
Investigations of Structural, Electronic and Magnetic Properties of MnSe under High Pressure


Abstract
:1. Introduction
2. Methods
3. Results and Discussion
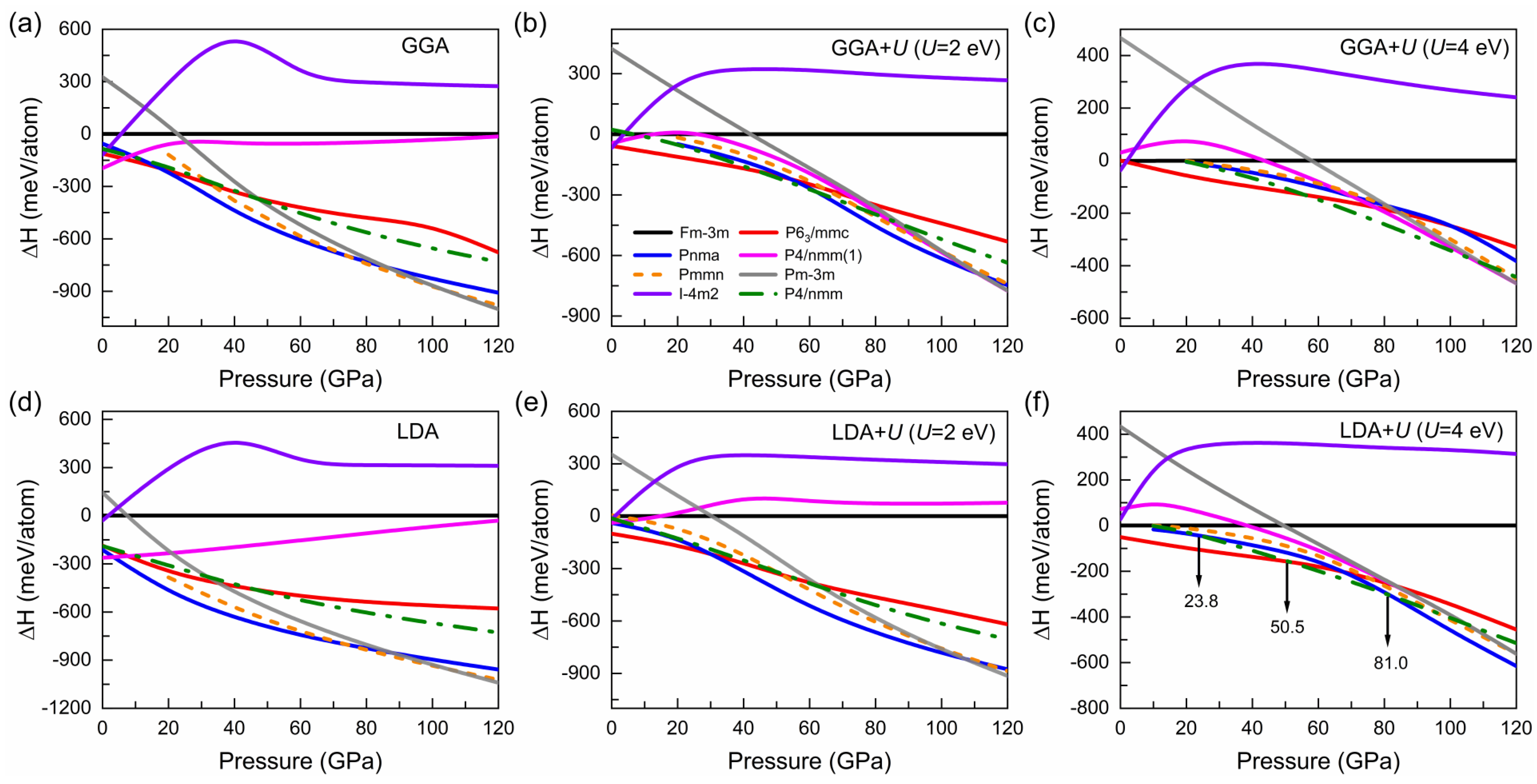
