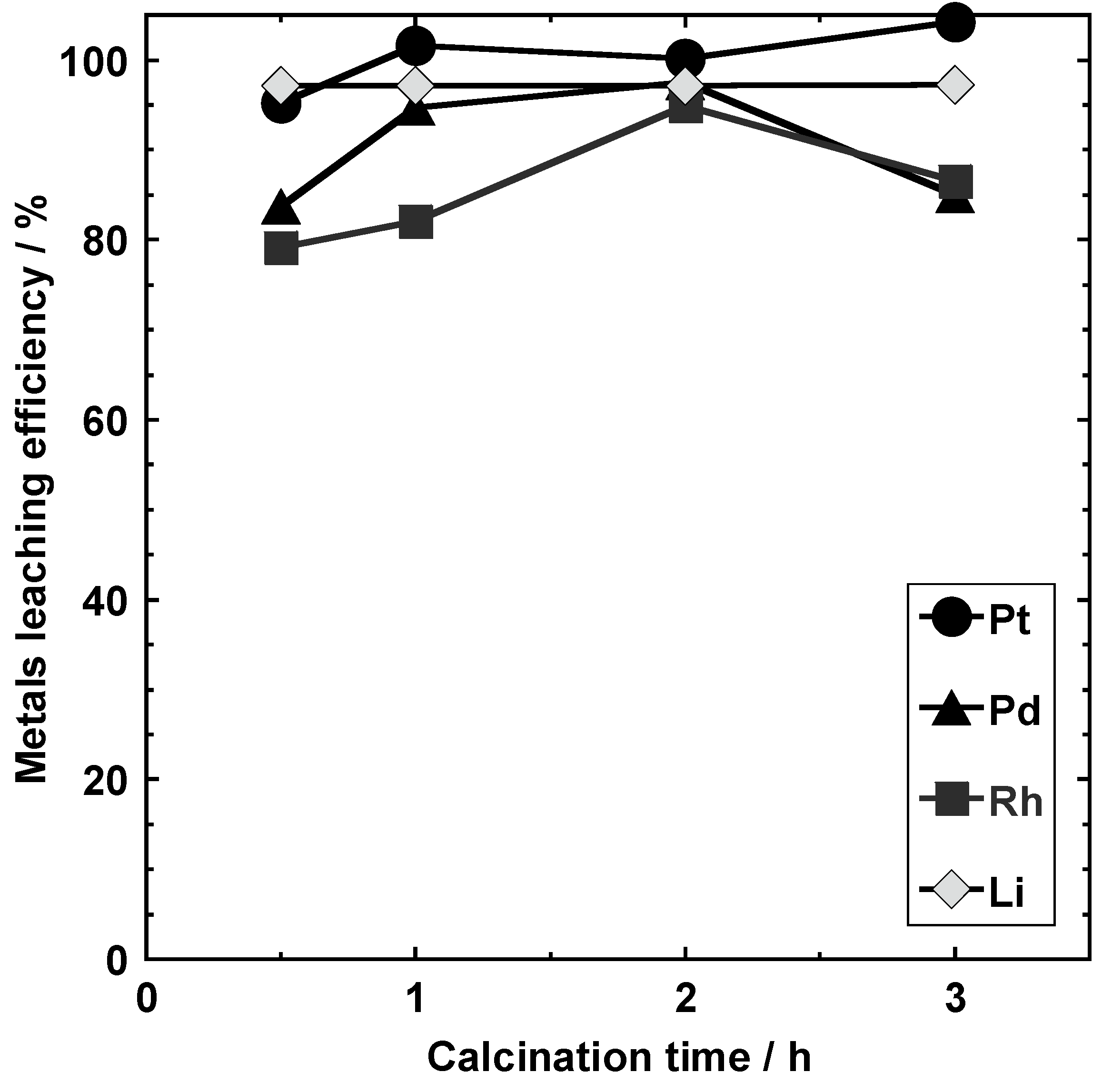
3.3.3. Calcination Time (Conditions I-E-1, I-E-2, I-E-3, and I-E-4)
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Johnson Matthey Plc. Pgm Market Report. May 2021. Available online: http://www.platinum.matthey.com/documents/new-item/pgm-market-reports/pgm-market-report-may-21.pdf (accessed on 2 July 2021).
