Abstract
The nanostructured β″ precipitates are critical for the strength of Al-Mg-Si-(Cu) aluminum alloys. However, there are still controversial reports about the composition of Cu-containing β″ phases. In this work, first-principles calculations based on density functional theory were used to investigate the composition, mechanical properties, and electronic structure of Cu-containing β″ phases. The results predict that the Cu-containing β″ precipitates with a stoichiometry of Mg4+xAl2−xCuSi4 (x = 0, 1) are energetically favorable. As the concentration of Cu atoms increases, Cu-containing β″ phases with different compositions will appear, such as Mg4AlCu2Si4 and Mg4Cu3Si4. The replacement order of Cu atoms in β″ phases can be summarized as one Si3/Al site → two Si3/Al sites → two Si3/Al sites and one Mg1 site. The calculated elastic constants of the considered β″ phases suggest that they are all mechanically stable, and all β″ phases are ductile. When Cu atoms replace Al atoms at Si3/Al sites in β″ phases, the values of bulk modulus (B), shear modulus (G), and Young’s modulus (E) all increase. The calculation of the phonon spectrum shows that Mg4+xAl2−xCuSi4 (x = 0, 1) are also dynamically stable. The electronic structure analysis shows that the bond between the Si atom and the Cu atom has a covalent like property. The incorporation of the Cu atom enhances the electron interaction between the Mg2 and the Si3 atom so that the Mg2 atom also joins the Si network, which may be one of the reasons why Cu atoms increase the structure stability of the β″ phases.
1. Introduction
Heat treatable Al-Mg-Si(-Cu) alloys in the 6xxx series are a common category of structural materials used in the construction and transportation industries. These alloys can be customized to have a desirable combination of properties, such as good formability, high specific strength, and corrosion resistance [1,2,3]. After proper aging treatment, the strength of the alloy can be greatly improved. This is mainly due to the precipitates that can contribute to the strengthening mechanisms by hindering the dislocation movement [4,5], particle strengthening σp [6], and coherency of the particles [7]. The mechanical properties of these alloys can be greatly influenced by the composition, morphology, scale, and distribution of these solute atom nanostructures [8]. The precipitation sequence for Al-Mg-Si alloys is generally considered to be [9,10]:
The supersaturated solid solution is denoted by the abbreviation SSSS. The Guinier-Preston zones (GP-zones) were first discovered in the Al-Cu system by Guinier [11] and Preston [12]. The GP-zones mainly refer to the nanoprecipitate phases formed in the early stage of aging, which is characterized by a certain ordered structure and completely coherent with the matrix.
Among the precipitates [4,13,14,15] formed in the aged Al-Mg-Si alloys, needle-like β″ precipitate is the most effective strengthening phase [14] responsible for the peak-hardening effect [16]. The β″ phase is a metastable precipitate phase, which is semi-coherent with the Al matrix in the needle cross-section, the space group is C2/m, a = 15.16 Å, b = 4.05 Å, c = 6.74 Å, and β = 105.3° [17,18]. The monoclinic β″ phase was originally proposed to have the composition of Mg5Si6 [17]. However, according to recent experimental and theoretical studies, the composition of β″ would fluctuate around Mg5Al2Si4 [19,20,21,22]. Furthermore, the most recent density functional theory (DFT) calculations inferred very minor formation enthalpy differences for β″-Mg5+xAl2−xSi4 (−1 < x < 1) [21]. These results indicate that the composition of β″ phase in Al matrix may change under certain conditions. For example, the dispersed nano-precipitates can be affected by the addition of Mg and/or Si, as well as other elements like Cu [5,23,24,25,26,27]. The addition of Cu is demonstrated to increase the age-hardening response, and it promotes the generation of higher number density and smaller size precipitates [14,28,29,30,31,32]. Therefore, a certain amount of Cu is usually added into Al-Mg-Si alloys. The addition of Cu increases the complexity of the precipitation sequence [32,33]. The precipitation sequence of Al-Mg-Si-Cu alloys is reported as [34]:
Previous work used various experimental and theoretical methods to study the incorporation of Cu in β″, and analyzed the Cu atoms as foreign solute atoms in the phases [20]. Cu addition could further enhance the positive effect of pre-aging on bake hardening for Al-Mg-Si alloys [35]. It has been demonstrated by high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) that Cu is mainly confined to the Si3/Al sites (Si or Al atoms completely occupy) of the β″ structure [26,35,36,37], which as mentioned was also supported from DFT-based calculations [38]. The β″ precipitates in Al-Mg-Si-Cu alloy were detected with an average composition of 28.6Al-38.7Mg-26.5Si-5.17Cu (at. %) using atom probe tomography (APT) and high-resolution energy-dispersive X-ray (EDX) mapping [36]. Furthermore, the addition of Cu has no effect on the type of β″ precipitate, Cu atoms incorporate in β″ and some of Mg, Si and Al in β″ unit cell are substituted by Cu atoms [39].
As mentioned above, the β″ precipitation behavior in Al-Mg-Si-Cu alloys has been investigated using various characterization methods. However, the detailed structures and stabilities are still unclear of Cu-containing β″ phases in these alloys, and these structural refinements could be supported by first-principles results [40]. In addition, we predict energy-lowering site occupations and stoichiometries of the β″ phases, where experimental information is incomplete. Understanding the structure of Cu-containing β″ precipitates is essential to elucidate the precipitation sequence in heat-treatable Al-Mg-Si (-Cu) alloys.
In the present work, first-principles calculations based on density functional theory (DFT) [41] were used to study the Cu-containing β″ phases. Based on the structural information obtained by experimental methods, first-principles atomistic calculations can provide structural, chemical, and energetic information [40]. A large number of Cu-containing β″ structures were constructed searching for possible stable configurations and structural stability, kinetic stability, and mechanical stability were also considered. Finally, the characteristics of Cu atoms occupying sites were analyzed through the electronic structure.
2. Materials and Methods
2.1. Atomic Model
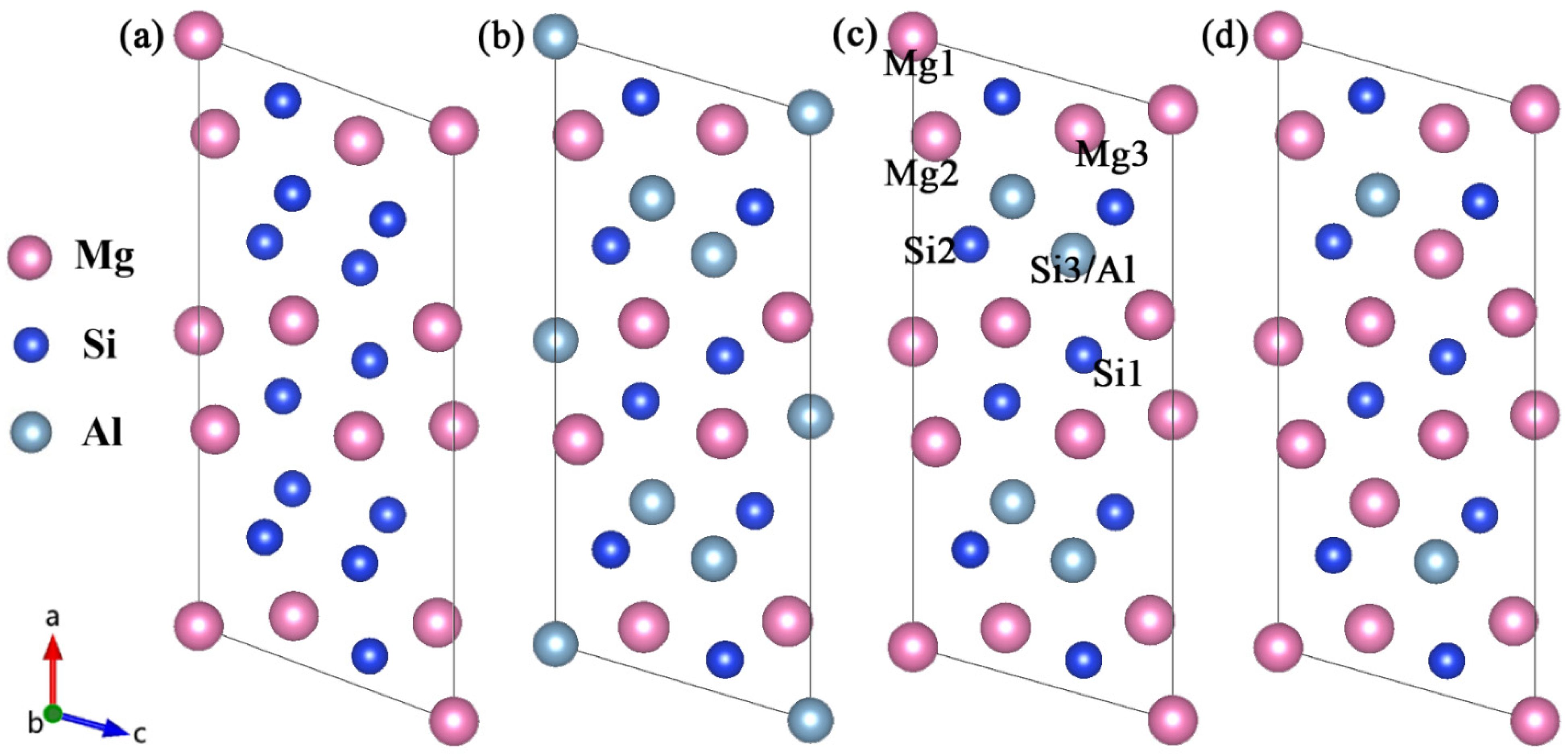
For the β″ phases, the formation enthalpies and lattice parameters of Mg4Al3Si4, Mg5Al2Si4, Mg6AlSi4, and Mg5Si6 were computed for each of the models of the crystal structures available in the literature [17,18,21], allowing a critical assessment of the validity of the models. Figure 1 shows four atomic models of the β″ without Cu. The Wyckoff site information of the energetically most favorable β″-Mg5Al2Si4 is shown in Table 1 [18,19].
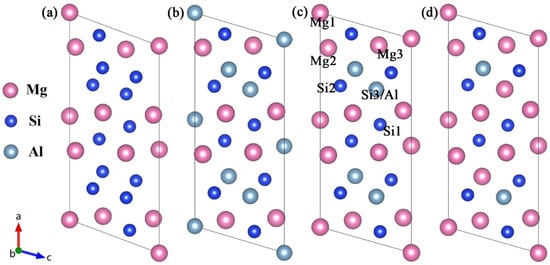
Figure 1.
