
Ab Initio Study on Dopant Relaxation Mechanism in Ti and Ce Cationically Substituted in Wurtzite Gallium Nitride


Abstract
:1. Introduction
2. Computational Details
3. Results and Discussion
3.1. Pure Gallium Nitride
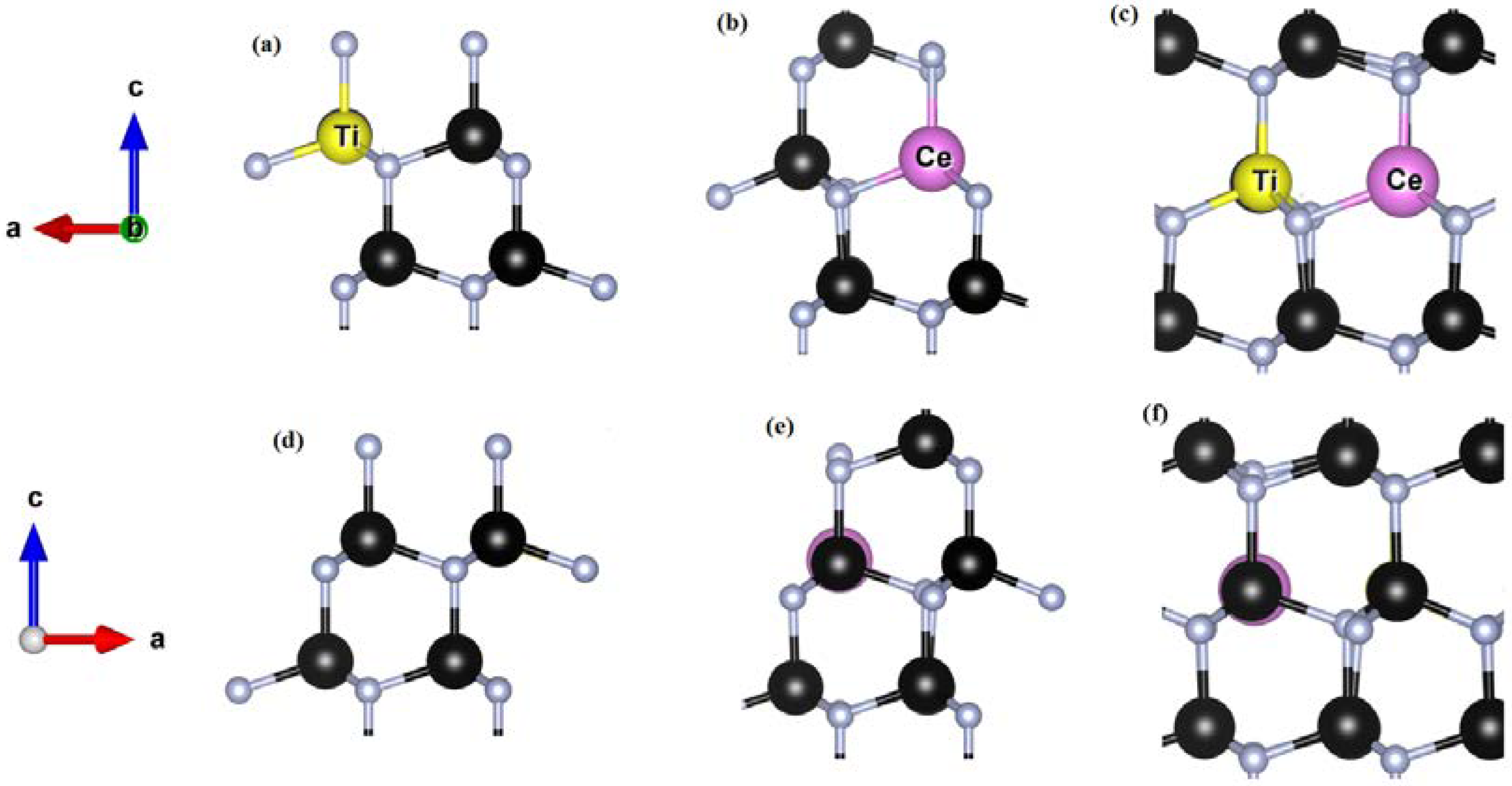
3.2. Ti-Doped GaN
3.3. Ce Doped GaN
3.4. Ti-Ce Codoped GaN
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Li, W.; Frenkel, A.I.; Woicik, J.C.; Ni, C.; Shah, S.I. Dopant location identification in Nd3+-doped Ti O2 nanoparticles. Phys. Rev. B 2005, 72, 155315. [Google Scholar] [CrossRef]
- Ali, I.; Shakoor, A.; Islam, M.U.; Saeed, M.; Ashiq, M.N.; Awan, M. Synthesis and characterization of hexagonal ferrite Co2Sr2Fe12O22 with doped polypyrrole composites. Curr. Appl. Phys. 2013, 13, 1090–1095. [Google Scholar] [CrossRef]
- Alsuwian, T.; Kousar, F.; Rasheed, U.; Imran, M.; Hussain, F.; Khalil, R.A.; Algadi, H.; Batool, N.; Khera, E.A.; Kiran, S.; et al. First principles investigation of physically conductive bridge filament formation of aluminum doped perovskite materials for neuromorphic memristive applications. Chaos Solitons Fractals 2021, 150, 111111. [Google Scholar] [CrossRef]
- Mahmood, B.K.; Kaygili, O.; Bulut, N.; Dorozhkin, S.V.; Ates, T.; Koytepe, S.; Gürses, C.; Ercan, F.; Kebiroglu, H.; Agid, R.S.; et al. Effects of strontium-erbium co-doping on the structural properties of hydroxyapatite: An Experimental and theoretical study. Ceram. Int. 2020, 46, 16354–16363. [Google Scholar] [CrossRef]
- Tsuzuki, T.; He, R.; Dodd, A.; Saunders, M. Challenges in determining the location of dopants, to study the influence of metal doping on the photocatalytic activities of ZnO nanopowders. Nanomaterials 2019, 9, 481. [Google Scholar] [CrossRef] [Green Version]
- Ullah, S.; Shi, Q.; Zhou, J.; Yang, X.; Ta, H.Q.; Hasan, M.; Ahmad, N.M.; Fu, L.; Bachmatiuk, A.; Rümmeli, M.H. Advances and trends in chemically doped graphene. Adv. Mater. Interfaces 2020, 7, 2000999. [Google Scholar] [CrossRef]
- Sudrajat, H.; Fadlallah, M.M.; Tao, S.; Kitta, M.; Ichikuni, N.; Onishi, H. Dopant site in indium-doped SrTiO3 photocatalysts. Phys. Chem. Chem. Phys. 2020, 22, 19178–19187. [Google Scholar] [CrossRef]
- Voyles, P.M.; Muller, D.A.; Grazul, J.L.; Citrin, P.H.; Gossmann, H.J.; Gossmann, L. Atomic-scale imaging of individual dopant atoms and clusters in highly n-type bulk Si. Nature 2002, 416, 826–829. [Google Scholar] [CrossRef]
- Euvrard, J.; Yan, Y.; Mitzi, D.B. Electrical doping in halide perovskites. Nat. Rev. Mater. 2021, 6, 531–549. [Google Scholar] [CrossRef]
