
A Kinetic Model Considering Catalyst Deactivation for Methanol-to-Dimethyl Ether on a Biomass-Derived Zr/P-Carbon Catalyst


 ,
,  ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Catalyst Preparation
2.2. Characterization
2.3. Catalyst Performance
3. Results and Discussion
3.1. Catalyst Properties
3.2. Catalyst Performance
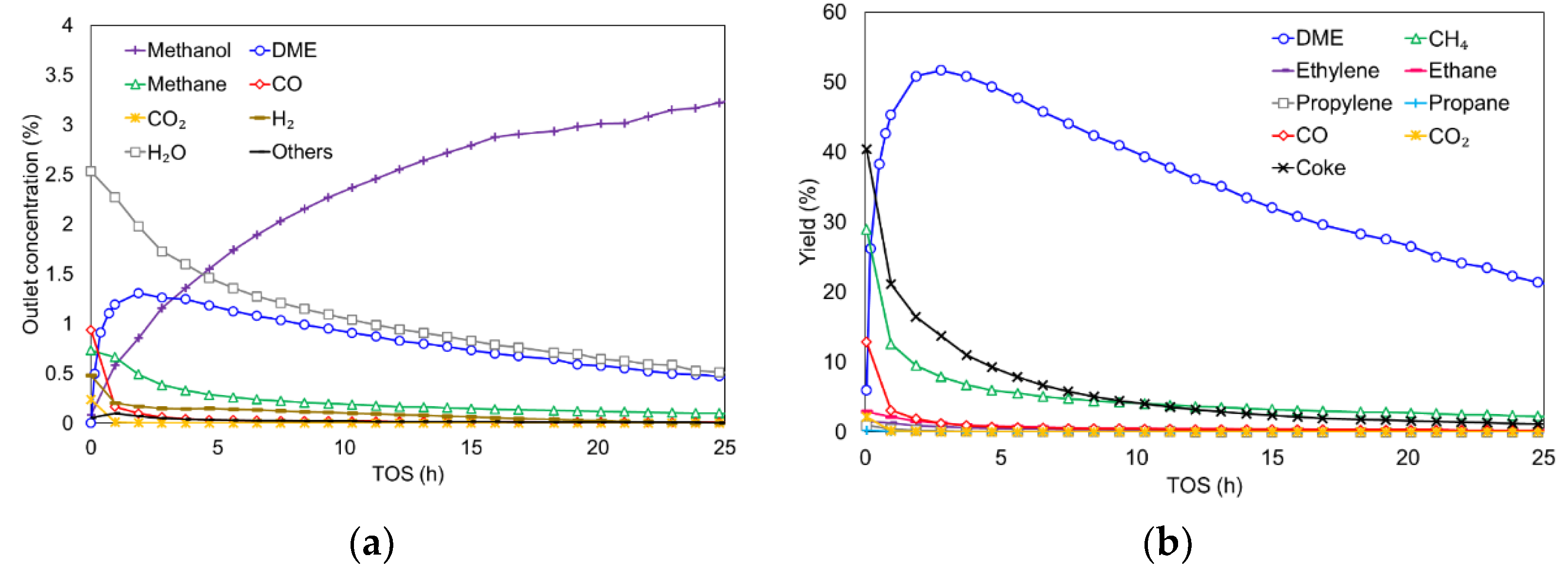
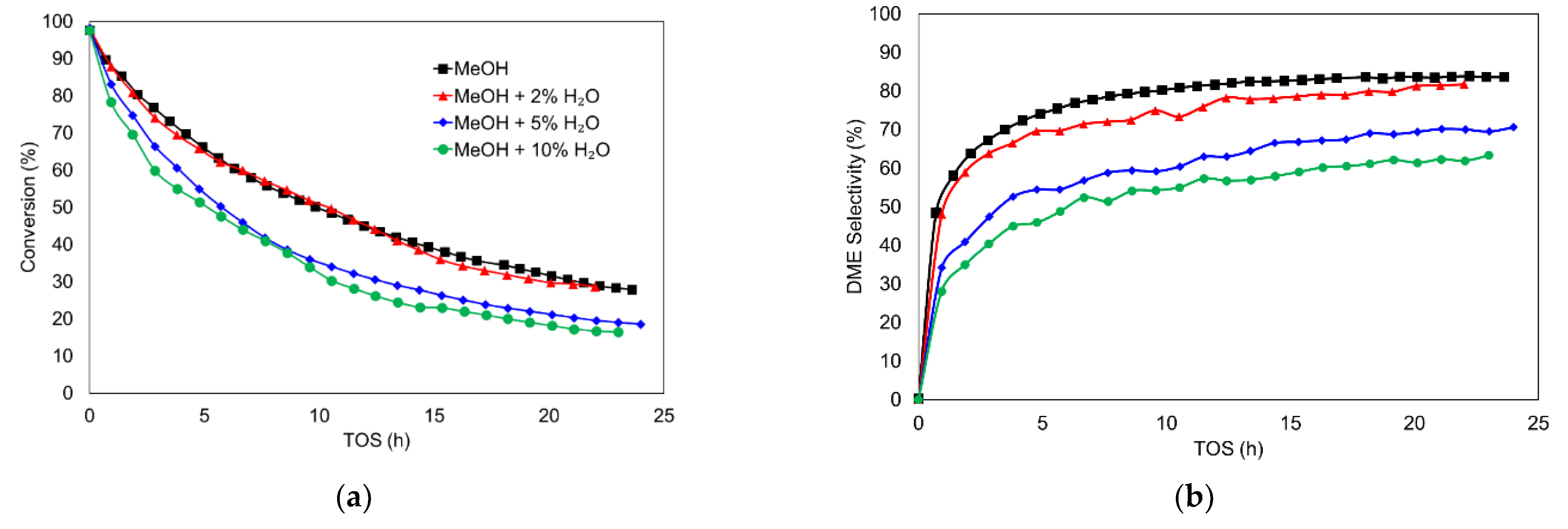
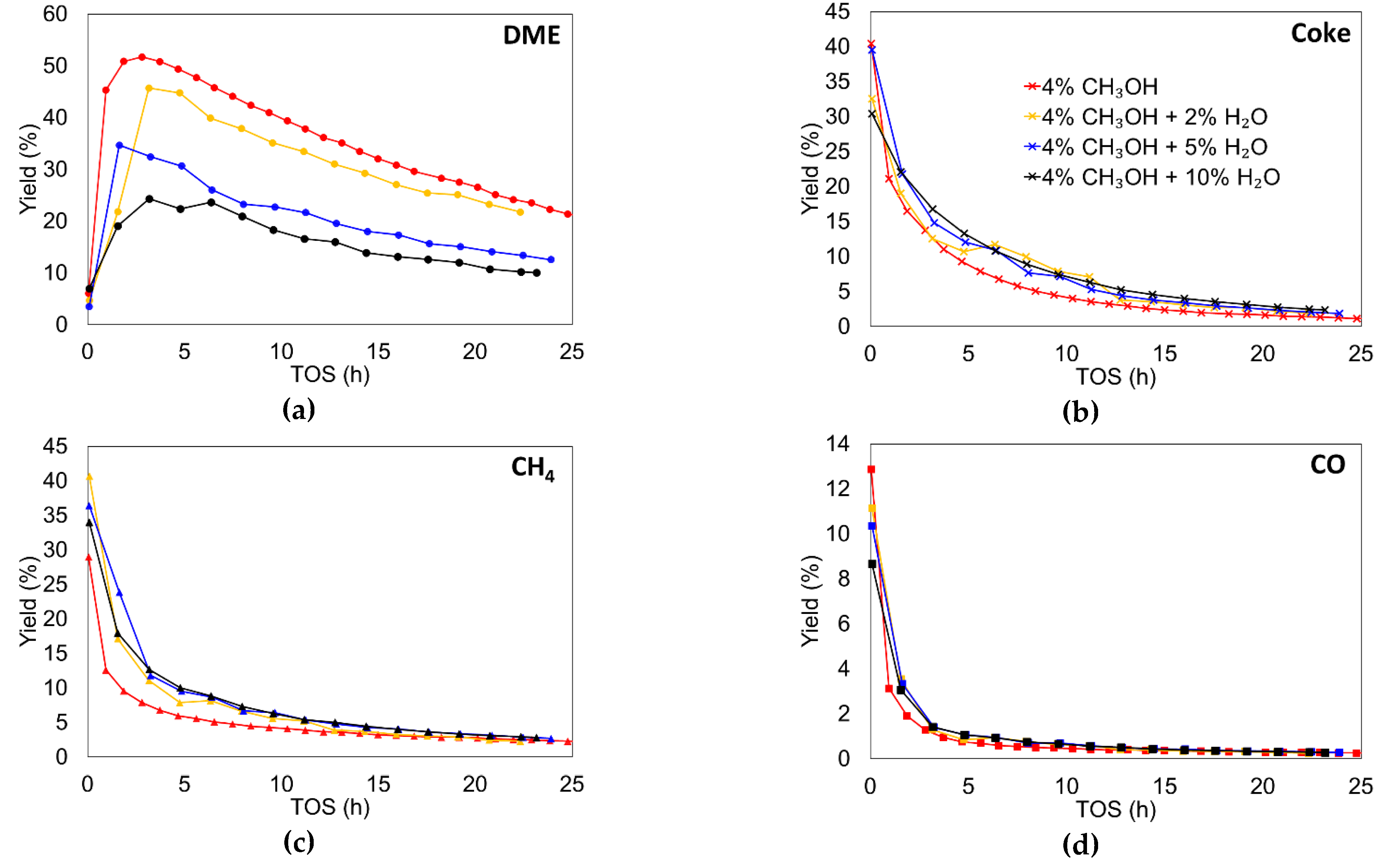
3.2.1. Effect of Inlet Water Vapor
3.2.2. Stoichiometric Study of MTD Reaction
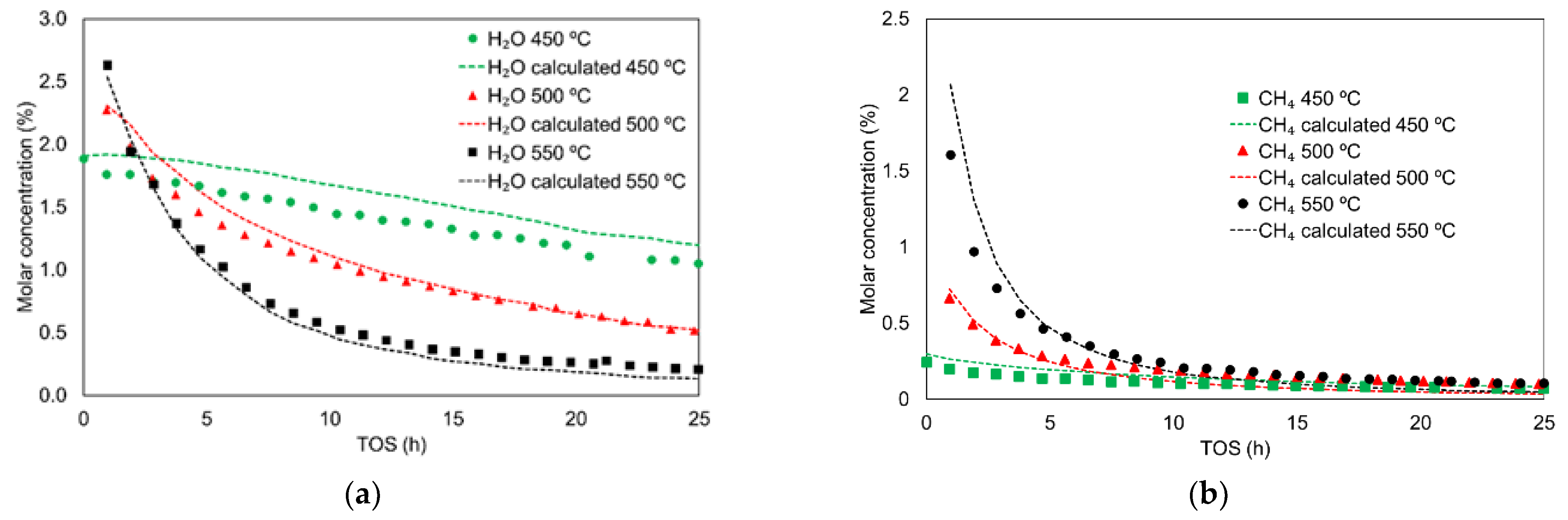
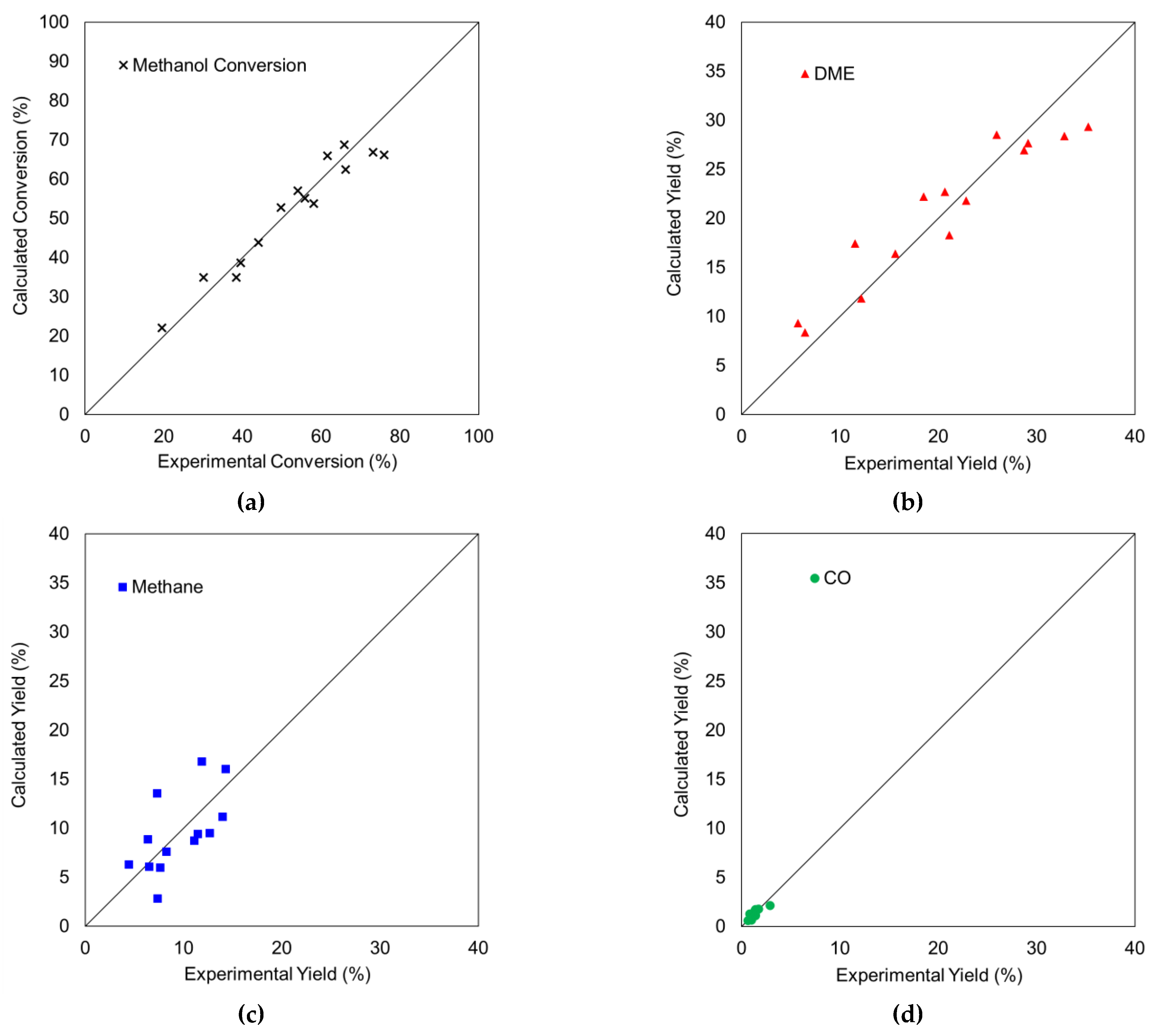
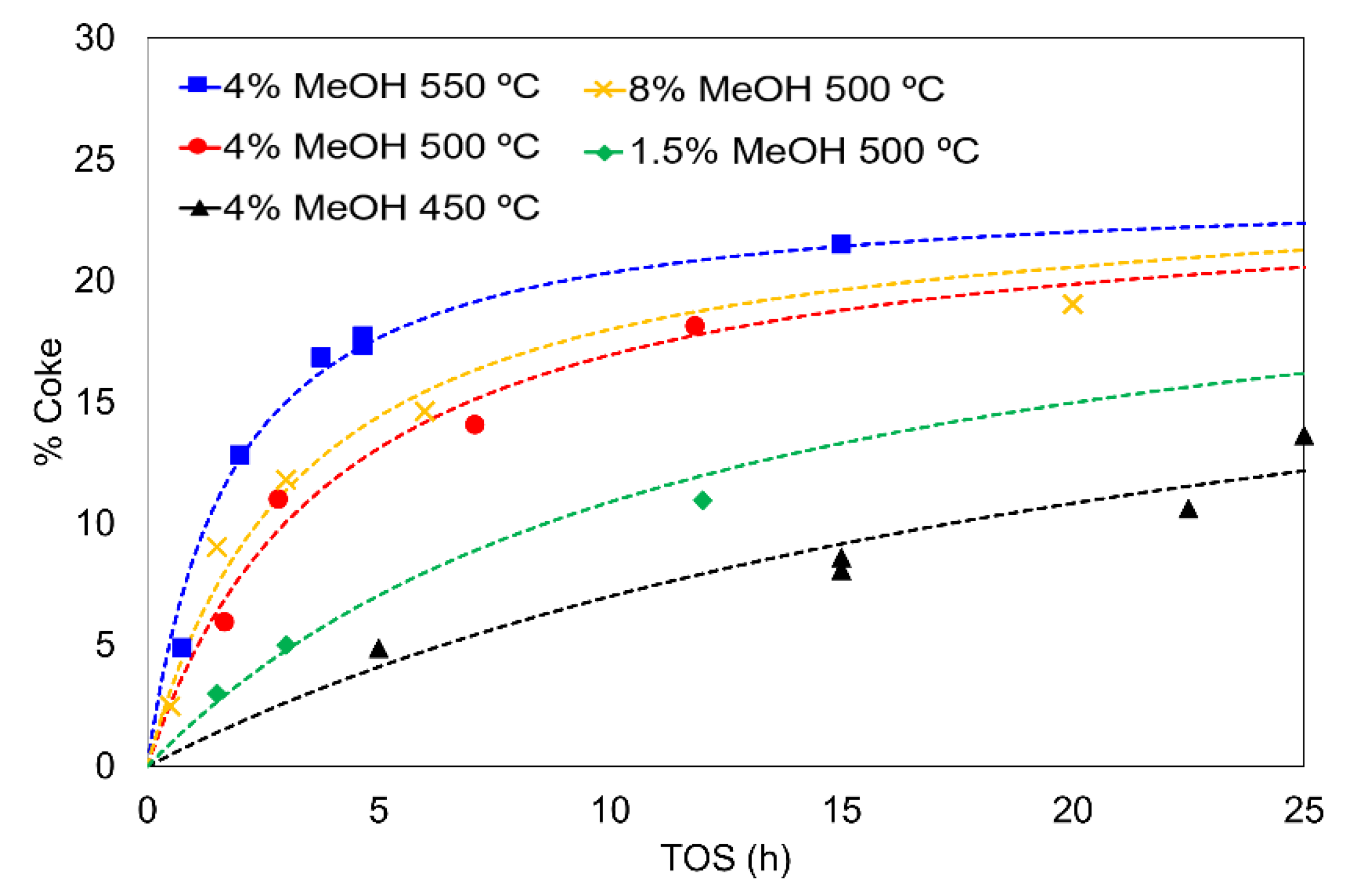
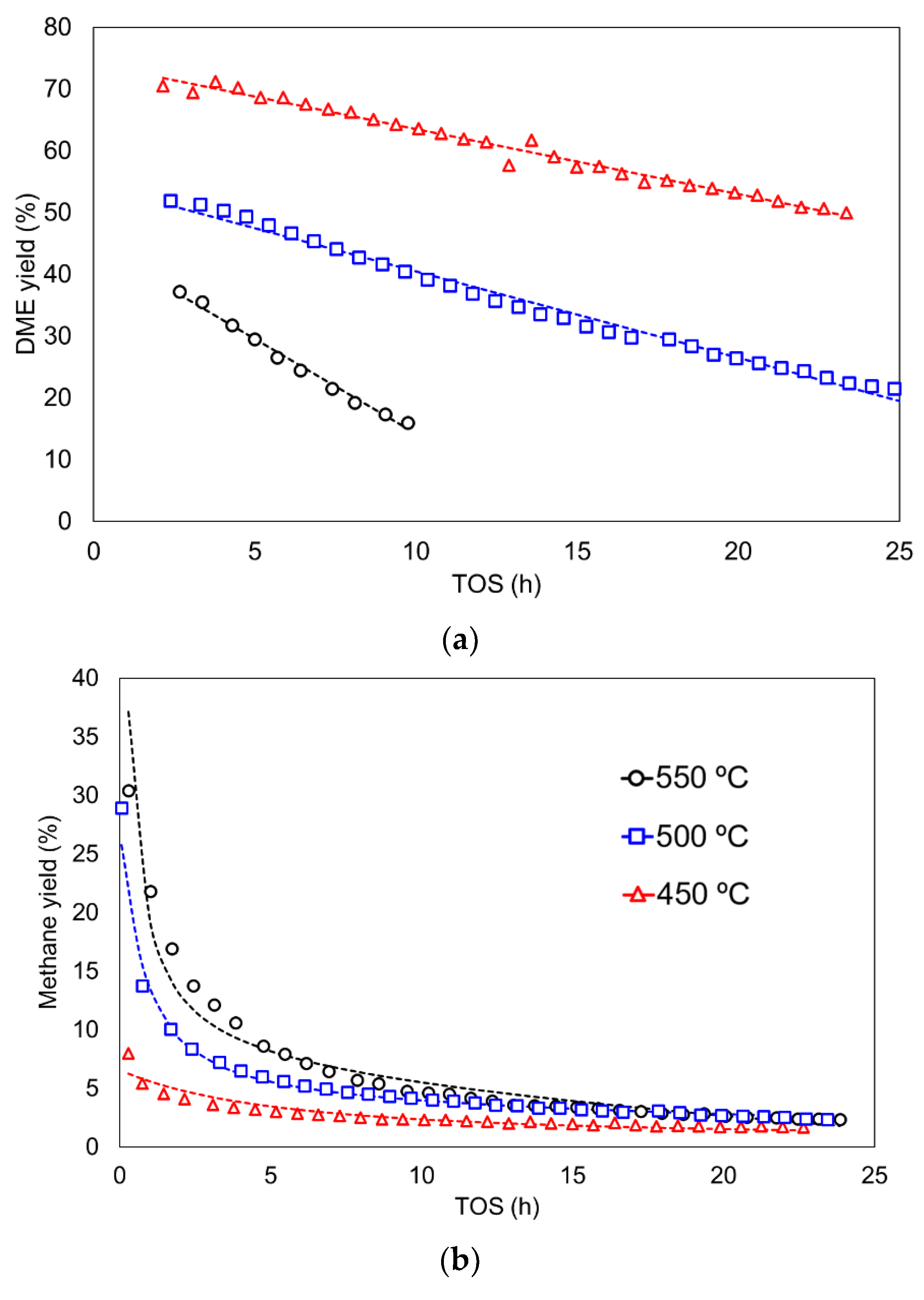
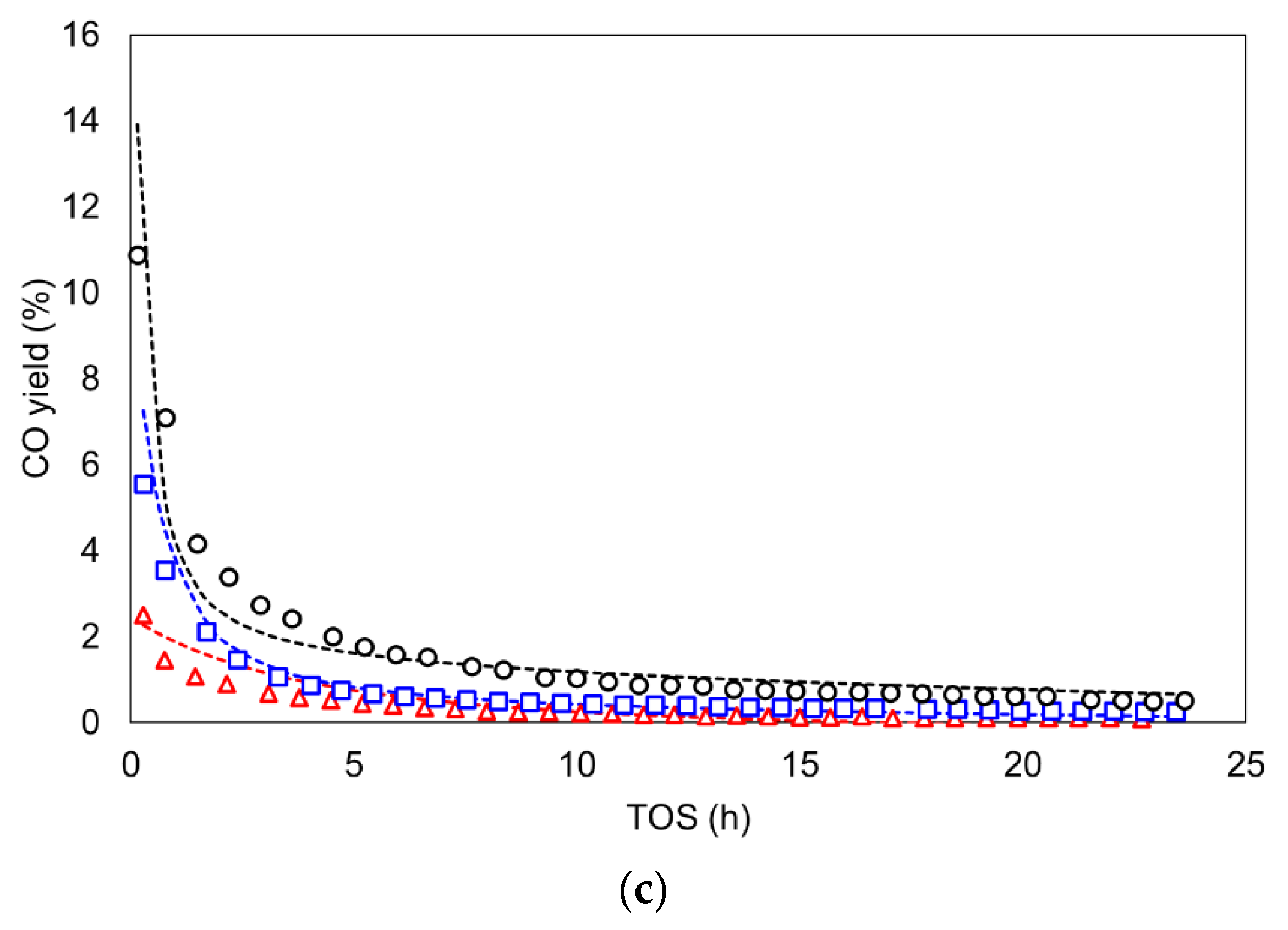
3.3. Kinetic Study including Deactivation
- A uniform distribution of active sites on the catalyst surface;
- Homogeneous distribution of the catalyst in the catalytic bed;
- Ideal flow, without radial gradients of concentration;
- Isotherm catalytic bed;
- Negligible heat and mass transfer limitations.
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Meinshausen, M.; Meinshausen, N.; Hare, W.; Raper, S.C.B.; Frieler, K.; Knutti, R.; Frame, D.J.; Allen, M.R. Greenhouse-gas emission targets for limiting global warming to 2 °C. Nature 2009, 458, 1158–1162. [Google Scholar] [CrossRef] [PubMed]
- Imarc Dimethyl Ether Market: Global Industry Trends, Share, Size, Growth, Opportunity and Forecast 2021–2026. Available online: https://www.imarcgroup.com/dimethyl-ether-market (accessed on 4 November 2021).
- Fortune Business Insight Dimethyl Ether Market Size, Share & COVID-19 Impact Analysis, By Application (LPG Blending, Aerosol Propellant, Transportation Fuel, and Others), and Regional Forectas, 2021–2028. Available online: https://www.fortunebusinessinsights.com/dimethyl-ether-market-104309 (accessed on 18 August 2021).
- Lu, M.; Fu, Z.; Yuan, X.; Sun, G.; Jia, G. Study of the reduced kinetic mechanism of methane/dimethyl ether combustion. Fuel 2021, 303, 121308. [Google Scholar] [CrossRef]
- Poto, S.; Gallucci, F.; Fernanda Neira d’Angelo, M. Direct conversion of CO2 to dimethyl ether in a fixed bed membrane reactor: Influence of membrane properties and process conditions. Fuel 2021, 302, 121080. [Google Scholar] [CrossRef]
