

On the Surface Modification of LLZTO with LiF via a Gas-Phase Approach and the Characterization of the Interfaces of LiF with LLZTO as Well as PEO+LiTFSI

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
CO32− + 2 HF → H2O + CO2 + 2 F−
2. Experimental Section
2.1. LLZTO Thin Film Deposition
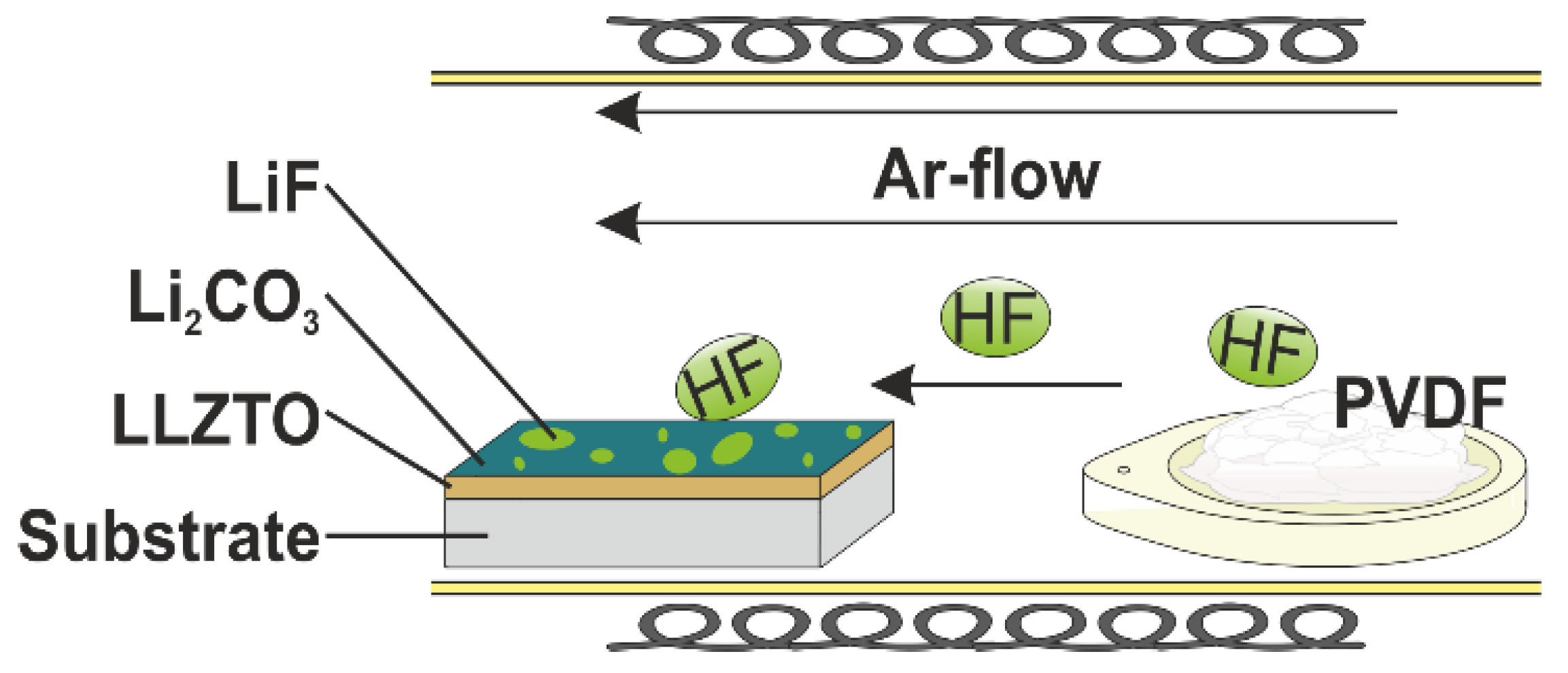
2.2. LiF Coating via Gas-Phase Fluorination
2.3. LiF Thin Films via Sputtering
2.4. PEO and LiTFSI Evaporation
2.5. LLZTO Pellet Preparation
2.6. X-ray Photoelectron Spectroscopy (XPS)
2.7. X-ray Diffraction (XRD)
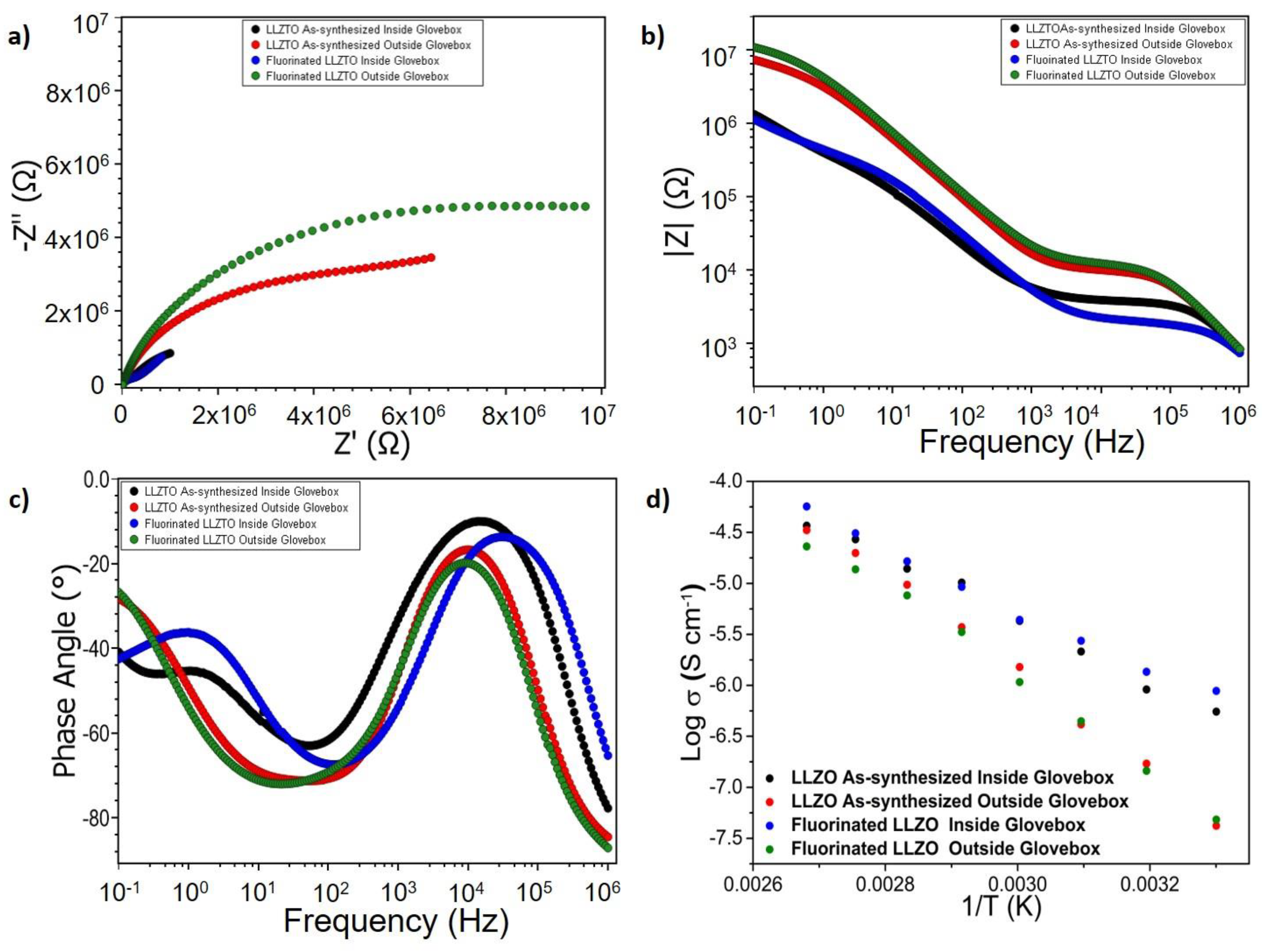
2.8. Electrochemical Impedance Spectroscopy
3. Results and Discussion
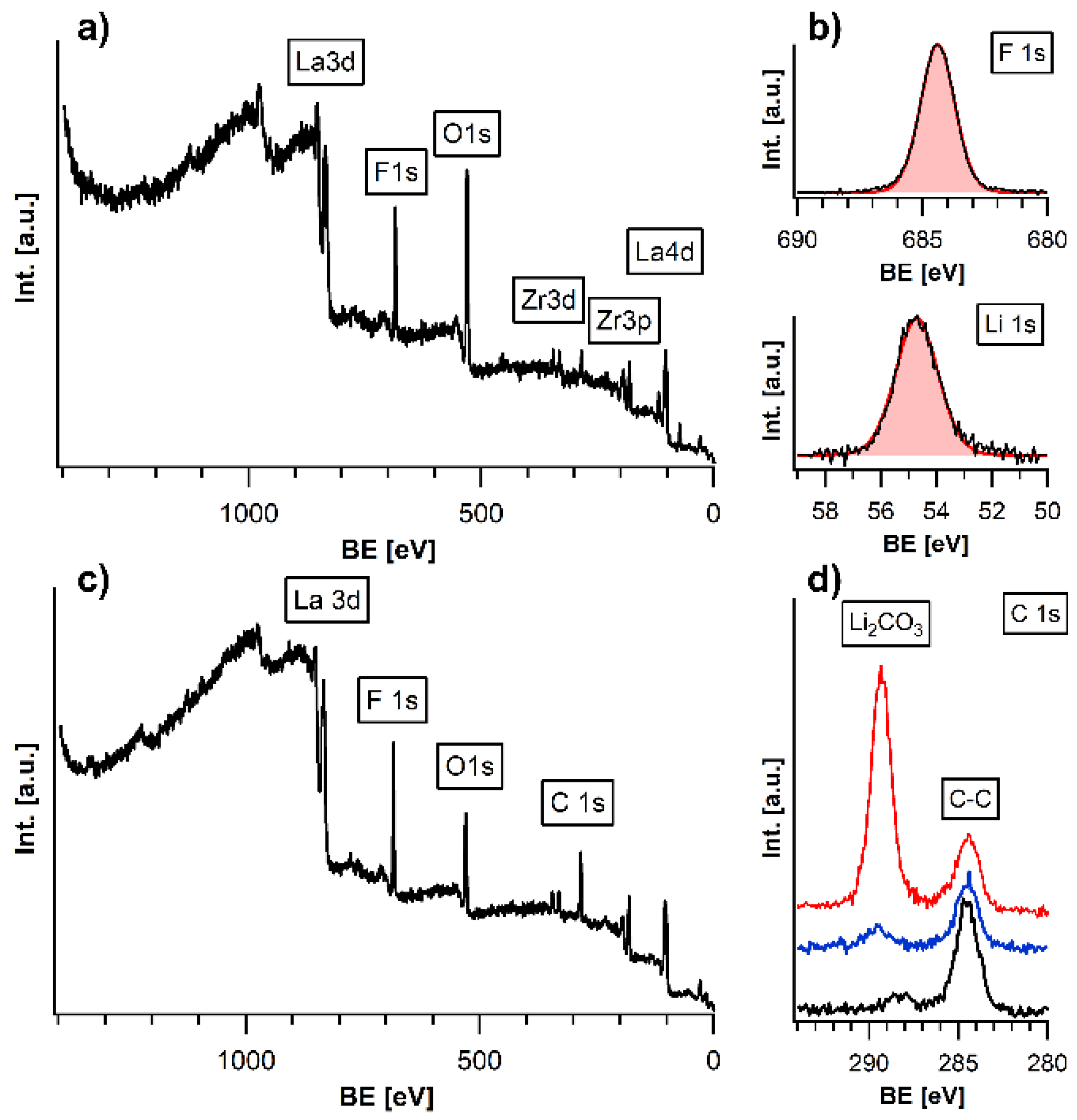
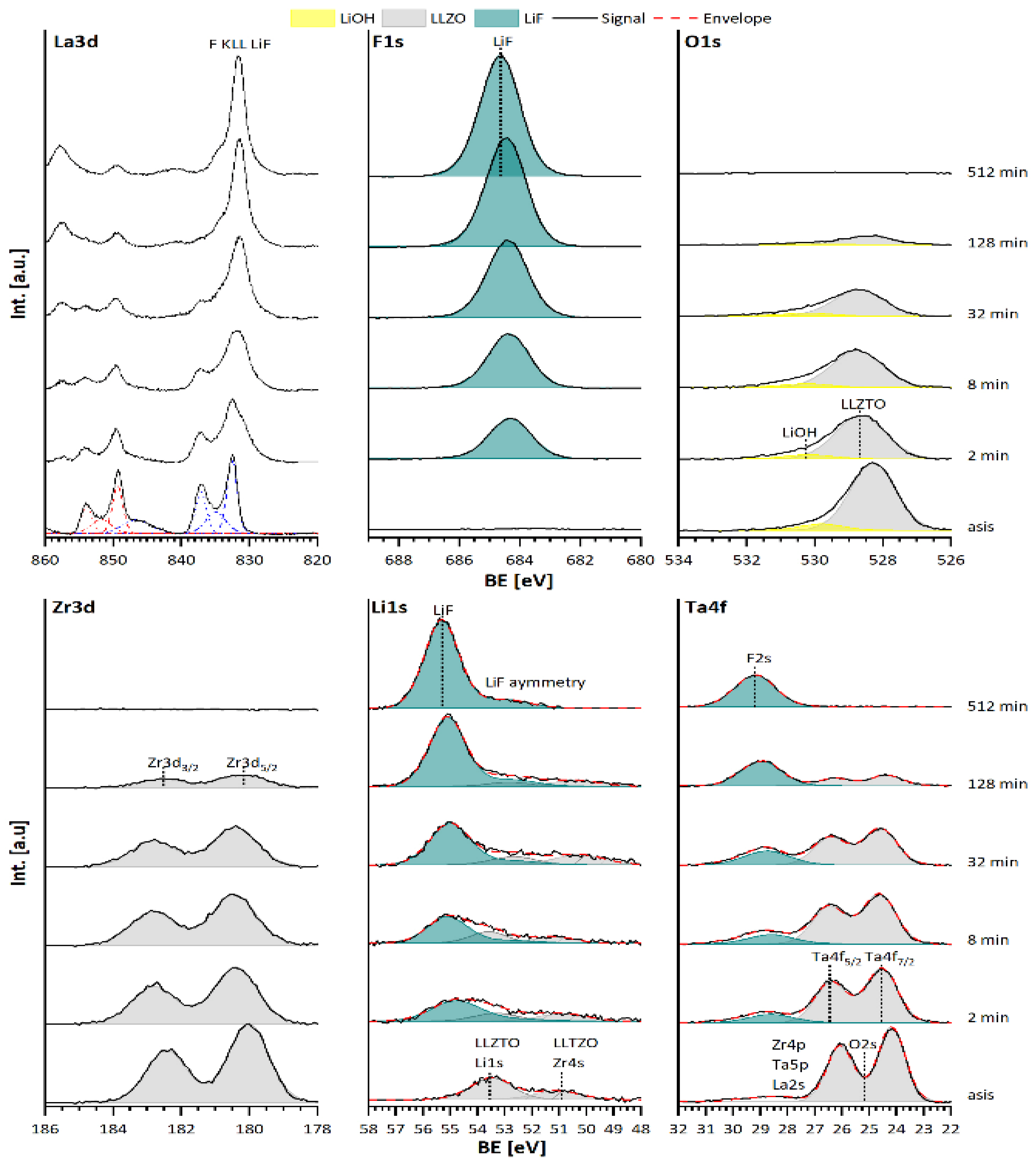
3.1. Surface Modification and Protection of LLZTO Thin Films via a Gas-Phase Fluorination Approach
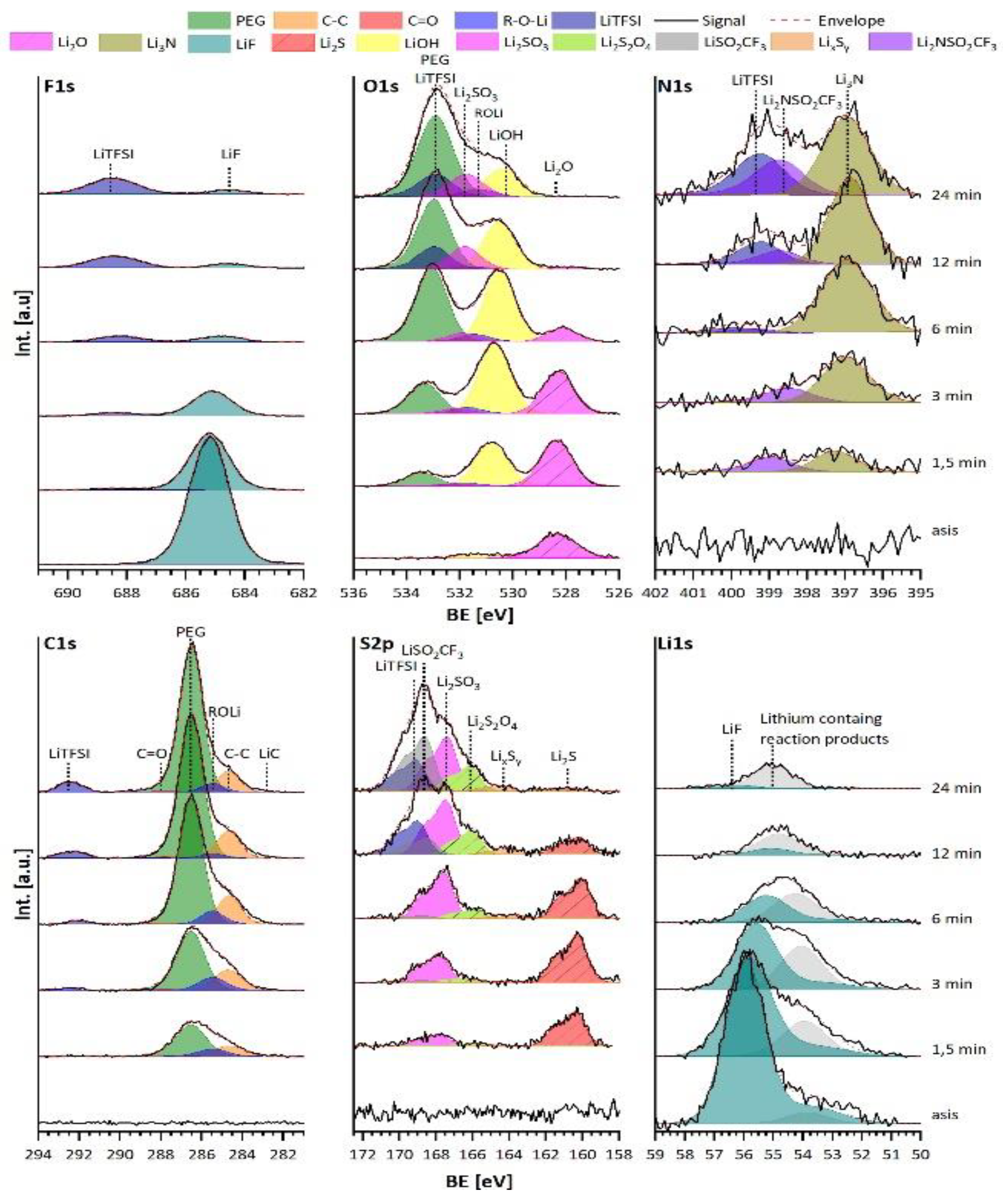
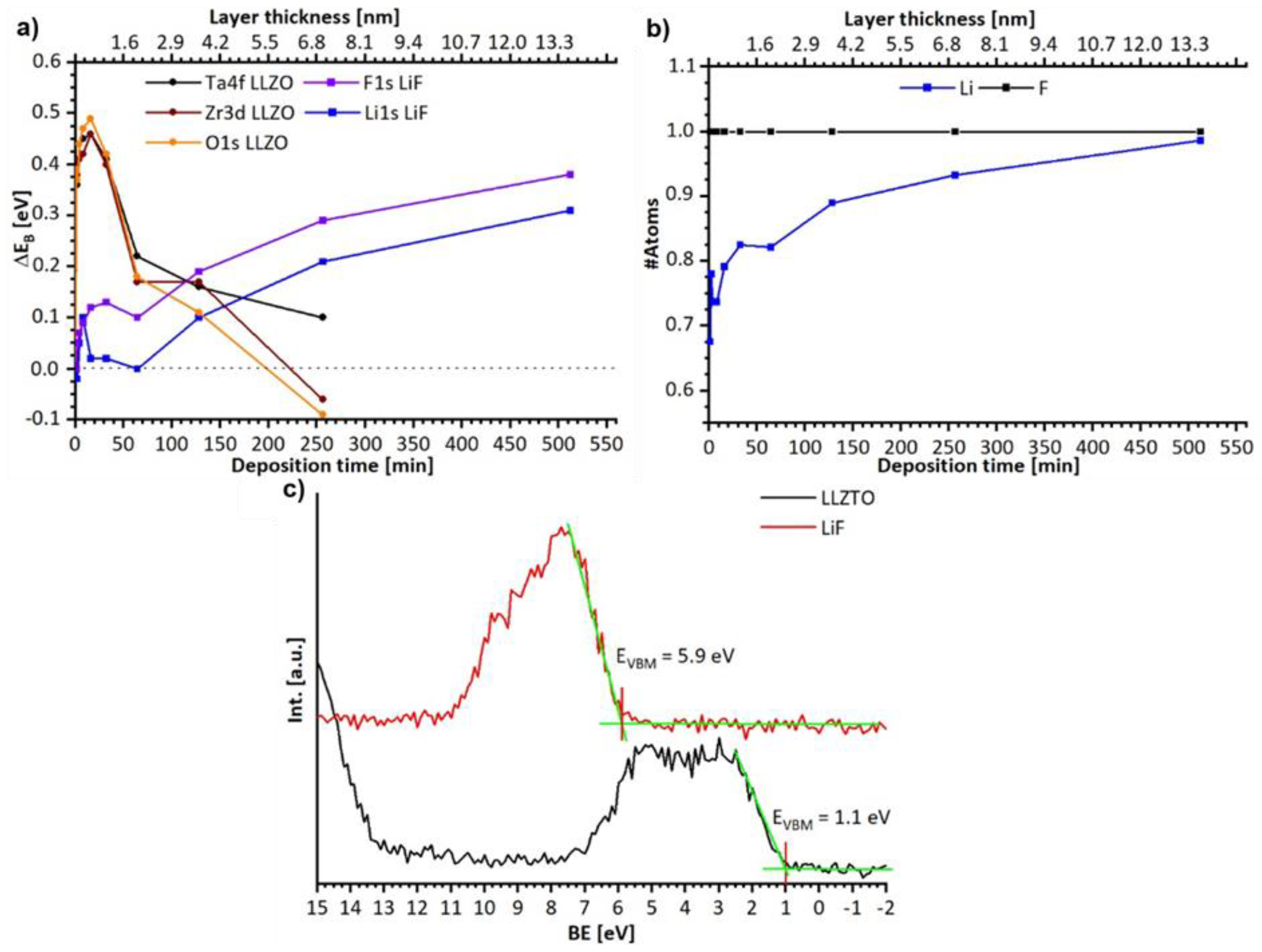
3.2. Analysis of the Stability of the LLZTO and PEO/LiTFSI Interfaces towards LiF
3.2.1. Discussion of the Binding Energy Shift at the LLZTO-LiF Interface
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Murugan, R.; Thangadurai, V.; Weppner, W. Fast lithium ion conduction in garnet-type Li7La3Zr2O12. Angew. Chem. Int. Ed. Engl. 2007, 46, 7778–7781. [Google Scholar] [CrossRef] [PubMed]
- Thangadurai, V.; Pinzaru, D.; Narayanan, S.; Baral, A.K. Fast Solid-State Li Ion Conducting Garnet-Type Structure Metal Oxides for Energy Storage. J. Phys. Chem. Lett. 2014, 6, 292–299. [Google Scholar] [CrossRef] [PubMed]
- Fingerle, M.; Loho, C.; Ferber, T.; Hahn, H.; Hausbrand, R. Evidence of the chemical stability of the garnet-type solid electrolyte Li5La3Ta2O12 towards lithium by a surface science approach. J. Power Sources 2017, 366, 72–79. [Google Scholar] [CrossRef]