- Wilke, K.; Breuer, H.D. The influence of transition metal doping on the physical and photocatalytic properties of titania. J. Photochem. Photobiol. A Chem. 1999, 121, 49–53. [Google Scholar] [CrossRef]
- Takeuchi, M.; Yamashita, H.; Matsuoka, M.; Anpo, M.; Hirao, T.; Itoh, N.; Iwamoto, N. Photocatalytic decomposition of NO under visible light irradiation on the Cr-ion-implanted TiO2 thin film photocatalyst. Catal. Lett. 2000, 67, 135–137. [Google Scholar] [CrossRef]
- Frayret, C.; Villesuzanne, A.; Pouchard, M.; Mauvy, F.; Bassat, J.M.; Grenier, J.-C. Identifying doping strategies to optimize the oxide ion conductivity in ceria-based materials. J. Phys. Chem. C 2010, 114, 19062–19076. [Google Scholar] [CrossRef]
- Moram, M.A.; Vickers, M.E. X-ray diffraction of III-nitrides. Rep. Prog. Phys. 2009, 72, 036502. [Google Scholar] [CrossRef]
- Hellenbrandt, M. The inorganic crystal structure database (ICSD)—present and future. Crystallogr. Rev. 2004, 10, 17–22. [Google Scholar] [CrossRef]
- Neugebauer, J.; Walle, C.G. Native defects and impurities in GaN. In Advances in Solid State Physics; Springer: Berlin/Heidelberg, Germany, 1996; Volume 35, pp. 25–44. [Google Scholar]
- Lany, S.; Zunger, A. Accurate prediction of defect properties in density functional supercell calculations. Model. Simul. Mater. Sci. Eng. 2009, 17, 084002. [Google Scholar] [CrossRef] [Green Version]
- Segev, D.; Van de Walle, C.G. Origins of Fermi-level pinning on GaN and InN polar and nonpolar surfaces. Europhys. Lett. 2006, 76, 305. [Google Scholar] [CrossRef]
- Sanson, A.; Zaltron, A.; Argiolas, N.; Sada, C.; Bazzan, M.; Schmidt, W.G.; Sanna, S. Polaronic deformation at the Fe2+/3+ impurity site in Fe: LiNbO3 crystals. Phys. Rev. B 2015, 91, 094109. [Google Scholar] [CrossRef]
- Zoroddu, A.; Bernardini, F.; Ruggerone, P.; Fiorentini, V. First-principles prediction of structure, energetics, formation enthalpy, elastic constants, polarization, and piezoelectric constants of AlN, GaN, and InN: Comparison of local and gradient-corrected density-functional theory. Phys. Rev. B 2001, 64, 045208. [Google Scholar] [CrossRef] [Green Version]
- Moses, P.G.; Miao, M.; Yan, Q.; Van de Walle, C.G. Hybrid functional investigations of band gaps and band alignments for AlN, GaN, InN, and InGaN. J. Chem. Phys. 2011, 134, 084703. [Google Scholar] [CrossRef]
- Majid, A.; Javed, M.; Rana, U.A.; Khan, S.U.D. Ti Ga–VN complexes in GaN: A new prospect of carrier mediated ferromagnetism. RSC Adv. 2015, 5, 87437–87444. [Google Scholar] [CrossRef]