- Kim, D.; Park, G.; Choi, B.; Kim, Y.B. Reaction characteristics of dimethyl ether (DME) steam reforming catalysts for hydrogen production. Int. J. Hydrogen Energy 2017, 42, 29210–29221. [Google Scholar] [CrossRef]
- Saravanan, K.; Ham, H.; Tsubaki, N.; Bae, J.W. Recent progress for direct synthesis of dimethyl ether from syngas on the heterogeneous bifunctional hybrid catalysts. Appl. Catal. B Environ. 2017, 217, 494–522. [Google Scholar] [CrossRef]
- Akarmazyan, S.S.; Panagiotopoulou, P.; Kambolis, A.; Papadopoulou, C.; Kondarides, D.I. Methanol dehydration to dimethylether over Al2O3 catalysts. Appl. Catal. B Environ. 2014, 145, 136–148. [Google Scholar] [CrossRef]
- Kim, S.; Kim, Y.T.; Zhang, C.; Kwak, G.; Jun, K.-W. Effect of Reaction Conditions on the Catalytic Dehydration of Methanol to Dimethyl Ether Over a K-modified HZSM-5 Catalyst. Catal. Lett. 2017, 147, 792–801. [Google Scholar] [CrossRef]
- Alharbi, W.; Kozhevnikova, E.F.; Kozhevnikov, I.V. Dehydration of Methanol to Dimethyl Ether over Heteropoly Acid Catalysts: The Relationship between Reaction Rate and Catalyst Acid Strength. ACS Catal. 2015, 5, 7186–7193. [Google Scholar] [CrossRef]
- Volkov, V.V.; Novitskii, E.G.; Dibrov, G.A.; Samokhin, P.V.; Kipnis, M.A.; Yaroslavtsev, A.B. Catalytic conversion of methanol to dimethyl ether on polymer/ceramic composite membranes. Catal. Today 2012, 193, 31–36. [Google Scholar] [CrossRef]
- Cheng, S.; Zhl Peng, G.; Clearfield, A. Literature Cited Decomposition of Alcohols over Zirconium and Titanium Phosphates. Ind. Eng. Chem. Prod. Res. Dev 1984, 23, 219–225. [Google Scholar] [CrossRef]
- Moreno-Castilla, C.; Carrasco-Marín, F.; Parejo-Pérez, C.; López Ramón, M. Dehydration of methanol to dimethyl ether catalyzed by oxidized activated carbons with varying surface acidic character. Carbon 2001, 39, 869–875. [Google Scholar] [CrossRef]
- Valero-Romero, M.J.; Calvo-Muñoz, E.M.; Ruiz-Rosas, R.; Rodríguez-Mirasol, J.; Cordero, T. Phosphorus-Containing Mesoporous Carbon Acid Catalyst for Methanol Dehydration to Dimethyl Ether. Ind. Eng. Chem. Res. 2019, 58, 4042–4053. [Google Scholar] [CrossRef]
- Marsh, H.; Rodríguez-Reinoso, F. Activated Carbon; Elsevier: Amsterdam, The Netherlands, 2006; ISBN 9780080444635. [Google Scholar]