- Chen, L.; Li, Y.; Li, S.P.; Fan, L.Z.; Nan, C.W.; Goodenough, J.B. PEO/garnet composite electrolytes for solid-state lithium batteries: From “ceramic-in-polymer” to “polymer-in-ceramic”. Nano Energy 2018, 46, 176–184. [Google Scholar] [CrossRef]
- Li, Y.; Xu, B.; Xu, H.; Duan, H.; Lü, X.; Xin, S.; Zhou, W.; Xue, L.; Fu, G.; Manthiram, A.; et al. Hybrid Polymer/Garnet Electrolyte with a Small Interfacial Resistance for Lithium-Ion Batteries. Angew. Chem. Int. Ed. Engl. 2017, 56, 753–756. [Google Scholar] [CrossRef] [PubMed]
- Larraz, G.; Orera, A.; Sanjuán, M.L. Cubic phases of garnet-type Li7La3Zr2O12: The role of hydration. J. Mater. Chem. A 2013, 1, 11419–11428. [Google Scholar] [CrossRef] [Green Version]
- Sharafi, A.; Yu, S.; Naguib, M.; Lee, M.; Ma, C.; Meyer, H.M.; Nanda, J.; Chi, M.; Siegel, D.J.; Sakamoto, J. Impact of air exposure and surface chemistry on Li–Li7La3Zr2O12 interfacial resistance. J. Mater. Chem. A 2017, 5, 13475–13487. [Google Scholar] [CrossRef]
- Waidha, A.I.; Ferber, T.; Donzelli, M.; Hosseinpourkahvaz, N.; Vanita, V.; Dirnberger, K.; Ludwigs, S.; Hausbrand, R.; Jaegermann, W.; Clemens, O. Compositional Dependence of Li-Ion Conductivity in Garnet-Rich Composite Electrolytes for All-Solid-State Lithium-Ion Batteries—Toward Understanding the Drawbacks of Ceramic-Rich Composites. ACS Appl. Mater. Interfaces 2021, 13, 31111–31128. [Google Scholar] [CrossRef]
- Besli, M.M.; Usubelli, C.; Metzger, M.; Pande, V.; Harry, K.; Nordlund, D.; Sainio, S.; Christensen, J.; Doeff, M.M.; Kuppan, S. Effect of Liquid Electrolyte Soaking on the Interfacial Resistance of Li7La3Zr2O12 for All-Solid-State Lithium Batteries. ACS Appl. Mater. Interfaces 2020, 12, 20605–20612. [Google Scholar] [CrossRef]