- Xiong, Z.; Shi, S.; Jiang, F. Ti in GaN: Ordering ferromagnetically from first-principles study. Chem. Phys. Lett. 2007, 443, 92–94. [Google Scholar] [CrossRef]
- Jana, S.; Srivastava, B.B.; Pradhan, N. Correlation of dopant states and host bandgap in dual-doped semiconductor nanocrystals. J. Phys. Chem. Lett. 2011, 2, 1747–1752. [Google Scholar] [CrossRef]
- Zuniga-Perez, J.; Consonni, V.; Lymperakis, L.; Kong, X.; Trampert, A.; Fernández-Garrido, S.; Oliver, B.; Hubert, R.; Stacia, K.; Karine, H.; et al. Polarity in GaN and ZnO: Theory, measurement, growth, and devices. Appl. Phys. Rev. 2016, 3, 041303. [Google Scholar] [CrossRef] [Green Version]
- Sivakumar, P.; Akkera, H.S.; Reddy, T.R.K.; Bitla, Y.; Ganesh, V.; Mohan, P.K.; Srinivas, R.; Madhu, k. Effect of Ti doping on structural, optical and electrical properties of SnO2 transparent conducting thin films deposited by sol-gel spin coating. Opt. Mater. 2021, 113, 110845. [Google Scholar] [CrossRef]
- Zhu, J.; Liu, F.; Stringfellow, G.B.; Wei, S. Strain-enhanced doping in semiconductors: Effects of dopant size and charge state. Phys. Rev. Lett. 2010, 105, 195503. [Google Scholar] [CrossRef]
- Freysoldt, C.; Grabowski, B.; Hickel, T.; Neugebauer, J.; Kresse, G.; Anderson, J.; Van de Walle, C.G. First-principles calculations for point defects in solids. Rev. Mod. Phys. 2014, 86, 253. [Google Scholar] [CrossRef]
- Vanpoucke, E.P.D.; Bultinck, P.; Cottenier, S.; Speybroeck, V.V.; Driessche, I.V. Aliovalent doping of CeO2: DFT study of oxidation state and vacancy effects. J. Mater. Chem. A 2014, 2, 13723–13737. [Google Scholar] [CrossRef] [Green Version]
- Ingason, A.S.; Mockute, A.; Dahlqvist, M.; Magnus, F.; Olafsson, S.; Arnalds, U.B.; Alling, B.; Abrikosov, I.A.; Hjörvarsson, B.; Persson, P.O.Å.; et al. Magnetic self-organized atomic laminate from first principles and thin film synthesis. Phys. Rev. Lett. 2013, 110, 195502. [Google Scholar] [CrossRef]
- Cui, Z.; Ke, X.; Li, E.; Liu, T. Electronic and optical properties of titanium-doped GaN nanowires. Mater. Des. 2016, 96, 409–415. [Google Scholar] [CrossRef]
- Mullica, D.F.; Milligan, W.O.; Grossie, D.A.; Beall, G.W.; Boatner, L.A. Ninefold coordination LaPO4: Pentagonal interpenetrating tetrahedral polyhedron. Inorg. Chim. Acta 1984, 95, 231–236. [Google Scholar] [CrossRef]
- Pan, J.; Dong, Z.; Wang, B.; Jiang, Z.; Zhao, C.; Wang, J.; Song, C.; Zheng, Y.; Li, C. The enhancement of photocatalytic hydrogen production via Ti3+ self-doping black TiO2/g-C3N4 hollow core-shell nano-heterojunction. Appl. Catal. B Environ. 2019, 242, 92–99. [Google Scholar] [CrossRef]