- Johnson Matthey Plc. Platinum 2013 Interim Review. Available online: http://www.platinum.matthey.com/documents/market-review/2013-interim/full-review/english.pdf (accessed on 2 July 2021).
- International Energy Agency. Trends in electric mobility. In Global EV Outlook 2020; IEA Publications: Paris, France, 2020; Chapter 1; Volume 2020, pp. 39–85. Available online: https://www.iea.org/reports/globa-ev-outlook-2020 (accessed on 2 July 2021).
- International Energy Agency. Prospects for Electric Mobility Deployment to 2030. In Global EV Outlook 2020; IEA Publications: Paris, France, 2020; Chapter 3; Volume 2020, pp. 153–184. Available online: https://www.iea.org/reports/global-ev-outlook-2020 (accessed on 2 July 2021).
- Meeting for the New Era of Automobiles. Available online: https://www.meti.go.jp/committee/kenkyukai/seizou/jidousha_shinjidai/pdf/001_01_00.pdf (accessed on 19 July 2021).
- Crundwell, F.; Moats, M.; Ramachandran, V.; Robinson, T.; Davenport, W.G. Refining of the Platinum-Group Metals. In Extractive Metallurgy of Nickel, Cobalt and Platinum Group Metals; Elsevier: Oxford, UK, 2011; Chapter 37; pp. 489–534. [Google Scholar] [CrossRef]
- Barnes, J.E.; Edwards, J.D. Solvent Extraction at Inco’s Acton Precious Metal Refinery. Chem. Ind. 1982, 6, 151–155. [Google Scholar]
- Kasuya, R.; Miki, T.; Morikawa, H.; Tai, Y. Synthesis of Alkali Metal Platinates and Their Dissolution Behavior in Hydrochloric Acid. J. Ceram. Soc. Jpn. 2013, 121, 884–890. [Google Scholar] [CrossRef] [Green Version]
- Kasuya, R.; Miki, T.; Tai, Y. Preparation of Li2PtO3 and Its Dissolution Properties in Hydrochloric Acid. J. Ceram. Soc. Jpn. 2013, 121, 261–264. [Google Scholar] [CrossRef] [Green Version]
- Kasuya, R.; Miki, T.; Morikawa, H.; Tai, Y. Synthesis of Sodium Platinates and Their Dissolution Behaviors in Hydrochloric Acid: Effects of Lithium Carbonate Addition on Platinate Formation. Int. J. Miner. Process. 2014, 128, 33–39. [Google Scholar] [CrossRef]
- Kasuya, R.; Miki, T.; Morikawa, H.; Tai, Y. Dissolution Process of Palladium in Hydrochloric Acid: A Route via Alkali Metal Palladates. Metall. Mater. Trans. B 2015, 46, 2476–2483. [Google Scholar] [CrossRef]
- Kasuya, R.; Miki, T.; Morikawa, H.; Tai, Y. Dissolution of Platinum in Catalyst Materials Using Hydrochloric Acid: A New Method Based on the Use of Complex Oxides. Miner. Eng. 2016, 87, 25–31. [Google Scholar] [CrossRef]
- Kasuya, R.; Nomura, K.; Narita, H. Solubilization of Rhodium in Hydrochloric Acid Using an Alkali Metal Salt Method. Metall. Mater. Trans. B 2020, 51, 377–385. [Google Scholar] [CrossRef]
- U. S. Geological Survey. Mineral Commodity Summaries; United States Geological Survey: Reston, VA, USA, 2021; pp. 98–99. [CrossRef]
- International Energy Agency. The Role of Critical Minerals in Clean Energy Transit. Available online: https://www.iea.org/reports/the-role-of-critical-minerals-in-clean-energy-transitions (accessed on 19 July 2021).