- Huo, H.; Chen, Y.; Zhao, N.; Lin, X.; Luo, J.; Yang, X.; Liu, Y.; Guo, X.; Sun, X. In-situ formed Li2CO3-free garnet/Li interface by rapid acid treatment for dendrite-free solid-state batteries. Nano Energy 2019, 61, 119–125. [Google Scholar] [CrossRef]
- Han, X.; Gong, Y.; Fu, K.; He, X.; Hitz, G.T.; Dai, J.; Pearse, A.; Liu, B.; Wang, H.; Rubloff, G.; et al. Negating interfacial impedance in garnet-based solid-state Li metal batteries. Nat. Mater. 2017, 16, 572–579. [Google Scholar] [CrossRef] [PubMed]
- Dubey, R.; Sastre, J.; Cancellieri, C.; Okur, F.; Forster, A.; Pompizii, L.; Priebe, A.; Romanyuk, Y.E.; Jeurgens, L.P.H.; Kovalenko, M.V.; et al. Building a Better Li-Garnet Solid Electrolyte/Metallic Li Interface with Antimony. Adv. Energy Mater. 2021, 11, 2102086. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, L.; Yu, C.; Yan, X.; Xu, B.; Wang, L.-M. Lithium halide coating as an effective intergrain engineering for garnet-type solid electrolytes avoiding high temperature sintering. Electrochim. Acta 2018, 289, 254–263. [Google Scholar] [CrossRef]
- Tang, S.; Chen, G.; Ren, F.; Wang, H.; Yang, W.; Zheng, C.; Gong, Z.; Yang, Y. Modifying an ultrathin insulating layer to suppress lithium dendrite formation within garnet solid electrolytes. J. Mater. Chem. A 2021, 9, 3576–3583. [Google Scholar] [CrossRef]
- Slater, P.R. PVDF as a reagent for the synthesis of K2NiF4-related inorganic oxide fluorides. J. Fluor. Chem. 2002, 117, 43–45. [Google Scholar] [CrossRef]
- Moon, E.J.; Choquette, A.K.; Huon, A.; Kulesa, S.Z.; Barbash, D.; May, S.J. Comparison of topotactic fluorination methods for complex oxide films. APL Mater. 2015, 3, 062511. [Google Scholar] [CrossRef] [Green Version]