- Wang, L.; Xie, R.; Suehiro, T.; Takeda, T.; Hirosaki, N. Down-conversion nitride materials for solid state lighting: Recent advances and perspectives. Chem. Rev. 2018, 118, 1951–2009. [Google Scholar] [CrossRef]
- Lee, I.; Choi, I.; Lee, C.; Shin, E.; Kim, D.; Noh, S.K.; Son, S.; Lim, K.Y.; Jae Lee, H. Stress relaxation in Si-doped GaN studied by Raman spectroscopy. J. Appl. Phys. 1998, 83, 5787–5791. [Google Scholar] [CrossRef]
- Martinez-Casado, R.; Dasmahapatra, A.; Sgroi, M.F.; Romero-Muñiz, C.; Herper, H.C.; Vekilova, O.Y.; Ferrari, A.M.; Pullini, D.; Desmarais, J.; Maschio, L. The CeFe11Ti permanent magnet: A closer look at the microstructure of the compound. J. Phys. Condens. Matter 2019, 31, 505505. [Google Scholar] [CrossRef] [PubMed]
- Guedel, H.U.; Furrer, A.; Blank, H. Exchange interactions in rare-earth-metal dimers. Neutron spectroscopy of cesium ytterbium halides, Cs3Yb2Cl9 and Cs3Yb2Br9. Inorg. Chem. 1990, 29, 4081–4084. [Google Scholar] [CrossRef]
- Ayala, P.; Arenal, R.; Rümmeli, M.; Rubio, A.; Pichler, T. The doping of carbon nanotubes with nitrogen and their potential applications. Carbon 2010, 48, 575–586. [Google Scholar] [CrossRef]
- Hernández, S.; Cuscó, R.; Artús, L.; Nogales, E.; Martin, R.W.; O’Donnell, K.P.; Halambalakis, G.; Briot, O.; Lorenz, K.; Alves, E. Lattice order in thulium-doped GaN epilayers: In situ doping versus ion implantation. Opt. Mater. 2006, 28, 771–774. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Configuration | Before Relaxation | After Relaxation | ||||||
|---|---|---|---|---|---|---|---|---|
| Ti-N Basal Bond Length (Å) | Ti-N Apical Bond Length (Å) | Ti-N-Ga Angle (Degree) | N-Ti-N Angle (Degree) | Ti-N Basal Bond Length (Å) | Ti-N Apical Bond Length (Å) | Ti-N-Ga Angle (Degree) | N-Ti-N Angle (Degree) | |
| Pure GaN | 1.654 | 1.652 | 109.36 | 109.36 | 1.983 | 1.991 | 109.84 | 109.84 |
| Ti substituted GaN | 1.670 | 1.706 | 108.92 | 112.40 | 1.995 | 2.017 | 109.68 | 110.54 |
| Ti displaced up Ga displaced down | 2.306 | 1.205 | 115.78 | 87.48 | 1.995 | 2.016 | 109.68 | 110.55 |
| Ti displaced down Ga displaced up | 1.845 | 2.706 | 115.78 | 119.63 | 1.995 | 2.017 | 109.68 | 110.56 |
| Ti displaced along a, b, c | 1.924 2.089 2.167 | 1.883 | 111.38 | 107.14 101.44 110.30 | 1.995 | 2.016 | 109.71 | 110.54 |