- Rodríguez-Reinoso, F. The role of carbon materials in heterogeneous catalysis. Carbon 1998, 36, 159–175. [Google Scholar] [CrossRef]
- Umeki, K.; Yamamoto, K.; Namioka, T.; Yoshikawa, K. High temperature steam-only gasification of woody biomass. Appl. Energy 2010, 87, 791–798. [Google Scholar] [CrossRef]
- Chen, W.H.; Lin, B.J. Hydrogen and synthesis gas production from activated carbon and steam via reusing carbon dioxide. Appl. Energy 2013, 101, 551–559. [Google Scholar] [CrossRef]
- García-Mateos, F.J.; Ruiz-Rosas, R.; Marqués, M.D.; Cotoruelo, L.M.; Rodríguez-Mirasol, J.; Cordero, T. Removal of paracetamol on biomass-derived activated carbon: Modeling the fixed bed breakthrough curves using batch adsorption experiments. Chem. Eng. J. 2015, 279, 18–30. [Google Scholar] [CrossRef] [Green Version]
- Cotoruelo, L.M.; Marqués, M.D.; Díaz, F.J.; Rodríguez-Mirasol, J.; Rodríguez, J.J.; Cordero, T. Lignin-based activated carbons as adsorbents for crystal violet removal from aqueous solutions. Environ. Prog. Sustain. Energy 2012, 31, 386–396. [Google Scholar] [CrossRef]
- Calvo-Muñoz, E.M.; García-Mateos, F.J.; Rosas, J.M.; Rodríguez-Mirasol, J.; Cordero, T. Biomass Waste Carbon Materials as adsorbents for CO2 Capture under Post-Combustion Conditions. Front. Mater. 2016, 3, 23. [Google Scholar] [CrossRef] [Green Version]
- Valero-Romero, M.J.; Rodríguez-Cano, M.Á.; Palomo, J.; Rodríguez-Mirasol, J.; Cordero, T. Carbon-Based Materials as Catalyst Supports for Fischer–Tropsch Synthesis: A Review. Front. Mater. 2021, 7, 455. [Google Scholar] [CrossRef]
- García-Mateos, F.J.; Cordero-Lanzac, T.; Berenguer, R.; Morallón, E.; Cazorla-Amorós, D.; Rodríguez-Mirasol, J.; Cordero, T. Lignin-derived Pt supported carbon (submicron)fiber electrocatalysts for alcohol electro-oxidation. Appl. Catal. B Environ. 2017, 211, 18–30. [Google Scholar] [CrossRef]
- Cordero-Lanzac, T.; Rodríguez-Mirasol, J.; Cordero, T.; Bilbao, J. Advances and Challenges in the Valorization of Bio-Oil: Hydrodeoxygenation Using Carbon-Supported Catalysts. Energy Fuels 2021. [Google Scholar] [CrossRef]