- Nair, A.; Wollstadt, S.; Witte, R.; Dasgupta, S.; Kehne, P.; Alff, L.; Komissinskiy, P.; Clemens, O. Synthesis and Characterisation of fluorinated epitaxial films of BaFeO2F: Tailoring magnetic anisotropy via a lowering of tetragonal distortion. RSC Adv. 2019, 9, 37136–37143. [Google Scholar] [CrossRef] [Green Version]
- Loho, C.; Darbandi, A.J.; Djenadic, R.; Clemens, O.; Hahn, H. CO2-Laser Flash Evaporation as Novel CVD Precursor Delivery System for Functional Thin Film Growth. Chem. Vap. Depos. 2014, 20, 152–160. [Google Scholar] [CrossRef]
- Loho, C.; Djenadic, R.; Bruns, M.; Clemens, O.; Hahn, H. Garnet-Type Li7La3Zr2O12 Solid Electrolyte Thin Films Grown by CO2-Laser Assisted CVD for All-Solid-State Batteries. J. Electrochem. Soc. 2017, 164, A6131–A6139. [Google Scholar] [CrossRef]
- Loho, C.; Djenadic, R.; Mundt, P.; Clemens, O.; Hahn, H. On processing-structure-property relations and high ionic conductivity in garnet-type Li5La3Ta2O12 solid electrolyte thin films grown by CO2-laser assisted CVD. Solid State Ion. 2017, 313, 32–44. [Google Scholar] [CrossRef]
- Contarini, S.; Rabalais, J. Ion bombardment-induced decomposition of Li and Ba sulfates and carbonates studied by X-ray photoelectron spectroscopy. J. Electron Spectrosc. Relat. Phenom. 1985, 35, 191–201. [Google Scholar] [CrossRef]
- Chase, M.W., Jr. NIST-JANAF Thermochemical Tables, Monograph 9; American Chemical Society: Washington, DC, USA, 1998; Volume 9, pp. 1–1951. [Google Scholar]
- Holleman, A.F.; Wiberg, N.; Wiberg, E. Lehrbuch der Anorganischen Chemie; De Gruyter: Berlin, Germany, 2007. [Google Scholar]