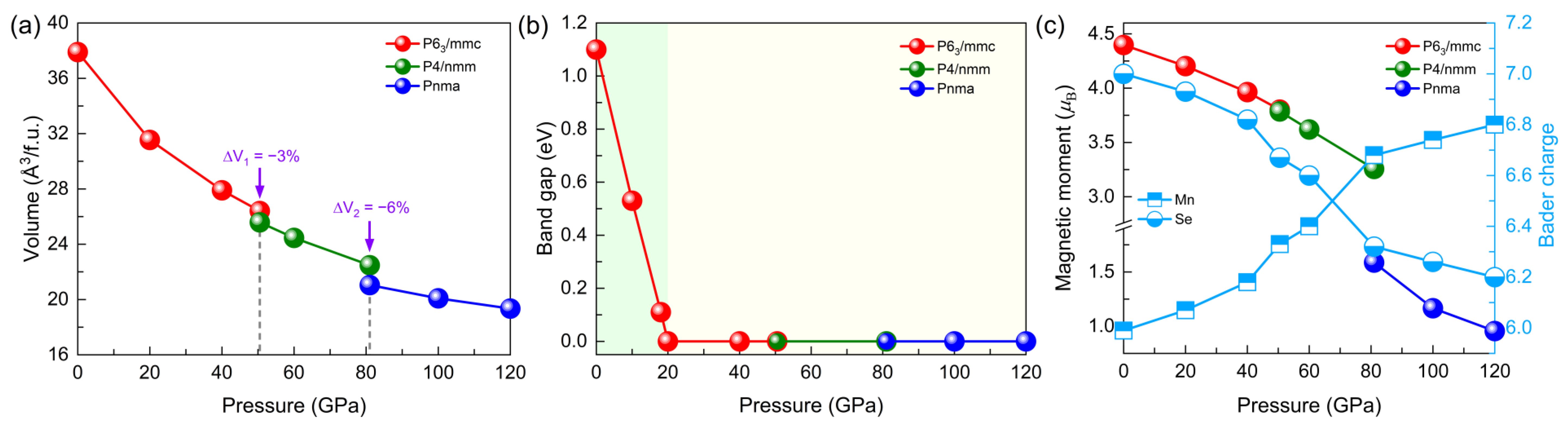
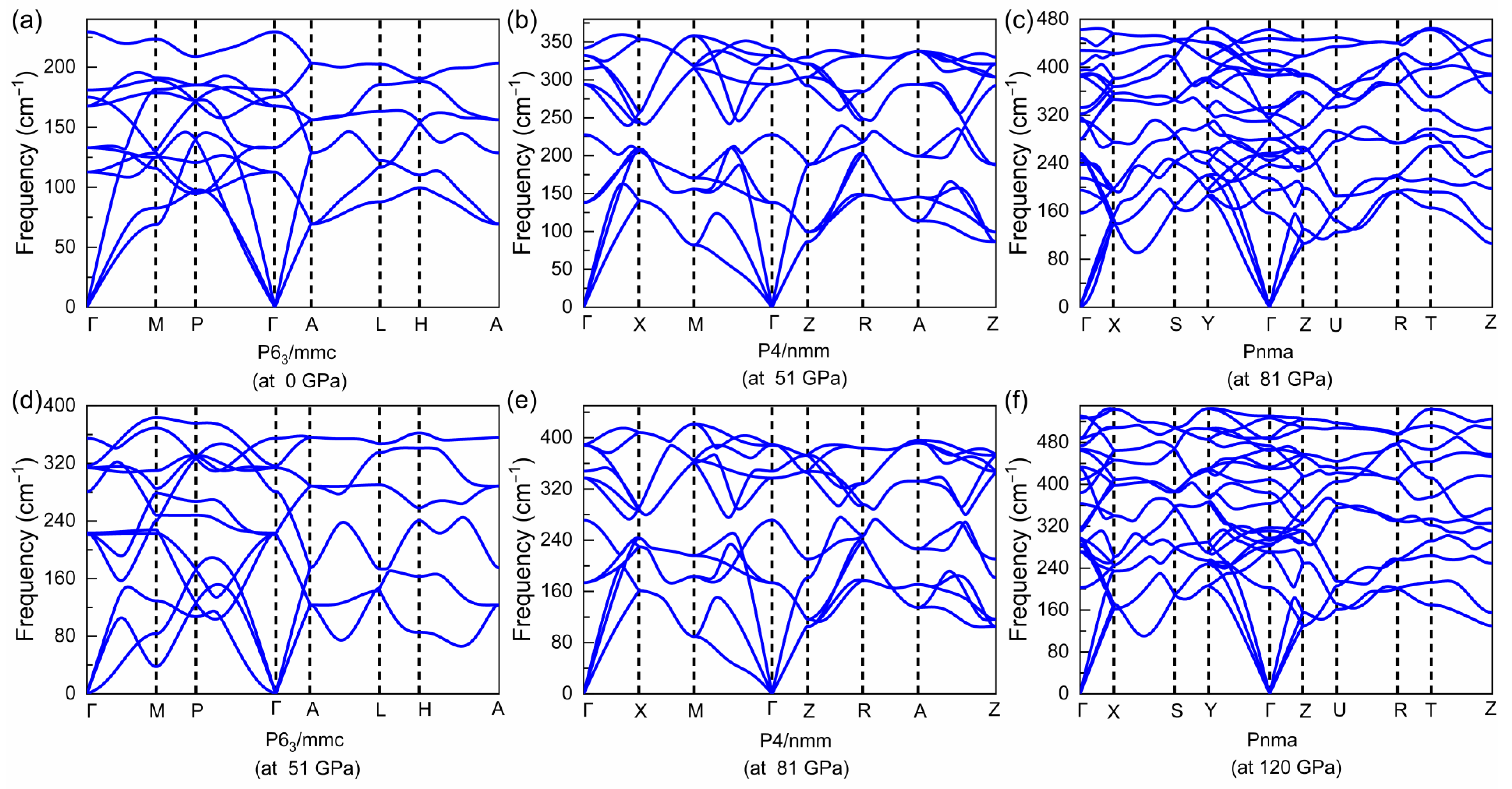