- García-Mateos, F.J.; Ruiz-Rosas, R.; Rosas, J.M.; Rodríguez-Mirasol, J.; Cordero, T. Phosphorus containing carbon (submicron)fibers as efficient acid catalysts. Catal. Today 2022, 383, 308–319. [Google Scholar] [CrossRef]
- Ibeh, P.O.; García-Mateos, F.J.; Ruiz-Rosas, R.; Rosas, J.M.; Rodríguez-Mirasol, J.; Cordero, T. Acid Mesoporous Carbon Monoliths from Lignocellulosic Biomass Waste for Methanol Dehydration. Materials 2019, 12, 2394. [Google Scholar] [CrossRef] [Green Version]
- Rosas, J.M.; Ruiz-Rosas, R.; Rodríguez-Mirasol, J.; Cordero, T. Kinetic study of the oxidation resistance of phosphorus-containing activated carbons. Carbon. 2012, 50, 1523–1537. [Google Scholar] [CrossRef]
- Palomo, J.; Rodríguez-Cano, M.A.; Rodríguez-Mirasol, J.; Cordero, T. On the kinetics of methanol dehydration to dimethyl ether on Zr-loaded P-containing mesoporous activated carbon catalyst. Chem. Eng. J. 2019, 378, 122198. [Google Scholar] [CrossRef]
- Torres-Liñán, J.; García-Rollán, M.; Rosas, J.M.; Rodríguez-Mirasol, J.; Cordero, T. Deactivation of a Biomass-Derived Zirconium-Doped Phosphorus-Containing Carbon Catalyst in the Production of Dimethyl Ether from Methanol Dehydration. Energy Fuels 2021, 35, 17225–17240. [Google Scholar] [CrossRef]
- Palomo, J.; Rodríguez-Mirasol, J.; Cordero, T. Methanol Dehydration to Dimethyl Ether on Zr-Loaded P-Containing Mesoporous Activated Carbon Catalysts. Materials 2019, 12, 2204. [Google Scholar] [CrossRef] [Green Version]
- Migliori, M.; Aloise, A.; Giordano, G. Methanol to dimethylether on H-MFI catalyst: The influence of the Si/Al ratio on kinetic parameters. Catal. Today 2014, 227, 138–143. [Google Scholar] [CrossRef]
- Trypolskyi, A.; Zhokh, A.; Gritsenko, V.; Chen, M.; Tang, J.; Strizhak, P. A kinetic study on the methanol conversion to dimethyl ether over H-ZSM-5 zeolite. Chem. Pap. 2021, 75, 3429–3442. [Google Scholar] [CrossRef]
- Raoof, F.; Taghizadeh, M.; Eliassi, A.; Yariopour, F. Effects of temperature and feed composition on catalytic dehydration of methanol to dimethyl ether over γ-alumina. Fuel 2008, 87, 2967–2971. [Google Scholar] [CrossRef]
- Bercic, G.; Levec, J. Intrinsic and global reaction rate of methanol dehydration over γ-alumina pellets. Ind. Eng. Chem. Res. 1992, 31, 1035–1040. [Google Scholar] [CrossRef]