Four atomic models of the β″ available in the literature [17,21]. (a) Mg5Si6 from Zandbergen [17]; (b) Mg4Al3Si4 and (c) the Mg5Al2Si4 from Hasting [19]; (d) Mg6AlSi4 from Ehlers [21]. The relative location of each site is marked in (c).

Table 1.
Wyckoff site information (x, y, z) in the β″-Mg5Al2Si4 phase [18,19]; atomic configuration is shown schematically in Figure 1c.
2.2. Computational Details
The first-principles calculations were performed utilize the plane wave pseudopotential method, as implemented in the highly efficient Vienna ab initio simulation package (VASP) [42,43], The electron-ion interactions were described through projector augmented wave (PAW) [44,45]. The exchange-correlation function were constructed by the generalized gradient approximation (GGA) of Perdew-Burke-Ernzerhof (PBE) [46]. All structures were fully relaxed with respect to atomic positions as well as all lattice parameters in order to find the lowest-energy structure. The electron wave function was expanded in plane waves up to a cutoff energy of 450 eV. The β′′phase was represented by a conventional cell with 22 atoms according to the experimental results, and 3 × 12 × 8 Γ-centered k-point meshes were employed in the Brilluion zone sampling and generated automatically by following the Monkhorst-Pack sampling scheme [47], while the 3 × 3 × 8 Γ-centered k-point meshes and 1 × 4 × 1 supercells were employed for calculation of “replacement energy” (the detailed definition is explained below). Atoms were relaxed until their residual forces converged to 0.01 eV/Å. The phonon spectra were obtained using the Phonopy package [48].
The four-parameter Birch–Murnaghan equation of state with its linear form [49] is employed to estimate the equilibrium total energy (E0), volume (V0),
where a, b, c, and d are fitting parameters. More details can be found in our previous work [50].
Compared with the energy of solid solution containing a Cu atom, the energy gain of the Cu atoms incorporated in β″ is referred to as “replacement energy”. In order to construct the Cu-containing β″ phases, it is necessary to determine the possible occupation sites of Cu in β″ phases. Additionally, computing the replacement energy (see Ref. [51]) can be used as a criterion for the possible occupation sites of solute atoms. There have been previous studies addressing the first-principles calculations for describing replacement energies of different sides. Since the replacement energy of Cu atoms at Mg2 and Mg3 sites were not shown in Saito’s work [38], one Cu atom was introduced into a 1 × 4 × 1 supercell and the preference of Cu atoms for each non-equivalent site in β″ was evaluated using the method described by Saito et al. [51], but with higher calculation precision.
To solve the compositional uncertainty preliminarily, the reported C2/m symmetries [18] were deliberately reduced to the level where only pairs of atoms (e.g., the two Cu atoms) were regarded as equivalent. This implies that space group P2/m was used throughout and there are 11 different sites within the unit cell. Besides, no partial occupancies were considered and vacancies were ignored. The replacement energy for Cu incorporation in β″ can be described as follows:
where H are the calculated enthalpy of the system, are the Cu-free structure, are the sides in , X are the solute atoms incorporated in the precipitates, and S are substitutional sites in the Al matrix. A certain atom X incorporates on site is referred to as “{X → }”. The formation enthalpy of solid solution (SS), , was used to find out the most energetically favorable configurations in the atomic models. Since there is no stable fcc structure for Mg and Si, their formation energies in relation to SS were determined as follows:
where Esub (Mg) and Esub (Si) are the enthalpies of substituting Al atoms by Mg and Si atoms, respectively. Esub (Mg) and Esub (Si) were calculated in a 3 × 3 × 3 Al supercell with one Mg/Si atom and 107 Al atoms with a k-point meshes of 5 × 5 × 5. The enthalpy of substituting a Mg atom was defined as:
where E (Al) is the enthalpy of a 3 × 3 × 3 Al supercell. The definition of Esub (Mg) was also feasible for Esub (Si).
Finally, in order to compare the structures with different Al content, the formation enthalpy can also be expressed in kJ/mol of solute atoms, instead of kJ/mol [52]. This transformation is achieved as follows: ΔHSS [kJ/mol solute] = ΔHSS [kJ/mol]/(xMg + xSi + xCu), where xMg and xSi and xCu are the atomic fractions of Mg and Si and Cu in the β″ phases MgaAlbCucSid (a = xMg, b = xAl, c = xCu, d = xSi). This is a common definition of formation enthalpy in the literature [9,21,52].
The elastic constant can be represented by a 6 × 6 matrix. Based on the symmetry of the crystal structure, the independent elastic constants of the monoclinic crystal are reduced to 13, as shown in Formula (5):