| Material/Details | Magnetic Moment (µB) | Ti-Ti NN Distance along All Axis | Ti-N Interatomic Distance (Å) | Ti-Ga Interatomic Distance (Å) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Per Ti | Total | a | b | c | Ti-N4 | Ti-N6 | Ti-N8 | Ti-N16 | Ti-Ga3 | Ti-Ga5 | Ti-Ga6 | Ti-Ga7 | Ti-Ga8 | Ti-Ga15 | |
| Pure GaN | 0 | -- | 6.494 | 6.494 | 10.562 | 1.983 | 1.983 | 1.983 | 1.991 | 3.238 | 3.238 | 3.238 | 3.238 | 3.238 | 5.281 |
| Ti-doped (displ.) | 0.683 | 0.709 | 6.518 | 6.518 | 10.583 | 1.995 | 1.995 | 1.995 | 2.016 | 3.280 | 3.279 | 3.242 | 3.279 | 3.241 | 5.322 |
| Ti-doped (U = 0) | 0.684 | 0.710 | 6.517 | 6.517 | 10.584 | 1.995 | 1.995 | 1.995 | 2.017 | 3.280 | 3.280 | 3.241 | 3.280 | 3.241 | 5.324 |
| Ti-doped (U = +1.0) | 0.714 | 0.736 | 6.518 | 6.518 | 10.585 | 2.000 | 2.000 | 2.000 | 2.024 | 3.282 | 3.282 | 3.242 | 3.282 | 3.242 | 5.325 |
| Ti-doped (U = +1.5) | 0.741 | 0.750 | 6.517 | 6.517 | 10.584 | 1.995 | 1.995 | 1.995 | 2.017 | 3.280 | 3.280 | 3.241 | 3.280 | 3.241 | 5.324 |
| Ti-doped (U = +2.0) | 0.774 | 0.786 | 6.520 | 6.520 | 10.587 | 2.010 | 2.010 | 2.010 | 2.039 | 3.285 | 3.285 | 3.244 | 3.285 | 3.244 | 5.327 |
| Ti-doped (U = +2.5) | 0.804 | 0.811 | 6.521 | 6.521 | 10.589 | 2.015 | 2.015 | 2.015 | 2.047 | 3.287 | 3.287 | 3.246 | 3.287 | 3.246 | 5.329 |
| Ti-doped (U = +3.0) | 0.833 | 0.835 | 6.522 | 6.522 | 10.590 | 2.021 | 2.021 | 2.021 | 2.056 | 3.290 | 3.290 | 3.247 | 3.290 | 3.247 | 5.331 |
| Ti-doped (U = +3.5) | 0.861 | 0.858 | 6.523 | 6.523 | 10.592 | 2.026 | 2.026 | 2.026 | 2.064 | 3.292 | 3.292 | 3.248 | 3.292 | 3.248 | 5.334 |
| Ti-doped (U = +4.0) | 0.876 | 0.872 | 6.524 | 6.524 | 10.595 | 2.028 | 2.028 | 2.028 | 2.076 | 3.300 | 3.300 | 3.245 | 3.300 | 3.245 | 5.342 |
| Ti-doped (U = +4.5) | 0.972 | 0.912 | 6.524 | 6.524 | 10.595 | 2.040 | 2.040 | 2.040 | 2.092 | 3.298 | 3.298 | 3.249 | 3.298 | 3.249 | 5.338 |
| Ti-doped (U = +5.0) | 0.946 | 0.882 | 6.526 | 6.526 | 10.601 | 2.038 | 2.038 | 2.038 | 2.105 | 3.311 | 3.311 | 3.243 | 3.311 | 3.243 | 5.244 |
| Ti-doped (U = +5.5) | 0.986 | 0.895 | 6.527 | 6.527 | 10.605 | 2.047 | 2.047 | 2.047 | 2.105 | 3.305 | 3.305 | 3.256 | 3.305 | 3.256 | 5.261 |
| Ti-doped (U = +6.0) | 1.032 | 0.911 | 6.527 | 6.527 | 10.610 | 2.055 | 2.055 | 2.055 | 2.110 | 3.302 | 3.302 | 3.266 | 3.302 | 3.266 | 5.272 |
| Ti-doped (U = +6.5) | 1.093 | 0.933 | 6.528 | 6.528 | 10.615 | 2.063 | 2.063 | 2.063 | 2.117 | 3.301 | 3.301 | 3.274 | 3.301 | 3.274 | 5.281 |
| Ti-doped (U = +7.0) | 1.187 | 0.969 | 6.529 | 6.529 | 10.620 | 2.076 | 2.076 | 2.076 | 2.132 | 3.301 | 3.301 | 3.279 | 3.301 | 3.279 | 5.288 |