- Li, J.; Wang, G.; Xu, Z. Environmentally Friendly Oxygen-Free Roasting/Wet Magnetic Separation Technology for In Situ Recycling Cobalt, Lithium Carbonate and Graphite from Spent LiCoO2/Graphite Lithium Batteries. J. Hazard. Mater. 2016, 302, 97–104. [Google Scholar] [CrossRef]
- Sit, K.; Li, P.K.C.; Ip, C.W.; Li, C.W.; Wan, L.; Lam, Y.F.; Lai, P.Y.; Fan, J.; Magnuson, D. Studies of the Energy and Power of Current Commercial Prismatic and Cylindrical Li-Ion Cells. J. Power Sources 2004, 125, 124–134. [Google Scholar] [CrossRef]
- Natarajan, S.; Boricha, A.B.; Bajaj, H.C. Recovery of Value-Added Products from Cathode and Anode Material of Spent Lithium-Ion Batteries. Waste Manag. 2018, 77, 455–465. [Google Scholar] [CrossRef]
- Swain, B. Recovery and Recycling of Lithium: A Review. Sep. Purif. Technol. 2017, 172, 388–403. [Google Scholar] [CrossRef]
- Heelan, J.; Gratz, E.; Zheng, Z.; Wang, Q.; Chen, M.; Apelian, D.; Wang, Y. Current and Prospective Li-Ion Battery Recycling and Recovery Processes. JOM 2016, 68, 2632–2638. [Google Scholar] [CrossRef] [Green Version]
- Bertuol, D.A.; Machado, C.M.; Silva, M.L.; Calgaro, C.O.; Dotto, G.L.; Tanabe, E.H. Recovery of Cobalt from Spent Lithium-Ion Batteries Using Supercritical Carbon Dioxide Extraction. Waste Manag. 2016, 51, 245–251. [Google Scholar] [CrossRef]
- Biswal, B.K.; Jadhav, U.U.; Madhaiyan, M.; Ji, L.; Yang, E.-H.; Cao, B. Biological Leaching and Chemical Precipitation Methods for Recovery of Co and Li from Spent Lithium-Ion Batteries. ACS Sustain. Chem. Eng. 2018, 6, 12343–12352. [Google Scholar] [CrossRef]
- da Cunha, J.M.; Klein, L.; Bassaco, M.M.; Tanabe, E.H.; Bertuol, D.A.; Dotto, G.L. Cobalt Recovery from Leached Solutions of Lithium-Ion Batteries Using Waste Materials as Adsorbents. Can. J. Chem. Eng. 2015, 93, 2198–2204. [Google Scholar] [CrossRef]
- Kuzuhara, S.; Ota, M.; Tsugita, F.; Kasuya, R. Recovering Lithium from the Cathode Active Material in Lithium-Ion Batteries via Thermal Decomposition. Metals 2020, 10, 433. [Google Scholar] [CrossRef] [Green Version]
- Larsson, F.; Andersson, P.; Blomqvist, P.; Mellander, B.E. Toxic Fluoride Gas Emissions from Lithium-Ion Battery Fires. Sci. Rep. 2017, 7, 10018. [Google Scholar] [CrossRef] [PubMed]
- Handel, P.; Fauler, G.; Kapper, K.; Schmuck, M.; Stangl, C.; Fischer, R.; Uhlig, F.; Koller, S. Thermal aging of electrolytes used in lithium-ion batteries—An investigation of the impact of protic impurities and different housing materials. J. Power Sources 2014, 267, 255–259. [Google Scholar] [CrossRef]
- Wang, M.; Tan, Q.; Liu, L.; Li, J. A Facile, Environmentally Friendly, and Low-Temperature Approach for Decomposition of Polyvinylidene Fluoride from the Cathode Electrode of Spent Lithium-ion Batteries. ACS Sustain. Chem. Eng. 2019, 7, 12799–12806. [Google Scholar] [CrossRef]
- Tanaka, F.; Lkhagvasuren, G.; Terakado, O.; Kuzuhara, S.; Kasuya, R. Dehydrofluorination behavior of poly(vinylidene fluoride) during thermal treatment using calcium carbonate. Metals 2021, 702, 178977. [Google Scholar] [CrossRef]
- National Institute of Standards & Technology. Certificate of Analysis, Standard Reference Material 2557, Used Auto Catalyst (Monolith). Available online: https://www-s.nist.gov/srmors/view_cert.cfm?srm=2557 (accessed on 2 July 2021).