The stress-strain method based on the generalized Hooke’s theorem is used to calculate the elastic constants of each crystal [53]. For more detailed stress-strain method description, please refer to [54]. The relationship between elastic constant Cijkl, stress tensor δkl, and strain tensor δkl can be expressed as:
The Hill model [55] is used to further obtain the bulk modulus (B), shear modulus (G), and Youngs modulus (E) of the crystal through the elastic constant. The Hill model takes into account that the calculation results of the Voigt model and the Reuss model will be high and low, respectively, and take the arithmetic mean of the values of the Voigt model and the Reuss model. For monoclinic crystal structure, the formula for calculating the bulk modulus (B) and shear modulus (G) of monoclinic crystals using Voigt model and Reuss model are [56]:
wherein:
The formula for calculating the bulk modulus (B), shear modulus (G), and elastic modulus (E) of monoclinic crystal by Hill model [55] is:
3. Results and Discussion
3.1. Structure Stability
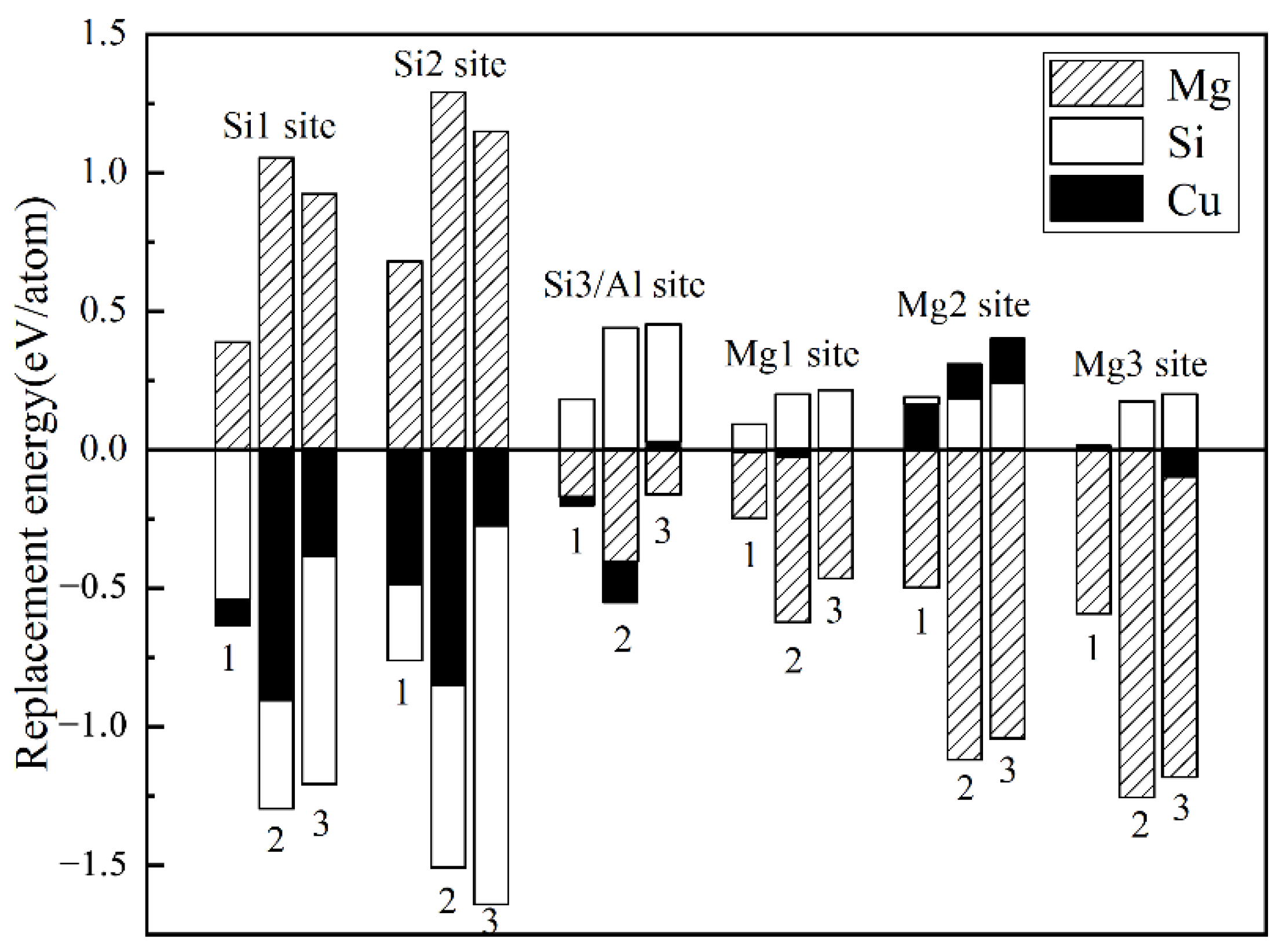
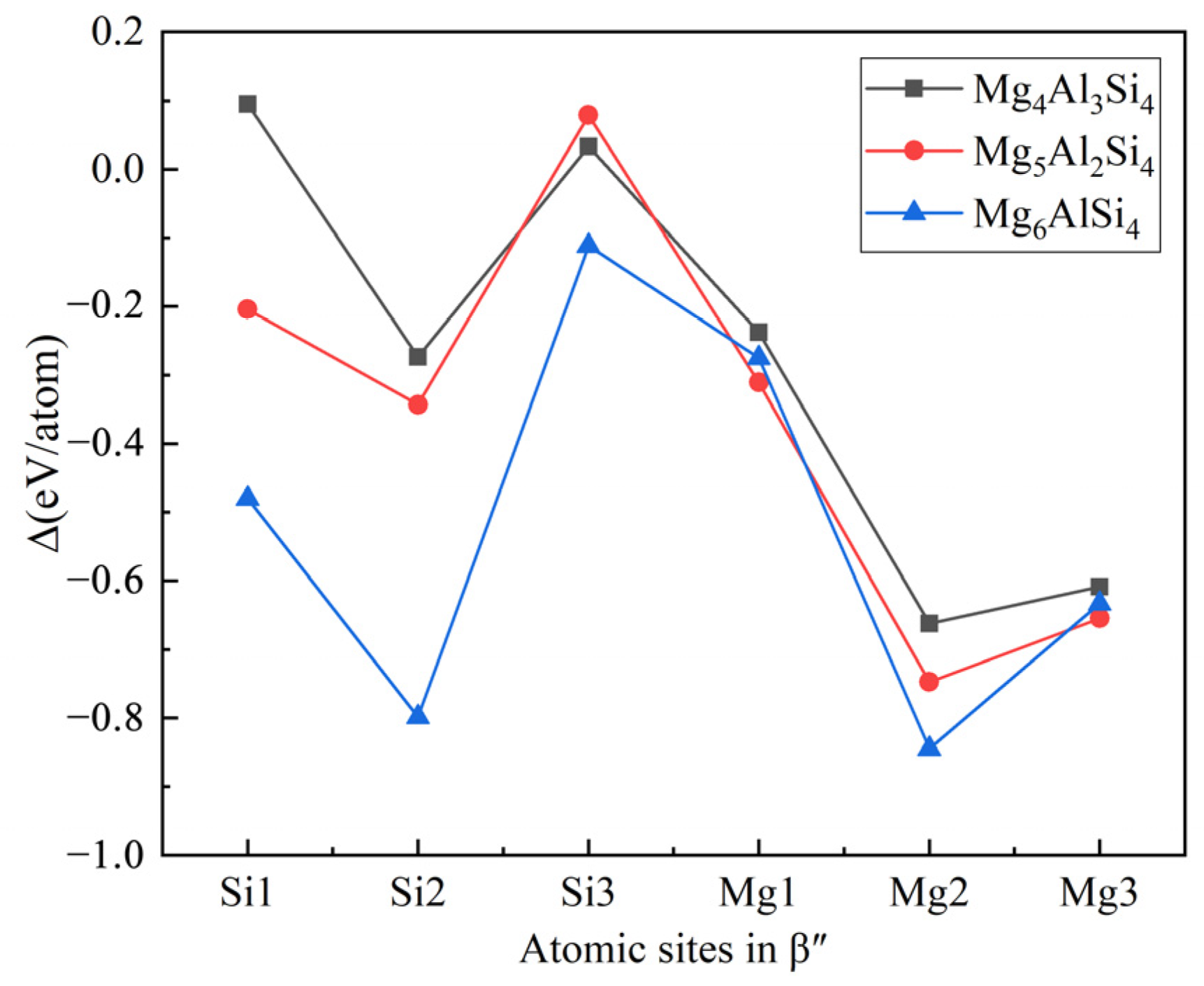
The replacement energy is shown in Figure 2 and alternative solute atoms Mg/Si were incorporated for comparison with Cu at different sites. In order to more intuitively express the competitive occupation sites of Cu atoms, the variable Δ is introduced and the Δ values of Cu atoms at different sites in different β″ configurations are shown in Figure 3. The Δ represents the “competitiveness” between Cu atoms and other solute atoms at each site, it is the difference between the lowest replacement energy of Mg/Si solute atoms and the replacement energy of Cu atoms. The larger the value of Δ, the more likely the Cu atom will occupy the site. Consequently, due to the low Cu occupancy in β″, only three designated Cu sites (Si1, Si3, Mg1, see Figure 1c) were allowed to host Cu atoms according to the relative value of replacement energy (refer to Figure 2). This conclusion is consistent with previous research [38].
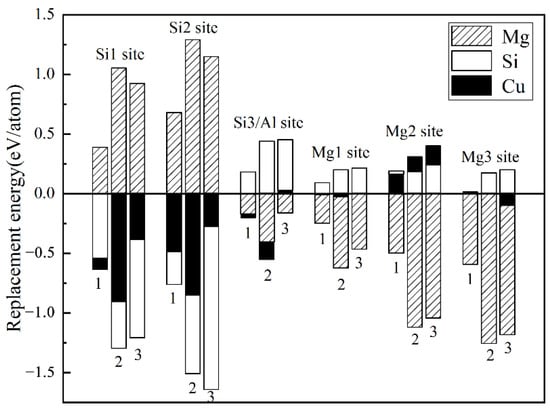
Figure 2.
Calculated replacement energies for Cu and alternative solute atoms Mg/Si on the different sites of three different β″ configurations. 1: Mg4Al3Si4, 2: Mg5Al2Si4, and 3: Mg6AlSi4. The position of each column represents a different position in a different configuration. Cu, Mg, and Si replacement energies are labelled with black, shaded, and white bars, respectively.
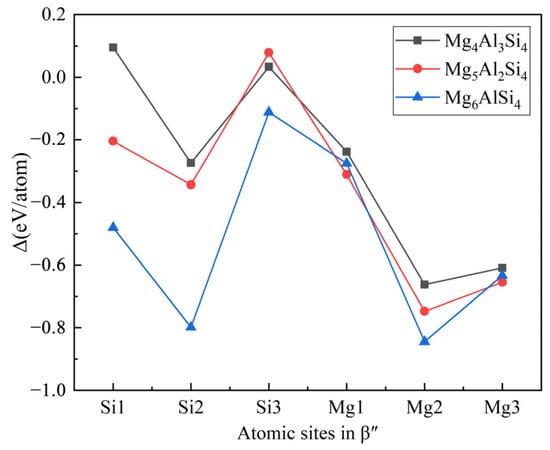
Figure 3.
The competitiveness (Δ values) of Cu atoms at different sites in different β″ configurations. The black square, red circle, and blue triangle represent Cu atoms in the Mg4Al3Si4, Mg5Al2Si4, and Mg6AlSi4 configurations, respectively.
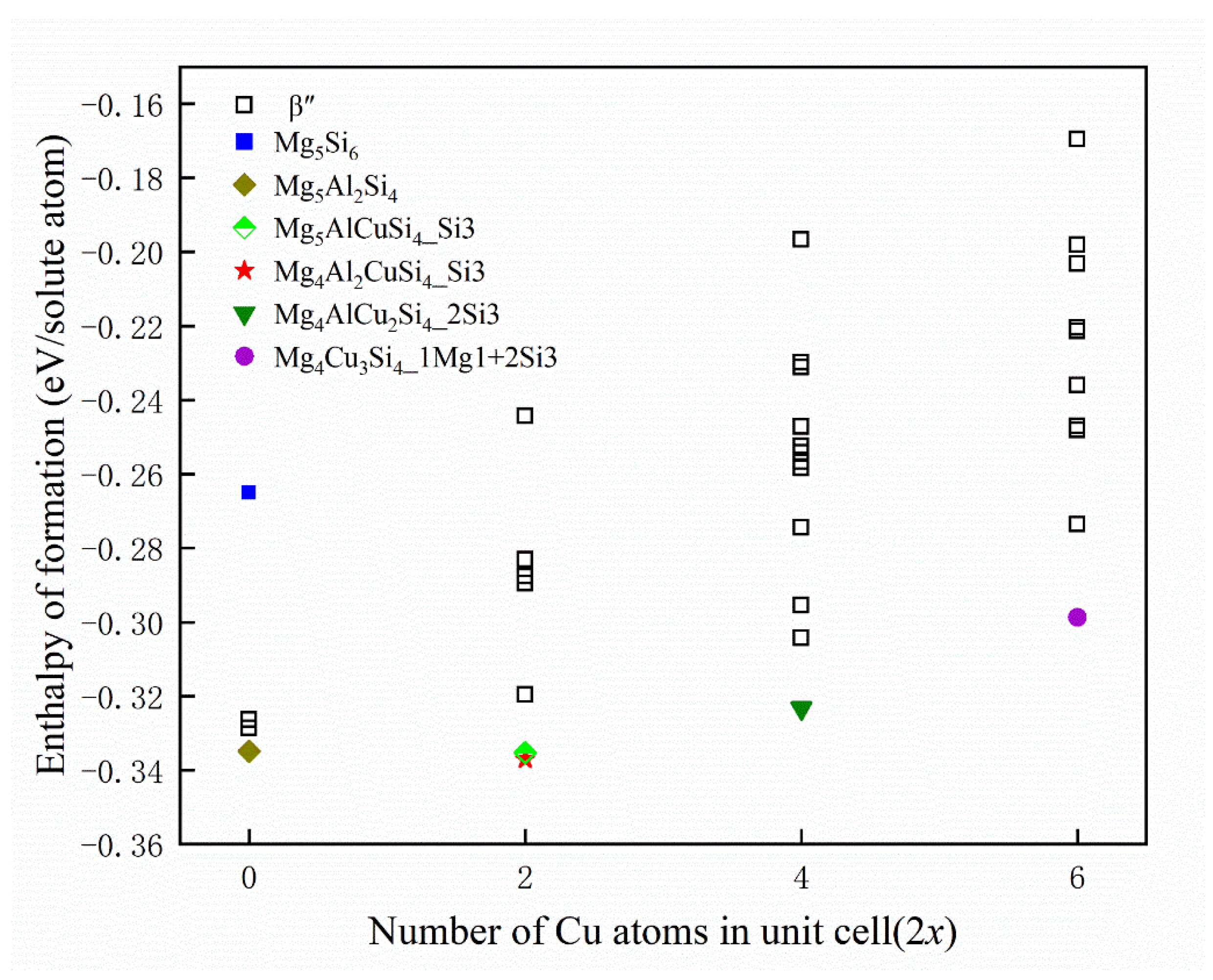
For checking the reliability of the calculations, Table 2 displays the structural parameters for selected β″ configuration without Cu atom, along with the results of earlier theoretical and experimental studies of β″. Available calculation results of formation enthalpies are shown in Table 3. The formation enthalpies of the 33 possible unit cells have been plotted in Figure 4, including the configuration without Cu atom. Since the given formation enthalpy of per solute atom (eV/solute atom) essentially presents the solute chemical potentials, the zero-temperature convex hull can be constructed to deduce the precipitation order of the system [57]. It can be seen that Cu occupying one column of each Si3 column pair is found to be the energetically most favorable option for the set of Mg4Al2CuSi4 compositions. While the formation enthalpy of Mg4Al2CuSi4 is −0.337 eV/solute atom, the formation enthalpy of Mg5AlCuSi4 is −0.335 eV/solute atom, which is similar to that of Mg4Al2CuSi4. This is consistent with the observed in previous experiments that Cu atoms mainly occupy Si3 sites [36]. The energy gained when replacing Mg/Si/Al with at the Wyckoff sites is clearly varying with x. When Cu atoms occupy two sites (that is, x = 2), Mg4AlCu2Si4 is the energetically most favorable phase, and Cu atoms occupy two Si3 columns. When Cu atoms occupy three sites, Mg4Cu3Si4 is the most stable structure, in which Cu atoms occupy one Mg1 site and two Si3 sites, which is consistent with experimental observations [36]. The results show that stoichiometry of Cu-containing β″ phase is suggested as Mg4Al3−xCuxSi4 (1 ≤ x ≤ 3). Since the formation enthalpy of Mg5AlCuSi4 is very close to that of Mg4Al2CuSi4, it can also be taken into account. This result emphasizes the possibility of fluctuations between various compositions as a function of the local alloying element concentration for the physical system during precipitated phases growth. Then the structural parameters of low energy configurations from Figure 4 also have been displayed in Table 2. As discussed above, sole minimization of the β″ phase formation enthalpy supports the well-defined Mg4+xAl2−xCuSi4 (x = 0, 1) unit cell shown in Figure 5.