- Waidha, A.; Vanita, V.; Clemens, O. PEO Infiltration of Porous Garnet-Type Lithium-Conducting Solid Electrolyte Thin Films. Ceramics 2021, 4, 421–436. [Google Scholar] [CrossRef]
- Schulz, N.; Hausbrand, R.; Wittich, C.; Dimesso, L.; Jaegermann, W. XPS-surface analysis of SEI layers on Li-ion cathodes: Part II. SEI-composition and formation inside composite electrodes. J. Electrochem. Soc. 2018, 165, A833. [Google Scholar] [CrossRef]
- Sun, B.; Xu, C.; Mindemark, J.; Gustafsson, T.; Edström, K.; Brandell, D. At the polymer electrolyte interfaces: The role of the polymer host in interphase layer formation in Li-batteries. J. Mater. Chem. A 2015, 3, 13994–14000. [Google Scholar] [CrossRef]
- Moulder, J.; Stickle, W.; Sobol, P.; Bomben, K.; Chastain, J.; King, R., Jr. Handbook of X-ray Photoelectron Spectroscopy: A Reference Book of Standard Spectra for Identification and Interpretation of XPS Data; Physical Electronics Inc.: Eden Prairie, MN, USA, 1995. [Google Scholar]
- Ferber, T.H.; Cangaz, Ş.; Jaegermann, W.; Hausbrand, R. Interface reactivity of in-situ formed LiCoO2-PEO solid-state interfaces investigated by X-ray photoelectron spectroscopy: Reaction products, energy level offsets and double layer formation. Appl. Surf. Sci. 2022, 571, 151218. [Google Scholar] [CrossRef]
- Eshetu, G.G.; Diemant, T.; Grugeon, S.; Behm, R.J.; Laruelle, S.; Armand, M.; Passerini, S. In-Depth Interfacial Chemistry and Reactivity Focused Investigation of Lithium–Imide- and Lithium–Imidazole-Based Electrolytes. ACS Appl. Mater. Interfaces 2016, 8, 16087–16100. [Google Scholar] [CrossRef]
- Wood, K.N.; Teeter, G. XPS on Li-Battery-Related Compounds: Analysis of Inorganic SEI Phases and a Methodology for Charge Correction. ACS Appl. Energy Mater. 2018, 1, 4493–4504. [Google Scholar] [CrossRef] [Green Version]