- Klusáček, K.; Schneider, P. Stationary catalytic kinetics via surface concentrations from transient data: Methanol dehydration. Chem. Eng. Sci. 1982, 37, 1523–1528. [Google Scholar] [CrossRef]
- Migliori, M.; Aloise, A.; Catizzone, E.; Giordano, G. Kinetic Analysis of Methanol to Dimethyl Ether Reaction over H-MFI Catalyst. Ind. Eng. Chem. Res. 2014, 53, 14885–14891. [Google Scholar] [CrossRef]
- Alamolhoda, S.; Kazemeini, M.; Zaherian, A.; Zakerinasab, M.R. Reaction kinetics determination and neural networks modeling of methanol dehydration over nano γ-Al2O3 catalyst. J. Ind. Eng. Chem. 2012, 18, 2059–2068. [Google Scholar] [CrossRef]
- Hosseininejad, S.; Afacan, A.; Hayes, R.E. Catalytic and kinetic study of methanol dehydration to dimethyl ether. Chem. Eng. Res. Des. 2012, 90, 825–833. [Google Scholar] [CrossRef]
- Gates, B.C.; Johanson, L.N. Langmuir-hinshelwood kinetics of the dehydration of methanol catalyzed by cation exchange resin. AIChE J. 1971, 17, 981–983. [Google Scholar] [CrossRef]
- An, W.; Chuang, K.T.; Sanger, A.R. Dehydration of Methanol to Dimethyl Ether by Catalytic Distillation. Can. J. Chem. Eng. 2004, 82, 948–955. [Google Scholar] [CrossRef]
- Lee, E.-Y.; Park, Y.-K.; Joo, O.-S.; Jung, K.-D. Methanol dehydration to produce dimethyl ether over g -Al 2 O 3. React. Kinet. Catal. Lett. 2006, 89, 115–121. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, H.; Ying, W.; Fang, D. Dehydration of methanol to dimethyl ether over γ-Al2O3 catalyst: Intrinsic kinetics and effectiveness factor. Can. J. Chem. Eng. 2013, 91, 1538–1546. [Google Scholar] [CrossRef]
- Mollavali, M.; Yaripour, F.; Atashi, H.; Sahebdelfar, S. Intrinsic Kinetics Study of Dimethyl Ether Synthesis from Methanol on γ-Al2O3 Catalysts. Ind. Eng. Chem. Res. 2008, 47, 3265–3273. [Google Scholar] [CrossRef]
- Ha, K.S.; Lee, Y.J.; Bae, J.W.; Kim, Y.W.; Woo, M.H.; Kim, H.S.; Park, M.J.; Jun, K.W. New reaction pathways and kinetic parameter estimation for methanol dehydration over modified ZSM-5 catalysts. Appl. Catal. A Gen. 2011, 395, 95–106. [Google Scholar] [CrossRef]
- Ortega, C.; Rezaei, M.; Hessel, V.; Kolb, G. Methanol to dimethyl ether conversion over a ZSM-5 catalyst: Intrinsic kinetic study on an external recycle reactor. Chem. Eng. J. 2018, 347, 741–753. [Google Scholar] [CrossRef]