Table 2.
First-principles (VASP-GGA) and experimental lattice parameters of β″ phases of Al-Mg-Si-(Cu) system. For the Cu-containing β″ phases, only the most stable crystal structures under different Cu concentrations are listed.
Table 3.
Formation enthalpies of β″ phases with different configurations in this work. The Cu occupied sites and its number are also listed in detail.
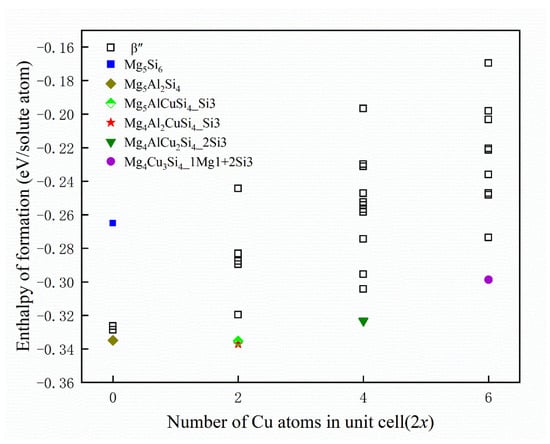
Figure 4.
Formation enthalpies of calculated β″ phases. When Cu atoms occupy x sites the number of Cu atoms in unit cell is corresponding to 2x. The lower energy configuration is marked with a specific shape, and Mg5Si6 is also marked as a reference.  represents the Mg5Si6,
represents the Mg5Si6,  represents the Mg5Al2Si4,
represents the Mg5Al2Si4,  represents the Mg5AlCuSi4 where Cu atoms occupy a Si3 site,
represents the Mg5AlCuSi4 where Cu atoms occupy a Si3 site,  represents the Mg4Al2CuSi4 where Cu atoms occupy a Si3 site,
represents the Mg4Al2CuSi4 where Cu atoms occupy a Si3 site,  represents the Mg4AlCu2Si4 where Cu atoms occupy two Si3 sites, and
represents the Mg4AlCu2Si4 where Cu atoms occupy two Si3 sites, and  represents the Mg4Cu3Si4 where Cu atoms occupy a Mg1 site and two Si3 sites.
represents the Mg4Cu3Si4 where Cu atoms occupy a Mg1 site and two Si3 sites.
represents the Mg5Si6, represents the Mg5Al2Si4, represents the Mg5AlCuSi4 where Cu atoms occupy a Si3 site, represents the Mg4Al2CuSi4 where Cu atoms occupy a Si3 site, represents the Mg4AlCu2Si4 where Cu atoms occupy two Si3 sites, and represents the Mg4Cu3Si4 where Cu atoms occupy a Mg1 site and two Si3 sites.
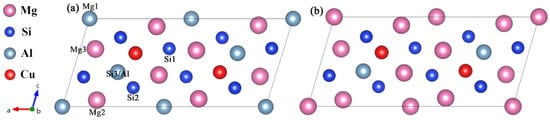
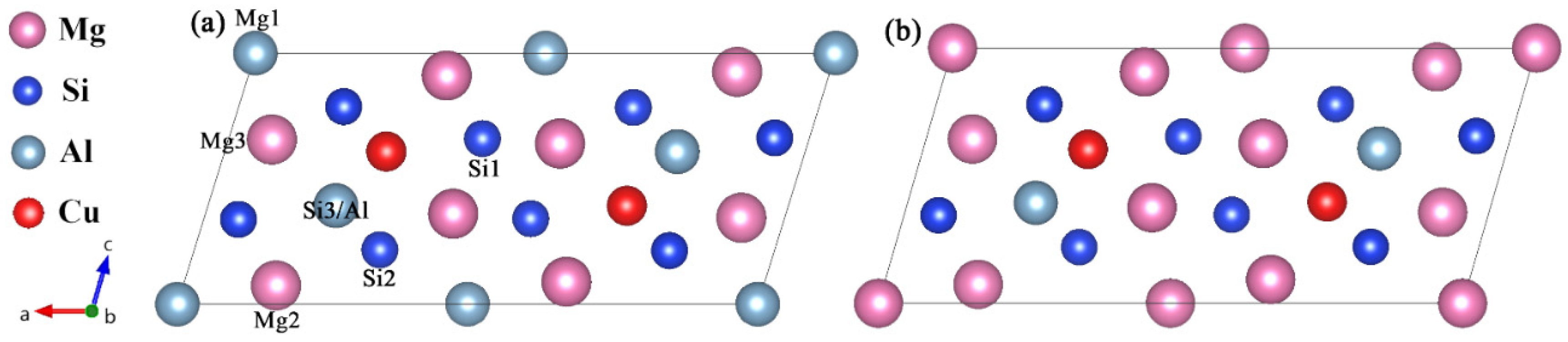
Figure 5.
The well-defined Mg4+xAl2−xCuSi4 (x = 0, 1) unit cell that sole minimization of the β″ phase formation enthalpy supports. (a) Mg4Al2CuSi4; (b) Mg5AlCuSi4. The relative location of each site is marked in (a).
3.2. Elastic Properties
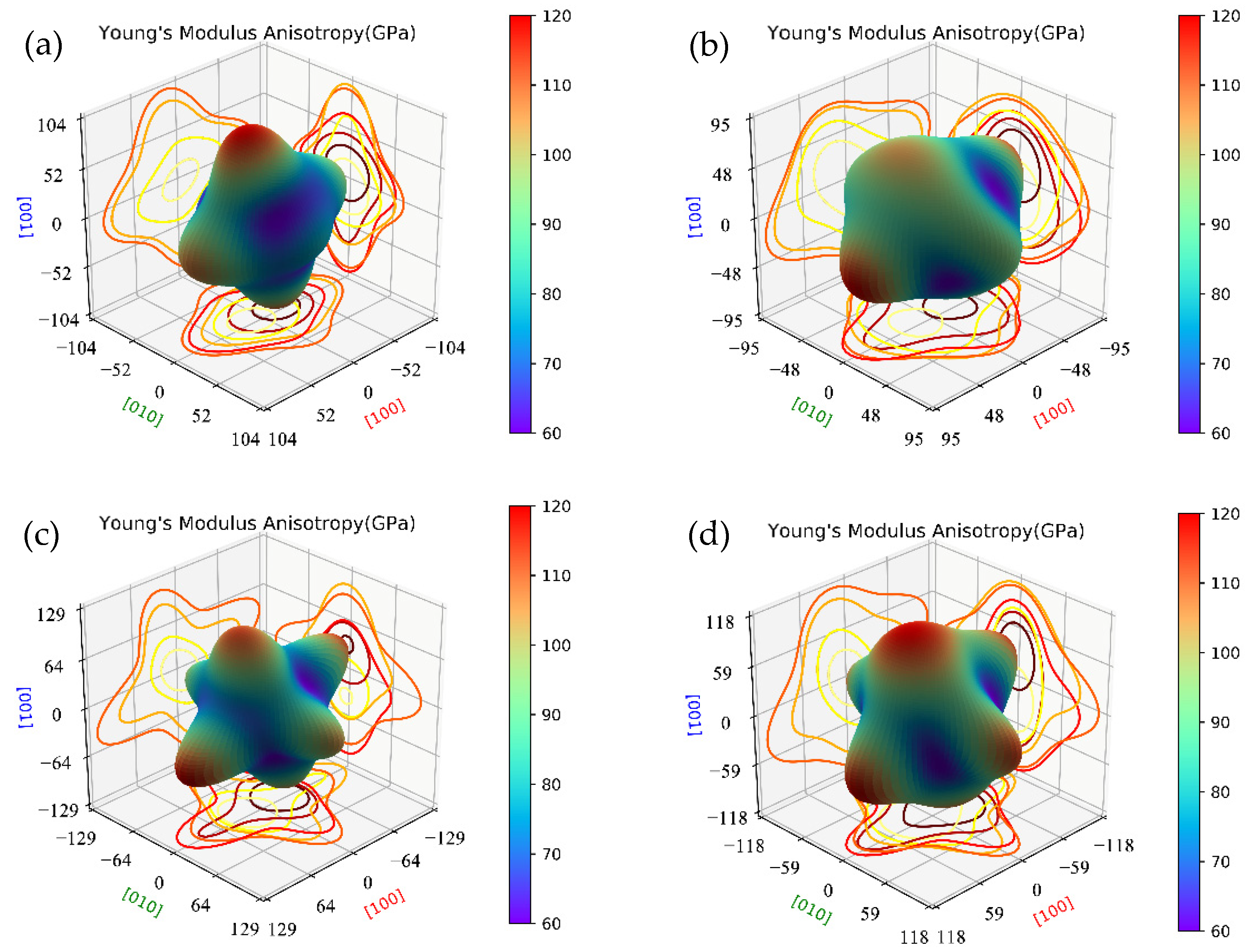
Here, we compare the mechanical properties of β″ with or without Cu atoms. The elastic constants of key β″ phases that are most likely to precipitate during aging were calculated by using fully relaxed crystal structures, and the results are listed in Table 4. According to the Born stability criterion [61], the elastic constants of Mg4Al2CuSi4 and Mg5AlCuSi4 all meet the stability criteria of monoclinic crystals. This further supports the stability of Mg4+xAl2−xCuSi4 (x = 0, 1) obtained from the formation enthalpy. The elastic constants C11, C22, and C33 are much greater than the other elastic constants in all calculated β″ phases, resulting in an obvious elastic anisotropy. In order to understand the anisotropic characteristics of these precipitation phases, the Young’s modulus anisotropies are evaluated by three-dimensional map as shown in Figure 6. Comparing Figure 6a and c, it can be seen that after Cu atoms substituted Al atoms on the Si3/Al sites, the Young’s modulus (E) anisotropy increases significantly; similar results are also shown in Figure 6b,d. This phenomenon indicates that the growth rate of the Cu-containing β″ phases may be faster than that of the β″ without Cu. It is consistent with the previous study that Cu can accelerates the age-hardening response [14,28,30].