- Ensling, D.; Stjerndahl, M.; Nytén, A.; Gustafsson, T.; Thomas, J.O. A comparative XPS surface study of Li2FeSiO4/C cycled with LiTFSI- and LiPF6-based electrolytes. J. Mater. Chem. 2009, 19, 82–88. [Google Scholar] [CrossRef]
- Choukourov, A.; Gordeev, I.; Polonskyi, O.; Artemenko, A.; Hanyková, L.; Krakovský, I.; Kylián, O.; Slavínská, D.; Biederman, H. Poly (ethylene oxide)-like Plasma Polymers Produced by Plasma-Assisted Vacuum Evaporation. Plasma Process. Polym. 2010, 7, 445–458. [Google Scholar] [CrossRef]
- Dedryvère, R.; Laruelle, S.; Grugeon, S.; Gireaud, L.; Tarascon, J.-M.; Gonbeau, D. XPS Identification of the Organic and Inorganic Components of the Electrode/Electrolyte Interface Formed on a Metallic Cathode. J. Electrochem. Soc. 2005, 152, A689–A696. [Google Scholar] [CrossRef]
- Kanamura, K.; Shiraishi, S.; Tamura, H.; Takehara, Z. X-Ray Photoelectron Spectroscopic Analysis and Scanning Electron Microscopic Observation of the Lithium Surface Immersed in Nonaqueous Solvents. J. Electrochem. Soc. 1994, 141, 2379–2385. [Google Scholar] [CrossRef]
- Pervez, S.A.; Vinayan, B.P.; Cambaz, M.A.; Melinte, G.; Diemant, T.; Braun, T.; Karkera, G.; Behm, R.J.; Fichtner, M. Electrochemical and compositional characterization of solid interphase layers in an interface-modified solid-state Li–sulfur battery. J. Mater. Chem. A 2020, 8, 16451–16462. [Google Scholar] [CrossRef]
- Fiedler, C.; Luerssen, B.; Rohnke, M.; Sann, J.; Janek, J. XPS and SIMS Analysis of Solid Electrolyte Interphases on Lithium Formed by Ether-Based Electrolytes. J. Electrochem. Soc. 2017, 164, A3742–A3749. [Google Scholar] [CrossRef]