- Pop, G.; Bozga, G.; Ganea, R.; Natu, N. Methanol Conversion to Dimethyl Ether over H-SAPO-34 Catalyst. Ind. Eng. Chem. Res. 2009, 48, 7065–7071. [Google Scholar] [CrossRef]
- Sierra, I.; Ereña, J.; Aguayo, A.T.; Ateka, A.; Bilbao, J. Kinetic Modelling for the Dehydration of Methanol to Dimethyl Ether over γ -Al2O3. Chem. Eng. Trans. 2013, 32, 613–618. [Google Scholar]
- Gao, Y.; Chen, S.L.; Wei, Y.; Wang, Y.; Sun, W.; Cao, Y.; Zeng, P. Kinetics of coke formation in the dimethyl ether-to-olefins process over SAPO-34 catalyst. Chem. Eng. J. 2017, 326, 528–539. [Google Scholar] [CrossRef]
- Simón, E.; Rosas, J.M.; Santos, A.; Romero, A. Study of the deactivation of copper-based catalysts for dehydrogenation of cyclohexanol to cyclohexanone. Catal. Today 2012, 187, 150–158. [Google Scholar] [CrossRef]
- Pérez-Uriarte, P.; Ateka, A.; Gayubo, A.G.; Cordero-Lanzac, T.; Aguayo, A.T.; Bilbao, J. Deactivation kinetics for the conversion of dimethyl ether to olefins over a HZSM-5 zeolite catalyst. Chem. Eng. J. 2017, 311, 367–377. [Google Scholar] [CrossRef]
- Levenspiel, O. Chemical Reaction Engineering, 3rd ed.; John Wiley and Sons Inc.: Hoboken, NJ, USA, 1999; Volume 38, ISBN 047125424X. [Google Scholar]
- Gayubo, A.G.; Aguayo, A.T.; Olazar, M.; Vivanco, R.; Bilbao, J. Kinetics of the irreversible deactivation of the HZSM-5 catalyst in the MTO process. Chem. Eng. Sci. 2003, 58, 5239–5249. [Google Scholar] [CrossRef]
- Fichtl, M.B.; Schlereth, D.; Jacobsen, N.; Kasatkin, I.; Schumann, J.; Behrens, M.; Schlögl, R.; Hinrichsen, O. Kinetics of deactivation on Cu/ZnO/Al2O3 methanol synthesis catalysts. Appl. Catal. A Gen. 2015, 502, 262–270. [Google Scholar] [CrossRef]
- Brunauer, S.; Emmett, P.H.; Teller, E. Adsorption of Gases in Multimolecular Layers. J. Am. Chem. Soc. 1938, 60, 309–319. [Google Scholar] [CrossRef]
- Harkins, W.D.; Jura, G. Surfaces of Solids. XIII. A Vapor Adsorption Method for the Determination of the Area of a Solid without the Assumption of a Molecular Area, and the Areas Occupied by Nitrogen and Other Molecules on the Surface of a Solid. J. Am. Chem. Soc. 1944, 66, 1366–1373. [Google Scholar] [CrossRef]