Table 4.
Calculated single crystal elastic stiffness constants (Cij′s) of the reported β″ phases and energy favorable Cu-containing β″ phases.
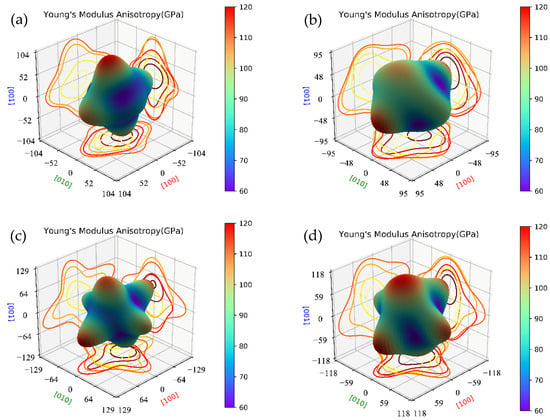
Figure 6.
The Young’s modulus anisotropies three-dimensional map of β″ phases. (a) Mg4Al3Si4; (b) Mg5Al2Si4; (c) Mg4Al2CuSi4; (d) Mg5AlCuSi4.
Based on the elastic constants in Table 4, the bulk modulus (B), shear modulus (G), and Young’s modulus (E) of polycrystalline are calculated by the Hill model [55], and the results are listed in Table 5. Comparing the values of E, G, and B of Mg4Al3Si4 and Mg4Al2CuSi4, it can be seen that the values of E, G, and B of β″ with Cu atoms are higher than that of β″ without Cu atoms. This relationship is also shown between Mg5Al2Si4 and Mg5AlCuSi4. In general, the Young’s modulus (E) can be used to measure the stiffness of the material. The stiffness of the material is greater with the increasing of Young’s modulus (E) [64]. It is obvious that the stiffness is enhanced after Cu incorporate into Si3 sites. Pugh [65] proposes using the ratio of the bulk and shear modulus, B/G, to predict brittle or ductile behavior of materials. According to the Pugh criterion, if B/G is more than 1.75, ductile behavior is expected; otherwise, the material would be brittle. From Table 4, the B/G values of calculated β″ phases are all larger than 1.75, therefore, all the compounds of β″ phase are ductile with or without Cu atoms and the ductility decreases after Cu atoms incorporate into β″. In addition, Poisson’s ratio v has been used to measure the shear stability of the lattice, which usually ranges from −1 to 0.5. The smaller the value, the stronger the ability of the crystal to maintain stability during shear deformation [66]. The value of Poisson’s ratio v > 0.26 means the ductility of the materials, and the Poisson’s ratio of metals is usually 0.25< v < 0.35 [67]. As one can see, all β″ configurations show ductility with minor differences. It is consistent with the conclusion based on Pugh criterion.
Table 5.
Calculated mechanic properties of the reported β″ phases and energy favorable Cu-containing β″ phases.
3.3. Phonon Spectra
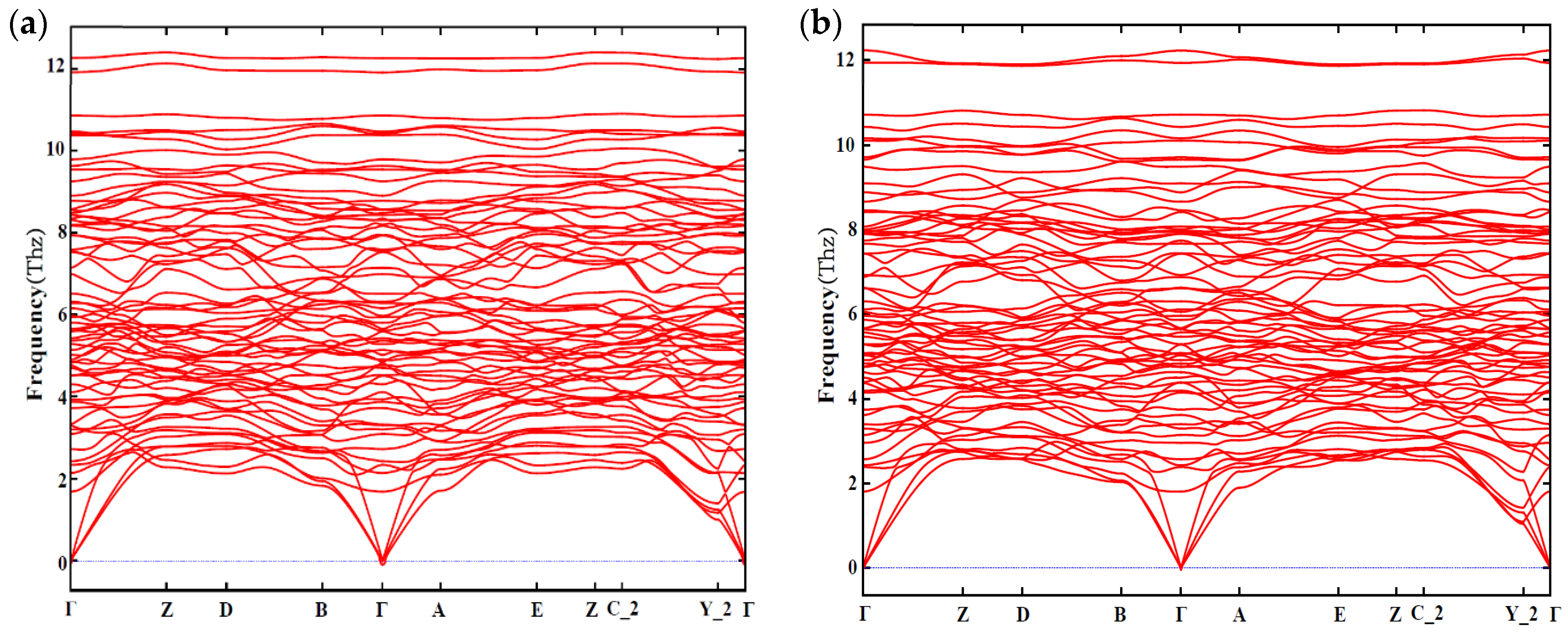
In addition, the dynamic stability is also taken into account. The phonon spectra of Mg4Al2CuSi4 and Mg5AlCuSi4 are shown in Figure 7. From Figure 7, one can see that there is no virtual frequency of configuration Mg4Al2CuSi4 and Mg5AlCuSi4, which is generally considered to be dynamically stable.
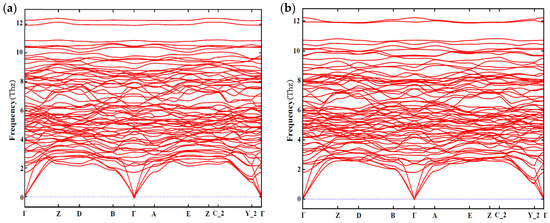
Figure 7.
The phonon spectrum along a highly symmetric K-points path of (a) Mg4Al2CuSi4; (b) Mg5AlCuSi4.