- Eijima, S.; Sonoki, H.; Matsumoto, M.; Taminato, S.; Mori, D.; Imanishi, N. Solid Electrolyte Interphase Film on Lithium Metal Anode in Mixed-Salt System. J. Electrochem. Soc. 2019, 166, A5421–A5429. [Google Scholar] [CrossRef]
- Lim, H.-D.; Park, H.; Kim, H.; Kim, J.; Lee, B.; Bae, Y.; Gwon, H.; Kang, K. A New Perspective on Li-SO2Batteries for Rechargeable Systems. Angew. Chem. 2015, 127, 9799–9803. [Google Scholar] [CrossRef]
- Sharova, V.; Moretti, A.; Diemant, T.; Varzi, A.; Behm, R.; Passerini, S. Comparative study of imide-based Li salts as electrolyte additives for Li-ion batteries. J. Power Sources 2018, 375, 43–52. [Google Scholar] [CrossRef]
- Nguyen, C.C.; Woo, S.-W.; Song, S.-W. Understanding the Interfacial Processes at Silicon–Copper Electrodes in Ionic Liquid Battery Electrolyte. J. Phys. Chem. C 2012, 116, 14764–14771. [Google Scholar] [CrossRef]
- Aurbach, D.; Weissman, I.; Schechter, A.; Cohen, H. X-ray photoelectron spectroscopy studies of lithium surfaces prepared in several important electrolyte solutions. A comparison with previous studies by Fourier transform infrared spectroscopy. Langmuir 1996, 12, 3991–4007. [Google Scholar] [CrossRef]
- Aurbach, D.; Zaban, A.; Ein-Eli, Y.; Weissman, I.; Chusid, O.; Markovsky, B.; Levi, M.; Levi, E.; Schechter, A.; Granot, E. Recent studies on the correlation between surface chemistry, morphology, three-dimensional structures and performance of Li and Li-C intercalation anodes in several important electrolyte systems. J. Power Sources 1997, 68, 91–98. [Google Scholar] [CrossRef]
- Yildirim, H.; Haskins, J.B.; Bauschlicher, C.W., Jr.; Lawson, J.W. Decomposition of ionic liquids at lithium interfaces. Ab initio molecular dynamics simulations. J. Phys. Chem. C 2017, 121, 28214–28234. [Google Scholar] [CrossRef]