- Thommes, M.; Kaneko, K.; Neimark, A.V.; Olivier, J.P.; Rodriguez-Reinoso, F.; Rouquerol, J.; Sing, K.S.W. IUPAC Technical Report Physisorption of gases, with special reference to the evaluation of surface area and pore size distribution (IUPAC Technical Report). Pure Appl. Chem. 2015, 87, 1051–1069. [Google Scholar] [CrossRef] [Green Version]
- Dubinin, M.M. The potential theory of adsorption of gases and vapors for adsorbents with energetically nonuniform surfaces. Chem. Rev. 1960, 60, 235–241. [Google Scholar] [CrossRef]
- Martinez-Espin, J.S.; Mortén, M.; Janssens, T.V.W.; Svelle, S.; Beato, P.; Olsbye, U. New insights into catalyst deactivation and product distribution of zeolites in the methanol-to-hydrocarbons (MTH) reaction with methanol and dimethyl ether feeds. Catal. Sci. Technol. 2017, 7, 2700–2716. [Google Scholar] [CrossRef]
- Vishwanathan, V.; Jun, K.W.; Kim, J.W.; Roh, H.S. Vapour phase dehydration of crude methanol to dimethyl ether over Na-modified H-ZSM-5 catalysts. Appl. Catal. A Gen. 2004, 276, 251–255. [Google Scholar] [CrossRef]
- Blaszkowski, S.R.; van Santen, R.A. The Mechanism of Dimethyl Ether Formation from Methanol Catalyzed by Zeolitic Protons. J. Am. Chem. Soc. 1996, 118, 5152–5153. [Google Scholar] [CrossRef] [Green Version]
- Xu, M.; Lunsford, J.H.; Goodman, D.W.; Bhattacharyya, A. Synthesis of dimethyl ether (DME) from methanol over solid-acid catalysts. Appl. Catal. A Gen. 1997, 149, 289–301. [Google Scholar] [CrossRef]
- Gayubo, A.G.; Aguayo, A.T.; Morán, A.L.; Olazar, M.; Bilbao, J. Role of water in the kinetic modeling of catalyst deactivation in the MTG process. AIChE J. 2002, 48, 1561–1571. [Google Scholar] [CrossRef]
- Figueiredo, J.; Pereira, M.F.; Freitas, M.M.; Órfão, J.J. Modification of the surface chemistry of activated carbons. Carbon 1999, 37, 1379–1389. [Google Scholar] [CrossRef]
- Froment, G.F.; Bischoff, K.B.; de Wilde, J. Chemical Reactor Analysis and Design, 3rd ed.; John Wiley & Sons: Hoboken, NJ, USA, 2011; ISBN 978-0-470-56541-4. [Google Scholar]
- Zhang, G.; Zhang, X.; Bai, T.; Chen, T.; Fan, W. Coking kinetics and influence of reaction-regeneration on acidity, activity and deactivation of Zn/HZSM-5 catalyst during methanol aromatization. J. Energy Chem. 2015, 24, 108–118. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| N2 Isotherm | CO2 Isotherm | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| At (m2/g) | ABET (m2/g) | Vt (cm3/g) | Vmes (cm3/g) | Vtot (cm3/g) | ADR (m2/g) | VDR (cm3/g) | ||||