3.4. Electronic Structure
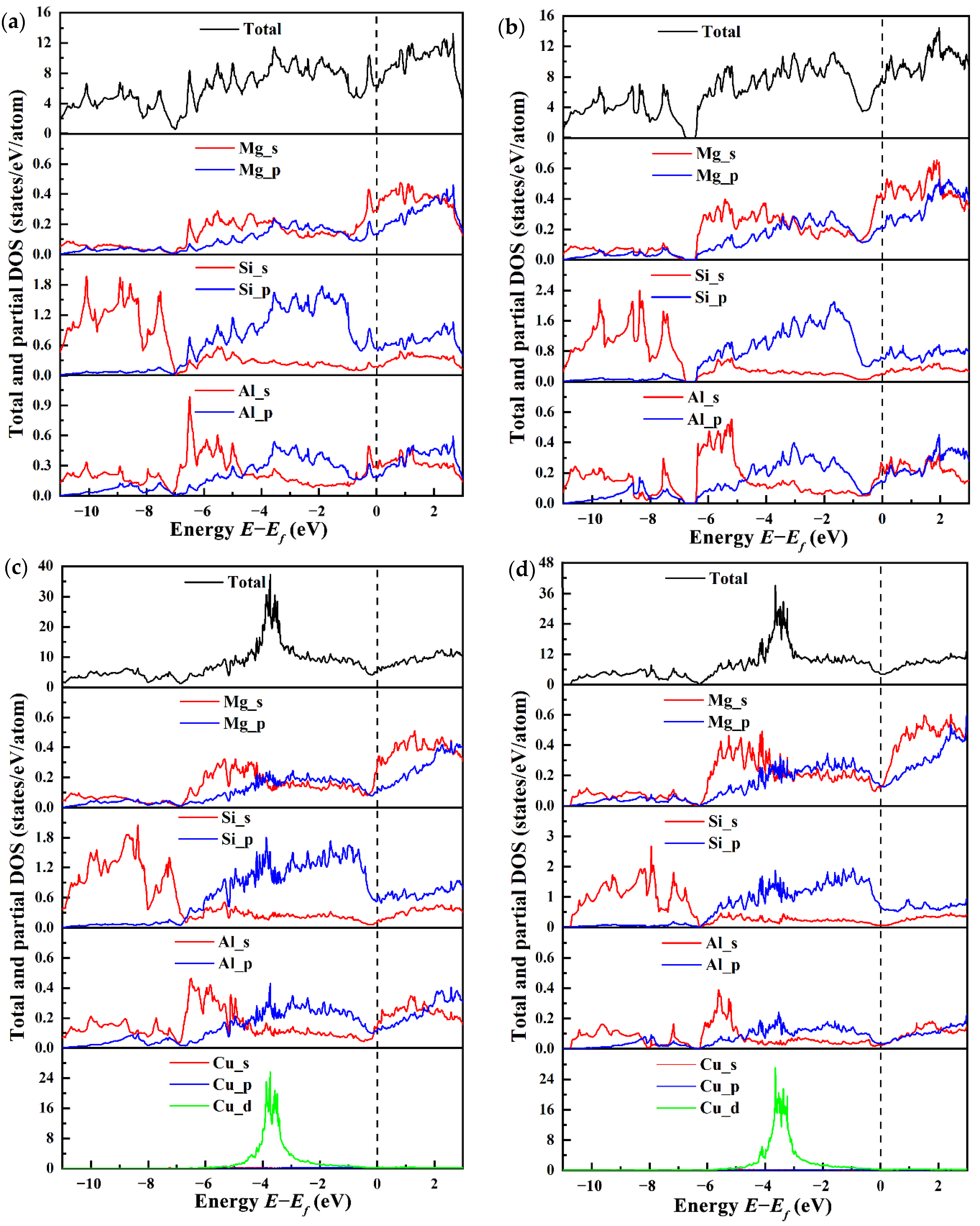
The total and partial electronic density of states (TDOSs and PDOSs) for four types of β″ configurations are calculated to explore the influence mechanism of electronic interaction on structural stability and mechanical properties, as shown in Figure 8, with the Fermi level set to zero. It is evident that incorporating Cu does not change the metallic characteristic of the β″ phase due to the finite DOS at the Fermi level. At the Fermi level, the TDOS for four types of β″ configurations at the Fermi level varies. The greatest n (Ef) is 7.41 states/eV/cell in Mg5Al2Si4, followed by 6.40 states/eV/cell in Mg4Al3Si4, 5.27 states/eV/cell in Mg4Al2CuSi4, and 4.18 states/eV/cell in Mg5AlCuSi4. This indicates that the Cu-containing β″ phases have a smaller n (Ef). In general, a smaller pseudo gap value n (Ef) corresponds to a more stable structure [68]. This indicates that Mg4+xAl2−xCuSi4 (x = 0, 1) are more stable than the β″ phases without Cu. The Si-s (range from around 11 eV to 7 eV) and Si-p states (from around 7 eV to the Fermi level) dominate the TDOS of Mg4Al3Si4 and Mg5Al2Si4 below the Fermi level. In between (ranging from about −7 eV up to −4 eV) regimes, a mixture of s and p character exists, indicating strong hybridization. Especially from −7 eV to −5 eV, the shapes of Si-s and Si-p are very similar, indicating that there is a strong interaction between Si atoms. This may be the origin for the formation of the Si-network; the Si-network acts as a stable skeleton of these phases [32,69]. One can see that Mg-s/Al-s and Si-p in the range from −7 to −4 eV, originating mainly from the s-p hybridization of Si atoms and Mg/Al atoms. The s-states and p-states of Al, Mg, and Si are strongly hybridized above the Fermi level. From Figure 8, it should be noted that, below the Fermi level, the Cu-d state is formed. The s/p orbitals of Mg, Si, and Al all interact with the Cu-d state, and there is obvious electron transfer. The Si-p orbital and the Cu-d orbital are hybridized to form a covalent like bonding, and more electrons are transferred to the new orbital formed by the p-d hybridization.
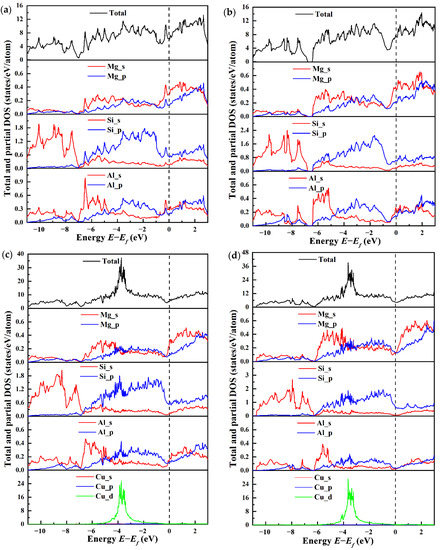
Figure 8.
The total and partial electronic density of states (PDOSs and TDOSs) for four β″ type compounds. (a) Mg4Al3Si4; (b) Mg5Al2Si4; (c) Mg4Al2CuSi4; (d) Mg5AlCuSi4.
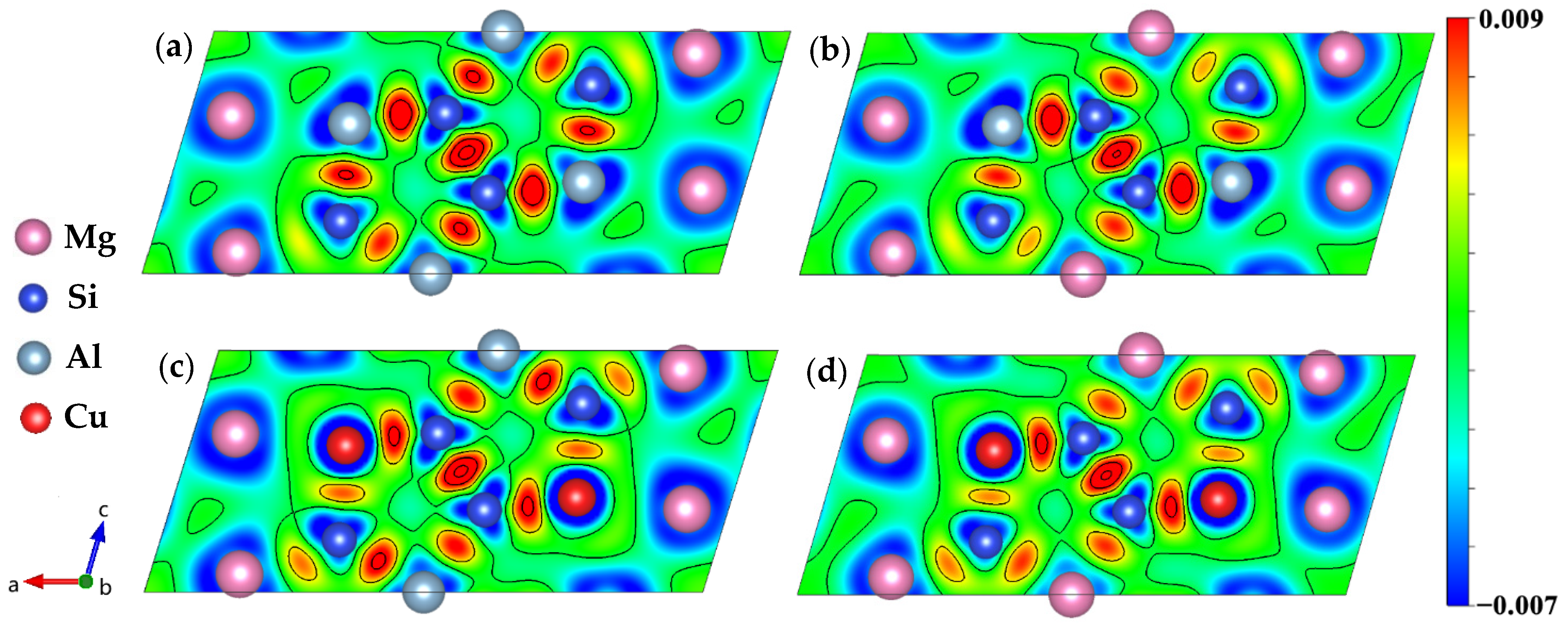
In order to gain a better understanding of the electronic structure of the studied system, the charge density distributions were used as an additional method. The charge-density difference between the (DFT) converged charge density and the isolated atomic charge densities were employed. Figure 9 shows the charge density difference contour plot for the (010) plane to analyze the interaction between Al, Mg, Si, and Cu atoms for the β″ phases. Here we clearly see that there has indeed been a transfer of charge to all the Si–Si bond regions, it is consistent with the analysis by Derlet et al. [69]. A dominant feature of Figure 9a,b is the concentration of charge between the Si1-S3/Al-Si2-Mg1-Si1 nearest neighbors, and to a lesser extent, between the Si3 and Mg1 nearest neighbors, indicating that covalency plays a role in this system, which was also reported in previous research [70]. Meanwhile, the charge distribution looks like a “charge loop”, which can lead to the formation of an “Si network”. Strong covalent bonds network can significantly increase the structural stability of β″ phases. Such a charge transfer to the bonding regions originates from the core regions of both atoms on the Mg and Si sites, in addition to the homogeneous interstitial region between the Mg atoms. The depletion of charge from the Mg3 sites indicates that for this system both metallicity and covalency are present in the bonding. Moreover, the charge transfer density between the Si3 and Si2 sites is slightly decreased, and the charge transfer density between the Mg2 and Si2 sites is increased, indicating the bonds between atoms on Mg2 and Si2 sites are covalent. As shown in Figure 9, the charge ionization of all Mg3 sites is strong, and when the Cu atom on the Si3 site charge ionization becomes stronger, it means both Mg and Cu valence electron are delocalized. The difference is that Mg uniformly provides charges to the surroundings to form a metallic environment [69], while the charges of Cu atoms are delocalized toward Si atoms in unit cells, forming a directional covalent like bond.
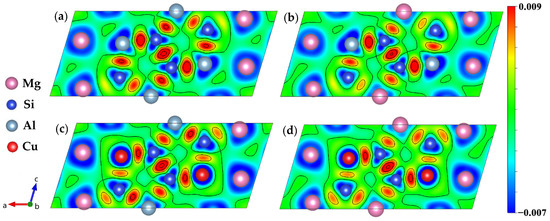
Figure 9.
Charge density difference of four calculated β″ phases. (a) Mg4Al3Si4; (b) Mg5Al2Si4; (c) Mg4Al2CuSi4; (d) Mg5AlCuSi4.