- Tsang, W.; Mak, C.; Wong, K. Epitaxial lithium fluoride films grown by pulsed laser deposition. Appl. Phys. A 2003, 77, 693–696. [Google Scholar] [CrossRef]
- Li, C.; Gu, L.; Maier, J. Enhancement of the Li Conductivity in LiF by Introducing Glass/Crystal Interfaces. Adv. Funct. Mater. 2012, 22, 1145–1149. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Donzelli, M.; Ferber, T.; Vanita, V.; Waidha, A.I.; Müller, P.; Mellin, M.; Hausbrand, R.; Jaegermann, W.; Clemens, O. On the Surface Modification of LLZTO with LiF via a Gas-Phase Approach and the Characterization of the Interfaces of LiF with LLZTO as Well as PEO+LiTFSI. Materials 2022, 15, 6900. https://doi.org/10.3390/ma15196900
Donzelli M, Ferber T, Vanita V, Waidha AI, Müller P, Mellin M, Hausbrand R, Jaegermann W, Clemens O. On the Surface Modification of LLZTO with LiF via a Gas-Phase Approach and the Characterization of the Interfaces of LiF with LLZTO as Well as PEO+LiTFSI. Materials. 2022; 15(19):6900. https://doi.org/10.3390/ma15196900
Chicago/Turabian StyleDonzelli, Manuel, Thimo Ferber, Vanita Vanita, Aamir Iqbal Waidha, Philipp Müller, Maximilian Mellin, René Hausbrand, Wolfram Jaegermann, and Oliver Clemens. 2022. "On the Surface Modification of LLZTO with LiF via a Gas-Phase Approach and the Characterization of the Interfaces of LiF with LLZTO as Well as PEO+LiTFSI" Materials 15, no. 19: 6900. https://doi.org/10.3390/ma15196900
APA StyleDonzelli, M., Ferber, T., Vanita, V., Waidha, A. I., Müller, P., Mellin, M., Hausbrand, R., Jaegermann, W., & Clemens, O. (2022). On the Surface Modification of LLZTO with LiF via a Gas-Phase Approach and the Characterization of the Interfaces of LiF with LLZTO as Well as PEO+LiTFSI. Materials, 15(19), 6900. https://doi.org/10.3390/ma15196900