| 279 | 1105 | 0.43 | 0.38 | 0.80 | 509 | 0.20 | ||||
| Atomic surface concentration (%) | ||||||||||
| C1s | O1s | P2p | Zr3d | P/Zr | ||||||
| 65.1 | 27.0 | 3.9 | 3.5 | 1.11 | ||||||
| Kinetic Parameter | Units | Constant Value at 500 °C | ||
|---|---|---|---|---|
| 1.3 | mol·gcat−1·s−1 | 65 | 5.3 × 10−5 | |
| 5.6 × 10−11 | atm | −123 | 0.01 | |
| 2.7 × 10−2 | mol·gcat−1·s−1 | 51 | 9.2 × 10−6 | |
| 6.6 | mol·gcat−1·s−1 | 15 | 0.7 | |
| 69.5 | - | 19 | 3.7 | |
| 3.0 | atm−1 | −15 | 31.1 | |
| 3.6 | atm−1 | −10 | 17.1 | |
| 0.01 | atm−1 | −41 | 8.5 |
| Deactivation Equation | OF | ||||
|---|---|---|---|---|---|
| (33) | 0.184 | 1.24 × 1010 | 135 | 0.88 | 0.019 |
| (34) | 0.043 | 5.14 × 108 | 122 | 0.96 | 0.021 |
| (35) | 0.038 | 2.61 × 109 | 130 | 0.79 | 0.015 |
| (36) | 5.98 × 1014 | 3.20 × 1021 | 87 | 0.88 | 0.053 |
| (37) | 4.16 × 107 | 1.91 × 1025 | 120 | 0.98 | 0.030 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Torres-Liñán, J.; Ruiz-Rosas, R.; Rosas, J.M.; Rodríguez-Mirasol, J.; Cordero, T. A Kinetic Model Considering Catalyst Deactivation for Methanol-to-Dimethyl Ether on a Biomass-Derived Zr/P-Carbon Catalyst. Materials 2022, 15, 596. https://doi.org/10.3390/ma15020596
Torres-Liñán J, Ruiz-Rosas R, Rosas JM, Rodríguez-Mirasol J, Cordero T. A Kinetic Model Considering Catalyst Deactivation for Methanol-to-Dimethyl Ether on a Biomass-Derived Zr/P-Carbon Catalyst. Materials. 2022; 15(2):596. https://doi.org/10.3390/ma15020596
Chicago/Turabian StyleTorres-Liñán, Javier, Ramiro Ruiz-Rosas, Juana María Rosas, José Rodríguez-Mirasol, and Tomás Cordero. 2022. "A Kinetic Model Considering Catalyst Deactivation for Methanol-to-Dimethyl Ether on a Biomass-Derived Zr/P-Carbon Catalyst" Materials 15, no. 2: 596. https://doi.org/10.3390/ma15020596
APA StyleTorres-Liñán, J., Ruiz-Rosas, R., Rosas, J. M., Rodríguez-Mirasol, J., & Cordero, T. (2022). A Kinetic Model Considering Catalyst Deactivation for Methanol-to-Dimethyl Ether on a Biomass-Derived Zr/P-Carbon Catalyst. Materials, 15(2), 596. https://doi.org/10.3390/ma15020596