According to the analysis of the thermodynamic results of replacement energies and formation enthalpies in Section 3.1 “Structure stability”, the stoichiometry of Cu-containing β″ phases in the precipitation sequence and the sequence of Cu atoms substituting sites in the β″ phases can be inferred. For the stable phases determined from the thermodynamics, the elastic properties of β″ phases with and without Cu were calculated in Section 3.2 “Elastic properties”, further supporting the proposed stoichiometry. Besides, the “Phonon spectra” study in Section 3.3 shows that they are also dynamically stable. In summary, the proposed compositions Mg4Al3−xCuxSi4 (1 ≤ x ≤ 3) are reasonable, which is consistent with the results observed in the experiment [36]. In the Section 3.4 “Electronic structure”, the origin for the stability of the Cu-containing β″ phases is analyzed from the perspective of electron interaction.
4. Conclusions
- (1)
- The calculation of the formation enthalpies of 33 Cu-containing β″ phases shows that the replacement order of Cu atoms in β″ phases can be summarized as one Si3/Al site → two Si3/Al sites → two Si3/Al sites and one Mg1 site.
- (2)
- The Cu atoms strongly favor occupying one of each pair of Si3/Al sites and the most stable Cu-containing β″ phases were expected to have a stoichiometry of Mg4+xAl2−xCuSi4 (x = 0, 1). In addition, taking into account the change of Cu content in β″ phases, the stoichiometry of Mg4Al3−xCuxSi4 (1 ≤ x ≤ 3) may precipitate.
- (3)
- The calculated mechanical properties show that all calculated β″ phases are mechanically stable. The incorporation of Cu atoms improves the values of bulk modulus (B), shear modulus (G), and Young’s modulus (E) of β″, respectively, and all β″ phases calculated show ductile behavior. Furthermore, the calculation of the phonon spectra shows that Mg4+xAl2−xCuSi4 (x = 0, 1) are dynamically stable.
- (4)
- The electronic structure results shows that the Cu atom will join the Si network, and the bond between the Si atom and the Cu atom has the covalent property. The incorporation of Cu atom increases the electron interaction between the Mg2 and the Si3 atom, which may be one of the reasons why the incorporation of Cu atom increases the stability of the β″ phase structure.
Author Contributions
Conceptualization, S.H. and J.W.; methodology, S.H. and J.W.; investigation, S.H., J.W.; resources, J.W., Q.W. and Y.D.; data curation, S.H. and J.W.; writing—original draft preparation, S.H.; writing—review and editing, J.W., D.Z., Y.K., Q.W. and Y.D.; visualization, S.H. and D.Z.; supervision, J.W. and Y.D.; project administration, J.W. and Y.D.; funding acquisition, J.W. and Y.K. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by National Key Research and Development Program of China, grant numbers 2019YFB2006500, and the National Natural Science Foundation of China, grant numbers 52171024, 51771234 and 51601228.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data are contained within the article.
Acknowledgments
This work was financially supported by National Key Research and Development Program of China (grant numbers: 2019YFB2006500), the National Natural Science Foundation of China (grant numbers 52171024, 51771234 and 51601228) are greatly acknowledged. First-principles calculations were partially carried out at the High Performance Computing of Central South University.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Li, Y.; Li, H.; Katgerman, L.; Du, Q.; Zhang, J.; Zhuang, L. Recent advances in hot tearing during casting of aluminium alloys. Prog. Mater. Sci. 2021, 117, 100741. [Google Scholar] [CrossRef]
- Abouarkoub, A.; Thompson, G.E.; Zhou, X.; Hashimoto, T.; Scamans, G. The influence of prolonged natural aging on the subsequent artificial aging response of the AA6111 automotive alloy. Met. Mater. Trans. A 2015, 46, 4380–4393. [Google Scholar] [CrossRef]
- Zhang, D.-L.; Wang, J.; Kong, Y.; Zou, Y.; Du, Y. First-principles investigation on stability and electronic structure of Sc-doped θ′/Al interface in Al-Cu alloys. Trans. Nonferrous Met. Soc. China 2021, 31, 3342–3355. [Google Scholar] [CrossRef]
- Chakrabarti, D.; Laughlin, E.D. Phase relations and precipitation in Al–Mg–Si alloys with Cu additions. Prog. Mater. Sci. 2004, 49, 389–410. [Google Scholar] [CrossRef]
- Gupta, A.; Lloyd, D.; Court, S. Precipitation hardening in Al–Mg–Si alloys with and without excess Si. Mater. Sci. Eng. A 2001, 316, 11–17. [Google Scholar] [CrossRef]
- Safyari, M.; Moshtaghi, M.; Hojo, T.; Akiyama, E. Mechanisms of hydrogen embrittlement in high-strength aluminum alloys containing coherent or incoherent dispersoids. Corros. Sci. 2021, 194, 109895. [Google Scholar] [CrossRef]
- Ardell, A.J. Precipitation hardening. Met. Mater. Trans. A 1985, 16, 2131–2165. [Google Scholar] [CrossRef]
- Polmear, I. In Aluminium alloys—A century of age hardening. Mater. Forum 2004, 28, 1–14. [Google Scholar]
- Van Huis, M.; Chen, J.; Sluiter, M.; Zandbergen, H. Phase stability and structural features of matrix-embedded hardening precipitates in Al–Mg–Si alloys in the early stages of evolution. Acta Mater. 2007, 55, 2183–2199. [Google Scholar] [CrossRef]
- Edwards, G.; Stiller, K.; Dunlop, G.; Couper, M. The precipitation sequence in Al–Mg–Si alloys. Acta Mater. 1998, 46, 3893–3904. [Google Scholar] [CrossRef]
- Guinier, A. Structure of age-hardened aluminium-copper alloys. Nat. Cell Biol. 1938, 142, 569–570. [Google Scholar] [CrossRef]
- Preston, G.D. Structure of age-hardened aluminium-copper alloys. Nat. Cell Biol. 1938, 142, 570. [Google Scholar] [CrossRef]
- Matsuda, K.; Ikeno, S.; Terayama, K.; Matsui, H.; Sato, T.; Uetani, Y. Comparison of precipitates between excess Si-type and balanced-type Al-Mg-Si alloys during continuous heating. Met. Mater. Trans. A 2005, 36, 2007–2012. [Google Scholar] [CrossRef]
- Miao, W.F.; Laughlin, D.E. Effects of Cu content and preaging on precipitation characteristics in aluminum alloy 6022. Met. Mater. Trans. A 2000, 31, 361–371. [Google Scholar] [CrossRef]
- Marioara, C.D.; Nordmark, H.; Andersen, S.J.; Holmestad, R. Post-β″ phases and their influence on microstructure and hardness in 6xxx Al-Mg-Si alloys. J. Mater. Sci. 2006, 41, 471–478. [Google Scholar] [CrossRef]
- Esmaeili, S.; Lloyd, D.; Poole, W. A yield strength model for the Al-Mg-Si-Cu alloy AA6111. Acta Mater. 2003, 51, 2243–2257. [Google Scholar] [CrossRef]
- Zandbergen, H.W.; Andersen, S.J.; Jansen, J. Structure determination of Mg5Si6 particles in Al by dynamic electron diffraction studies. Science 1997, 277, 1221–1225. [Google Scholar] [CrossRef]
- Andersen, S.J.; Zandbergen, H.; Jansen, J.; Træholt, C.; Tundal, U.; Reiso, O. The crystal structure of the β″ phase in Al–Mg–Si alloys. Acta Mater. 1998, 46, 3283–3298. [Google Scholar] [CrossRef]
- Hasting, H.S.; Frøseth, A.G.; Andersen, S.J.; Vissers, R.; Walmsley, J.C.; Marioara, C.D.; Danoix, F.; Lefebvre, W.; Holmestad, R. Composition of β″ precipitates in Al–Mg–Si alloys by atom probe tomography and first principles calculations. J. Appl. Phys. 2009, 106, 123527. [Google Scholar] [CrossRef]
- Pogatscher, S.; Antrekowitsch, H.; Leitner, H.; Sologubenko, A.; Uggowitzer, P. Influence of the thermal route on the peak-aged microstructures in an Al–Mg–Si aluminum alloy. Scr. Mater. 2013, 68, 158–161. [Google Scholar] [CrossRef]
- Ehlers, F.J. Ab initio interface configuration determination for β″ in Al–Mg–Si: Beyond the constraint of a preserved precipitate stoichiometry. Comput. Mater. Sci. 2014, 81, 617–629. [Google Scholar] [CrossRef] [Green Version]
- Ninive, P.H.; Strandlie, A.; Gulbrandsen-Dahl, S.; Lefebvre, W.; Marioara, C.D.; Andersen, S.J.; Friis, J.; Holmestad, R.; Løvvik, O.M. Detailed atomistic insight into the β″ phase in Al–Mg–Si alloys. Acta Mater. 2014, 69, 126–134. [Google Scholar] [CrossRef] [Green Version]
- Xiao, Q.; Liu, H.; Yi, D.; Yin, D.; Chen, Y.; Zhang, Y.; Wang, B. Effect of Cu content on precipitation and age-hardening behavior in Al-Mg-Si-xCu alloys. J. Alloys Compd. 2017, 695, 1005–1013. [Google Scholar] [CrossRef]
- Man, J.; Jing, L.; Jie, S.G. The effects of Cu addition on the microstructure and thermal stability of an Al–Mg–Si alloy. J. Alloys Compd. 2007, 437, 146–150. [Google Scholar] [CrossRef]
- Ding, L.; Jia, Z.; Liu, Y.; Weng, Y.; Liu, Q. The influence of Cu addition and pre-straining on the natural aging and bake hardening response of Al-Mg-Si alloys. J. Alloys Compd. 2016, 688, 362–367. [Google Scholar] [CrossRef]