| Ti-doped (U = +7.5) | 1.246 | 0.990 | 6.529 | 6.529 | 10.625 | 2.084 | 2.084 | 2.084 | 2.141 | 3.301 | 3.301 | 3.286 | 3.301 | 3.286 | 5.296 |
| Ti-doped (U = +8.0) | 1.306 | 1.009 | 6.530 | 6.530 | 10.631 | 2.092 | 2.092 | 2.092 | 2.151 | 3.301 | 3.301 | 3.293 | 3.301 | 3.293 | 5.304 |
| Material | Lattice Constants a = b (Å) | c/a Ratio | Ga-N (for GaN) Ti-N (for Ti:GaN) Bond Length (Å) | Angle (Degree) | Internal Parameter u (Å) | Formation Enthalpy/Defect Formation Energy (eV) | |||
|---|---|---|---|---|---|---|---|---|---|
| In-Plane | Out-of-Plane | Ga-N-Ga | N-Ti-N | Ti-N-Ga | |||||
| GaN (2 × 2 × 2) | 3.247 | 1.6263 | 1.983 | 1.991 | 109.84 | 109.84 | 109.84 | 0.375 | −1.055 |
| Ti2+:GaN (2 × 2 × 2) | 3.287 | 1.6284 | 2.008 | 2.025 | 109.89 | 110.79 | 109.58 | 0.375 0.377 | 5.325 |
| Ti3+:GaN (2 × 2 × 2) | 3.259 | 1.6238 | 1.987 | 1.988 | 110.00 | 110.54 | 109.68 | 0.375 0.377 | −0.065 |
| Ti4+:GaN (3 × 3 × 2) | 3.240 | 1.6240 | 1.939 | 1.937 | 109.90 | 110.46 | 111.10 | 0.382 0.368 | −0.598 |
| Ti4+:GaN (2 × 2 × 2) | 3.232 | 1.6190 | 1.940 | 1.927 | 108.85 | 110.50 | 110.50 | 0.380 0.370 | −0.432 |
| Material/Details | Magnetic Moment (µB) | Ce-Ce (nn Distance) along All Axis (Å) | Ce-N Interatomic Distance (Å) | Ce-Ga Interatomic Distance (Å) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Per Ce | Total | a | b | c | Ce-N2 | Ce-N4 | Ce-N8 | Ce-N12 | Ce-Ga2 | Ce-Ga3 | Ce-Ga4 | Ce-Ga7 | Ce-Ga8 | Ce-Ga15 | |
| Pure GaN | 0 | -- | 6.494 | 6.494 | 10.562 | 1.983 | 1.983 | 1.983 | 1.991 | 3.238 | 3.238 | 3.238 | 3.238 | 3.238 | 3.247 |
| Ce doped (displaced) | 0.533 | 0.595 | 6.554 | 6.567 | 10.606 | 2.190 | 2.203 | 2.198 | 2.272 | 3.296 | 3.307 | 3.297 | 3.372 | 3.345 | 3.335 |
| Ce doped MAG | 0.527 | 0.589 | 6.543 | 6.543 | 10.658 | 2.205 | 2.205 | 2.205 | 2.261 | 3.392 | 3.309 | 3.392 | 3.309 | 3.392 | 3.272 |
| Ce doped (U = +6.2) | 0.921 | 0.923 | 6.545 | 6.545 | 10.672 | 2.246 | 2.246 | 2.246 | 2.316 | 3.423 | 3.300 | 3.423 | 3.300 | 3.423 | 3.275 |
| Configuration | Before Relaxation | After Relaxation | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Basal Bond Length (Å) | Apical Bond Length (Å) | Ti-Ce Distance(Å) | Angle (Degree) | Basal Bond Length (Å) | Apical Bond Length (Å) | Ti-Ce Distance(Å) | Angle (Degree) | |||||||||
| Ti-N | Ce-N | Ti-N | Ce-N | N-Ce-N | N- Ti-N | Ce-N-Ti | Ti-N | Ce-N | Ti-N | Ce-N | N-Ce-N | N-Ti-N | Ce-N-Ti | |||
| Pure GaN | 1.654 | 1.654 | 1.652 | 1.652 | 2.699 | 109.36 | 109.36 | 109.36 | 1.983 | 1.983 | 1.991 | 1.991 | 3.247 | 109.84 | 109.8 | 109.84 |