- Pan, K.; Hu, C.; Sun, Z.; Xu, G.; Zhang, D.; Yu, L.; Wang, K.; Jiang, K. Structural and electrochemical characterization of LiMn2O4 and Li1.05Mn1.97Nb0.03O4 with excellent high-temperature cycling stability synthesized by a simple route. J. Appl. Electrochem. 2020, 50, 451–462. [Google Scholar] [CrossRef]
- Musić, S.; Filipović-Vinceković, N.; Sekovanić, L. Precipitation of Amorphous SiO2 Particles and Their Properties. Braz. J. Chem. Eng. 2011, 28, 89–94. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Element | Concentration (mg/kg) | Element | Concentration (mass%) |
|---|---|---|---|
| Pt | 1131 ± 11 | Al | 20 |
| Pd | 233.2 ± 1.9 | Si | 18 |
| Rh | 135.1 ± 1.9 | Mg | 6 |
| Pb | 13,931 ± 97 | Fe | 1.5 |
| Ce | 1.3 | ||
| Ni | 0.5 | ||
| Ba | 0.29 | ||
| Ca | 0.2 | ||
| La | 0.07 |
| Preparation Conditions | Starting Materials | Calcination Temperature (°C) | Holding Time (h) | |||
|---|---|---|---|---|---|---|
| Spent Autocatalyst (g) | Cordierite (g) | Li2CO3 (mg) | LiF (mg) | |||
| I-A | 1.0 | - | - | - | - | - |
| I-B | 1.0 | - | - | - | 800 | 3 |
| I-C-1 1 | 1.0 | - | 100 | - | 800 | 3 |
| I-C-2 | 1.0 | - | 75 | 25 | 800 | 3 |
| I-C-3 | 1.0 | - | 50 | 50 | 800 | 3 |
| I-C-4 | 1.0 | - | 25 | 75 | 800 | 3 |
| I-C-5 | 1.0 | - | - | 100 | 800 | 3 |
| I-D-1 | 1.0 | - | 100 | - | 600 | 3 |
| I-D-2 | 1.0 | - | 100 | - | 700 | 3 |
| I-D-3 1 | 1.0 | - | 100 | - | 800 | 3 |
| I-D-4 | 1.0 | - | 100 | - | 900 | 3 |
| I-E-1 | 1.0 | - | 100 | - | 800 | 0.5 |
| I-E-2 | 1.0 | - | 100 | - | 800 | 1 |
| I-E-3 | 1.0 | - | 100 | - | 800 | 2 |
| I-E-4 1 | 1.0 | - | 100 | - | 800 | 3 |
| II-A | - | 1.0 | - | - | 800 | 3 |
| II-B | - | 1.0 | 500 | - | 800 | 3 |
| II-C | - | 1.0 | - | 500 | 800 | 3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kuzuhara, S.; Ota, M.; Kasuya, R. Recovery of Platinum Group Metals from Spent Automotive Catalysts Using Lithium Salts and Hydrochloric Acid. Materials 2021, 14, 6843. https://doi.org/10.3390/ma14226843
Kuzuhara S, Ota M, Kasuya R. Recovery of Platinum Group Metals from Spent Automotive Catalysts Using Lithium Salts and Hydrochloric Acid. Materials. 2021; 14(22):6843. https://doi.org/10.3390/ma14226843
Chicago/Turabian StyleKuzuhara, Shunsuke, Mina Ota, and Ryo Kasuya. 2021. "Recovery of Platinum Group Metals from Spent Automotive Catalysts Using Lithium Salts and Hydrochloric Acid" Materials 14, no. 22: 6843. https://doi.org/10.3390/ma14226843