- Saito, T.; Marioara, C.D.; Andersen, S.J.; Lefebvre, W.; Holmestad, R. Aberration-corrected HAADF-STEM investigations of precipitate structures in Al–Mg–Si alloys with low Cu additions. Philos. Mag. 2014, 94, 520–531. [Google Scholar] [CrossRef] [Green Version]
- Saito, T.; Mørtsell, E.A.; Wenner, S.; Marioara, C.D.; Andersen, S.J.; Friis, J.; Matsuda, K.; Holmestad, R. Atomic structures of precipitates in Al-Mg-Si alloys with small additions of other elements. Adv. Eng. Mater. 2018, 20, 18. [Google Scholar] [CrossRef] [Green Version]
- Murayama, M.; Hono, K.; Miao, W.F.; Laughlin, D.E. The effect of Cu additions on the precipitation kinetics in an Al-Mg-Si alloy with excess Si. Met. Mater. Trans. A 2001, 32, 239–246. [Google Scholar] [CrossRef]
- Jia, Z.; Ding, L.; Cao, L.; Sanders, R.; Li, S.; Liu, Q. The influence of composition on the clustering and precipitation behavior of Al-Mg-Si-Cu alloys. Met. Mater. Trans. A 2016, 48, 459–473. [Google Scholar] [CrossRef]
- Zandbergen, M.; Cerezo, A.; Smith, G. Study of precipitation in Al–Mg–Si Alloys by atom probe tomography II. Influence of Cu additions. Acta Mater. 2015, 101, 149–158. [Google Scholar] [CrossRef]
- Bobel, A.; Kim, K.; Wolverton, C.; Walker, M.; Olson, G.B. Equilibrium composition variation of Q-phase precipitates in aluminum alloys. Acta Mater. 2017, 138, 150–160. [Google Scholar] [CrossRef]
- Ehlers, F.J.H.; Wenner, S.; Andersen, S.J.; Marioara, C.D.; Lefebvre, W.; Boothroyd, C.B.; Holmestad, R. Phase stabilization principle and precipitate-host lattice influences for Al–Mg–Si–Cu alloy precipitates. J. Mater. Sci. 2014, 49, 6413–6426. [Google Scholar] [CrossRef] [Green Version]
- Torsæter, M.; Ehlers, F.; Marioara, C.; Andersen, S.J.; Holmestad, R. Applying precipitate–host lattice coherency for compositional determination of precipitates in Al–Mg–Si–Cu alloys. Philos. Mag. 2012, 92, 3833–3856. [Google Scholar] [CrossRef]
- Marioara, C.D.; Andersen, S.J.; Stene, T.N.; Hasting, H.; Walmsley, J.; van Helvoort, A.T.J.; Holmestad, R. The effect of Cu on precipitation in Al–Mg–Si alloys. Philos. Mag. 2007, 87, 3385–3413. [Google Scholar] [CrossRef]
- Weng, Y.; Jia, Z.; Ding, L.; Liu, M.; Wu, X.; Liu, Q. Combined effect of pre-aging and Ag/Cu addition on the natural aging and bake hardening in Al-Mg-Si alloys. Prog. Nat. Sci. 2018, 28, 363–370. [Google Scholar] [CrossRef]
- Li, K.; Beche, A.; Song, M.; Sha, G.; Lu, X.; Zhang, K.; Du, Y.; Ringer, S.; Schryvers, D. Atomistic structure of Cu-containing β″ precipitates in an Al–Mg–Si–Cu alloy. Scr. Mater. 2014, 75, 86–89. [Google Scholar] [CrossRef]
- Sunde, J.K.; Marioara, C.D.; Holmestad, R. The effect of low Cu additions on precipitate crystal structures in overaged Al-Mg-Si(-Cu) alloys. Mater. Charact. 2020, 160, 110087. [Google Scholar] [CrossRef]
- Saito, T.; Ehlers, F.J.; Lefebvre, W.; Hernandez-Maldonado, D.; Bjørge, R.; Marioara, C.D.; Andersen, S.J.; Mørtsell, E.A.; Holmestad, R. Cu atoms suppress misfit dislocations at the β″/Al interface in Al–Mg–Si alloys. Scr. Mater. 2016, 110, 6–9. [Google Scholar] [CrossRef] [Green Version]
- Dong, L.; Chu, S.; Hu, B.; Zeng, X.; Chen, B. Atomic-scale investigation into precipitated phase thickening in Al-Si-Mg-Cu alloy. J. Alloys Compd. 2018, 766, 973–978. [Google Scholar] [CrossRef]
- Wolverton, C. Crystal structure and stability of complex precipitate phases in Al–Cu–Mg–(Si) and Al–Zn–Mg alloys. Acta Mater. 2001, 49, 3129–3142. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 1965, 140, 1133–1138. [Google Scholar] [CrossRef] [Green Version]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [Green Version]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Togo, A.; Tanaka, I. First principles phonon calculations in materials science. Scr. Mater. 2015, 108, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Shang, S.-L.; Wang, Y.; Kim, D.; Liu, Z.-K. First-principles thermodynamics from phonon and Debye model: Application to Ni and Ni3Al. Comput. Mater. Sci. 2010, 47, 1040–1048. [Google Scholar] [CrossRef]
- Wang, J.; Shang, S.-L.; Wang, Y.; Mei, Z.-G.; Liang, Y.; Du, Y.; Liu, Z.-K. First-principles calculations of binary Al compounds: Enthalpies of formation and elastic properties. Calphad 2011, 35, 562–573. [Google Scholar] [CrossRef]
- Saito, T.; Ehlers, F.; Lefebvre, W.; Hernandez-Maldonado, D.; Bjørge, R.; Marioara, C.D.; Andersen, S.J.; Holmestad, R. HAADF-STEM and DFT investigations of the Zn-containing β″ phase in Al–Mg–Si alloys. Acta Mater. 2014, 78, 245–253. [Google Scholar] [CrossRef] [Green Version]
- Van Huis, M.; Chen, J.; Zandbergen, H.; Sluiter, M. Phase stability and structural relations of nanometer-sized, matrix-embedded precipitate phases in Al–Mg–Si alloys in the late stages of evolution. Acta Mater. 2006, 54, 2945–2955. [Google Scholar] [CrossRef]
- Mehl, M.J.; Osburn, J.E.; Papaconstantopoulos, D.A.; Klein, B.M. Structural properties of ordered high-melting-temperature intermetallic alloys from first-principles total-energy calculations. Phys. Rev. B 1990, 41, 10311–10323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Du, Y.; Tao, X.; Ouyang, Y.; Zhang, L.; Chen, Q.; Engström, A. First-principles generated mechanical property database for multi-component Al alloys: Focusing on Al-rich corner. J. Min. Met. Sect. B Met. 2017, 53, 1–7. [Google Scholar] [CrossRef]
- Hill, R. The elastic behaviour of a crystalline aggregate. Proc. Phys. Soc. Sect. A 1952, 65, 349–354. [Google Scholar] [CrossRef]
- Wu, Z.-J.; Zhao, E.-J.; Xiang, H.-P.; Hao, X.-F.; Liu, X.-J.; Meng, J. Crystal structures and elastic properties of superhard IrN2 and IrN3 from first principles. Phys. Rev. B 2007, 76, 054115. [Google Scholar] [CrossRef]
- Wang, D.; Amsler, M.; Hegde, V.I.; Saal, J.E.; Issa, A.; Zhou, B.-C.; Zeng, X.; Wolverton, C. Crystal structure, energetics, and phase stability of strengthening precipitates in Mg alloys: A first-principles study. Acta Mater. 2018, 158, 65–78. [Google Scholar] [CrossRef]
- Ravi, C. First-principles study of crystal structure and stability of Al–Mg–Si–(Cu) precipitates. Acta Mater. 2004, 52, 4213–4227. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, Z.-K.; Chen, L.-Q.; Wolverton, C. First-principles calculations of β″-Mg5Si6/α-Al interfaces. Acta Mater. 2007, 55, 5934–5947. [Google Scholar] [CrossRef]
- Zhao, D.; Zhou, L.; Kong, Y.; Wang, A.; Wang, J.; Peng, Y.; Du, Y.; Ouyang, Y.; Zhang, W. Structure and thermodynamics of the key precipitated phases in the Al–Mg–Si alloys from first-principles calculations. J. Mater. Sci. 2011, 46, 7839–7849. [Google Scholar] [CrossRef]
- Grimvall, G.; Magyari-Köpe, B.; Ozolins, V.; Persson, K.A. Lattice instabilities in metallic elements. Rev. Mod. Phys. 2012, 84, 945–986. [Google Scholar] [CrossRef] [Green Version]
- Jain, A.C.P.; Marchand, D.; Glensk, A.; Ceriotti, M.; Curtin, W.A. Machine learning for metallurgy III: A neural network potential for Al-Mg-Si. Phys. Rev. Mater. 2021, 5, 053805. [Google Scholar] [CrossRef]
- Kobayashi, R.; Giofré, D.; Junge, T.; Ceriotti, M.; Curtin, W.A. Neural network potential for Al-Mg-Si alloys. Phys. Rev. Mater. 2017, 1, 053604. [Google Scholar] [CrossRef]
- Guan, Y.; Zhang, H.; Li, W. First-principles study on alloying stability, electronic structure, and mechanical properties of Al-based intermetallics. Phys. B Condens. Matter 2011, 406, 1149–1153. [Google Scholar] [CrossRef]
- Pugh, S. XCII. Relations between the elastic moduli and the plastic properties of polycrystalline pure metals. Lond. Edinb. Dublin Philos. Mag. J. Sci. 1954, 45, 823–843. [Google Scholar] [CrossRef]
- Ravindran, P.; Fast, L.; Korzhavyi, P.; Johansson, B.; Wills, J.; Eriksson, O. Density functional theory for calculation of elastic properties of orthorhombic crystals: Application to TiSi2. J. Appl. Phys. 1998, 84, 4891–4904. [Google Scholar] [CrossRef]
- Greaves, G.N.; Greer, A.L.; Lakes, R.S.; Rouxel, T. Poisson’s ratio and modern materials. Nat. Mater. 2011, 10, 823–837. [Google Scholar] [CrossRef]
- Ouyang, Y.; Liu, F.; Lu, T.; Tao, X.; Du, Y.; He, Y. First-principles investigation of the mechanical, electronic and thermophysical properties of Q-phase in Al–Mg–Si–Cu alloys. Comput. Mater. Sci. 2013, 67, 334–340. [Google Scholar] [CrossRef]
- Derlet, P.M.; Andersen, S.J.; Marioara, C.D.; Froseth, A.G. A first-principles study of the β″-phase in Al-Mg-Si alloys. J. Phys. Condens. Matter 2002, 14, 4011–4024. [Google Scholar] [CrossRef]
- Frøseth, A.G.; Høier, R.; Derlet, P.M.; Andersen, S.J.; Marioara, C.D. Bonding in MgSi and Al-Mg-Si compounds relevant to Al-Mg-Si alloys. Phys. Rev. B 2003, 67, 224106. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).