| Ce-Ti:GaN | 1.991 | 1.995 | 1.992 | 2.017 | 3.259 | 110.54 | 108.81 | 109.68 | 1.985 | 2.227 | 2.037 | 2.294 | 3.294 | 110.71 | 94.43 | 102.60 |
| Ce(up)-Ti(down): GaN | 2.306 | 1.845 | 3.410 | 4.183 | 3.525 | 119.63 | 108.48 | 115.78 | 1.984 | 2.227 | 2.037 | 2.293 | 3.290 | 110.69 | 94.40 | 102.59 |
| Material/Details | Magnetic Moment (µB) | Ce-Ce (nn Distance) along All Axis (Å) Ti-Ti (nn Distance) along All Axis (Å) | Ti-Ce (Å) | Ce-N Interatomic Distance Ti-N Interatomic Distance (Å) | Ce-Ga Interatomic Distance (Å) Ti-Ga Interatomic Distance (Å) | Ti-N-Ce Angle (°) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Per Ce | Per Ti | Total | a | b | c | Ce-N2Ti-N4 | Ce-N4Ti-N6 | Ce-N8Ti-N8 | Ce-N12Ti-N16 | Ce-Ga11Ti-Ga14 | Ce-Ga14Ti-Ga11 | Ce-Ga10Ti-Ga13 | |||
| Pure GaN | -- | -- | 0 | 6.494 | 6.494 | 10.562 | 3.247 | 1.983 | 1.983 | 1.983 | 1.991 | 5.281 | 6.199 | 3.247 | 109.84 |
| Ti-Ce:GaN (GGA) | 0.168 | −0.049 | 0.103 | 6.582 6.588 | 6.588 6.588 | 10.727 10.727 | 3.294 | 2.220 1.979 | 2.225 1.979 | 2.225 2.006 | 2.299 1.997 | 5.364 5.320 | 6.296 6.327 | 3.270 3.350 | 102.10 |
| Ti-Ce:GaN (GGA) FM (CeGa16 and TiGa12) | 0.690 | 0.589 | 1.356 | 6.580 6.580 | 6.591 6.591 | 10.729 10.729 | 3.291 | 2.228 1.998 | 2.228 1.985 | 2.228 1.985 | 2.294 2.037 | 5.380 5.372 | 6.297 6.299 | 3.259 3.321 | 102.61 |
| Ti-Ce:GaN (GGA) AFM (CeGa16 and TiGa12) | 0.691 | −0.589 | 0.101 | 6.580 6.580 | 6.591 6.591 | 10.729 10.729 | 3.292 | 2.228 1.998 | 2.228 1.985 | 2.228 1.985 | 2.294 2.037 | 5.380 5.372 | 6.306 5.357 | 3.333 3.270 | 102.60 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alkhedher, M.; Majid, A.; Bulut, N.; Elkhatib, S.E. Ab Initio Study on Dopant Relaxation Mechanism in Ti and Ce Cationically Substituted in Wurtzite Gallium Nitride. Materials 2022, 15, 3599. https://doi.org/10.3390/ma15103599
Alkhedher M, Majid A, Bulut N, Elkhatib SE. Ab Initio Study on Dopant Relaxation Mechanism in Ti and Ce Cationically Substituted in Wurtzite Gallium Nitride. Materials. 2022; 15(10):3599. https://doi.org/10.3390/ma15103599
Chicago/Turabian StyleAlkhedher, Mohammad, Abdul Majid, Niyazi Bulut, and Samah Elsayed Elkhatib. 2022. "Ab Initio Study on Dopant Relaxation Mechanism in Ti and Ce Cationically Substituted in Wurtzite Gallium Nitride" Materials 15, no. 10: 3599. https://doi.org/10.3390/ma15103599
APA StyleAlkhedher, M., Majid, A., Bulut, N., & Elkhatib, S. E. (2022). Ab Initio Study on Dopant Relaxation Mechanism in Ti and Ce Cationically Substituted in Wurtzite Gallium Nitride. Materials, 15(10), 3599. https://doi.org/10.3390/ma15103599