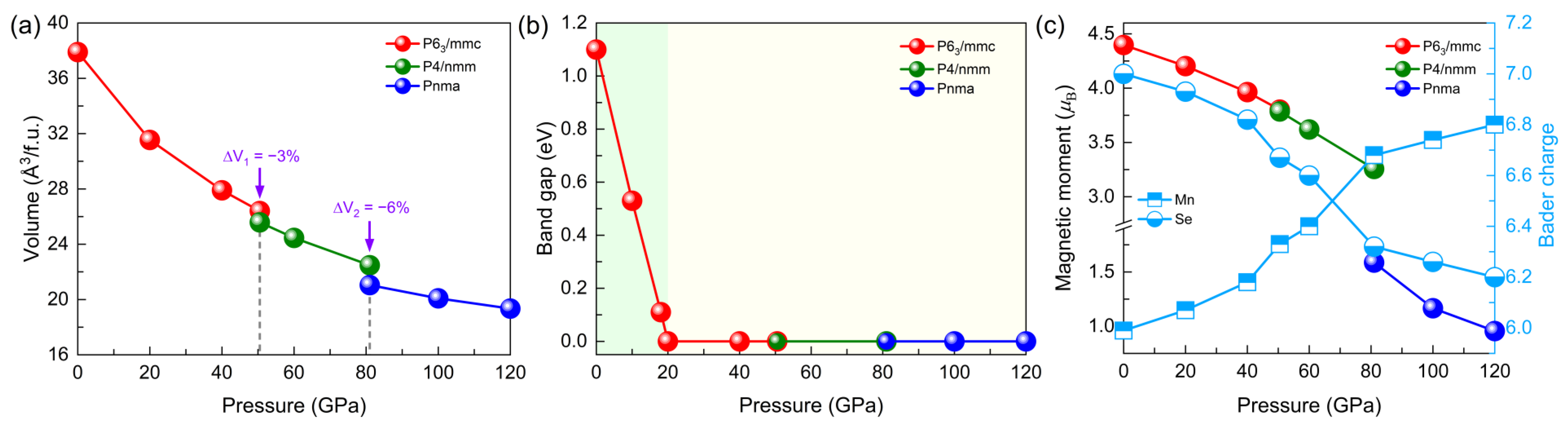
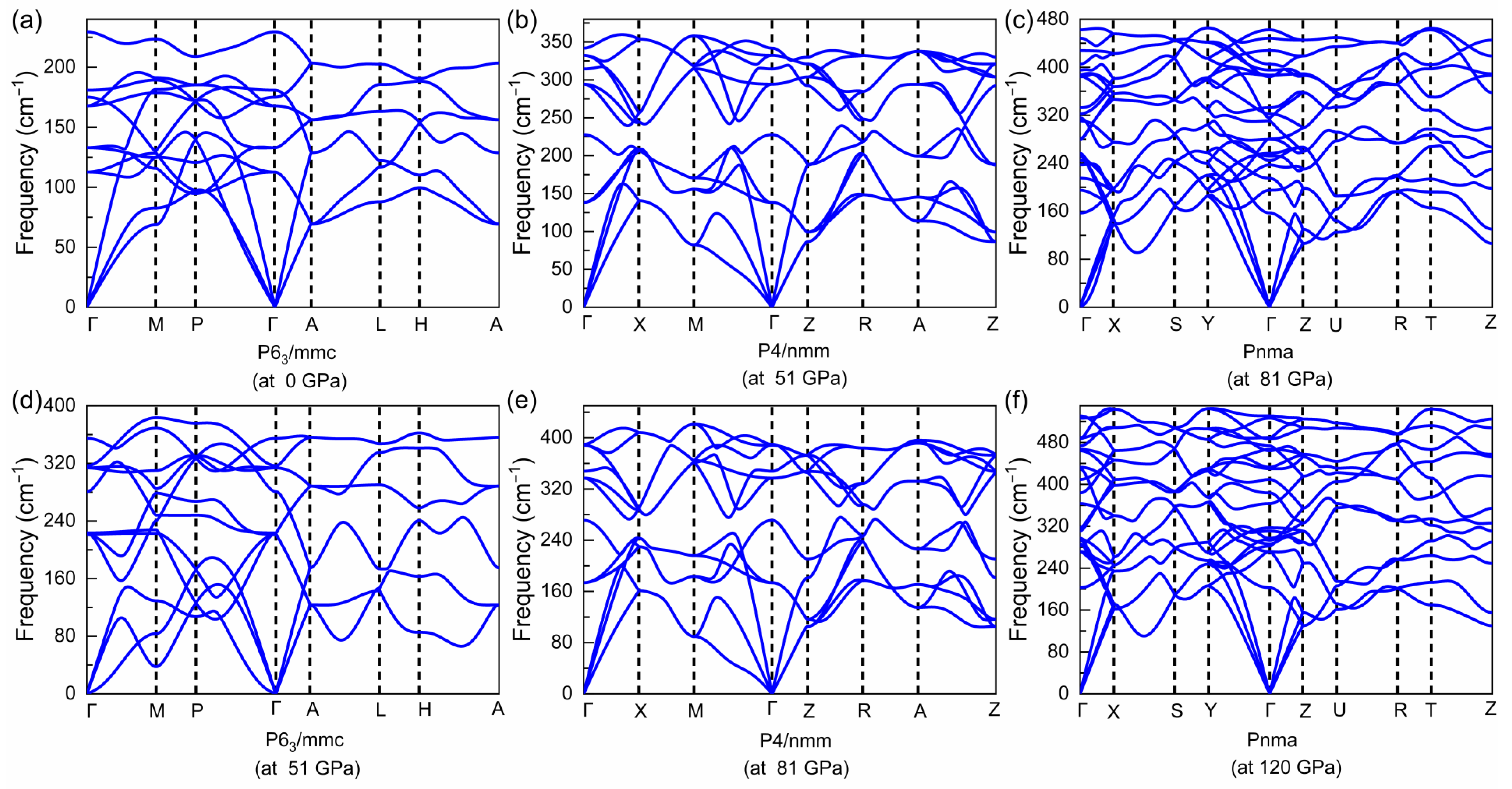
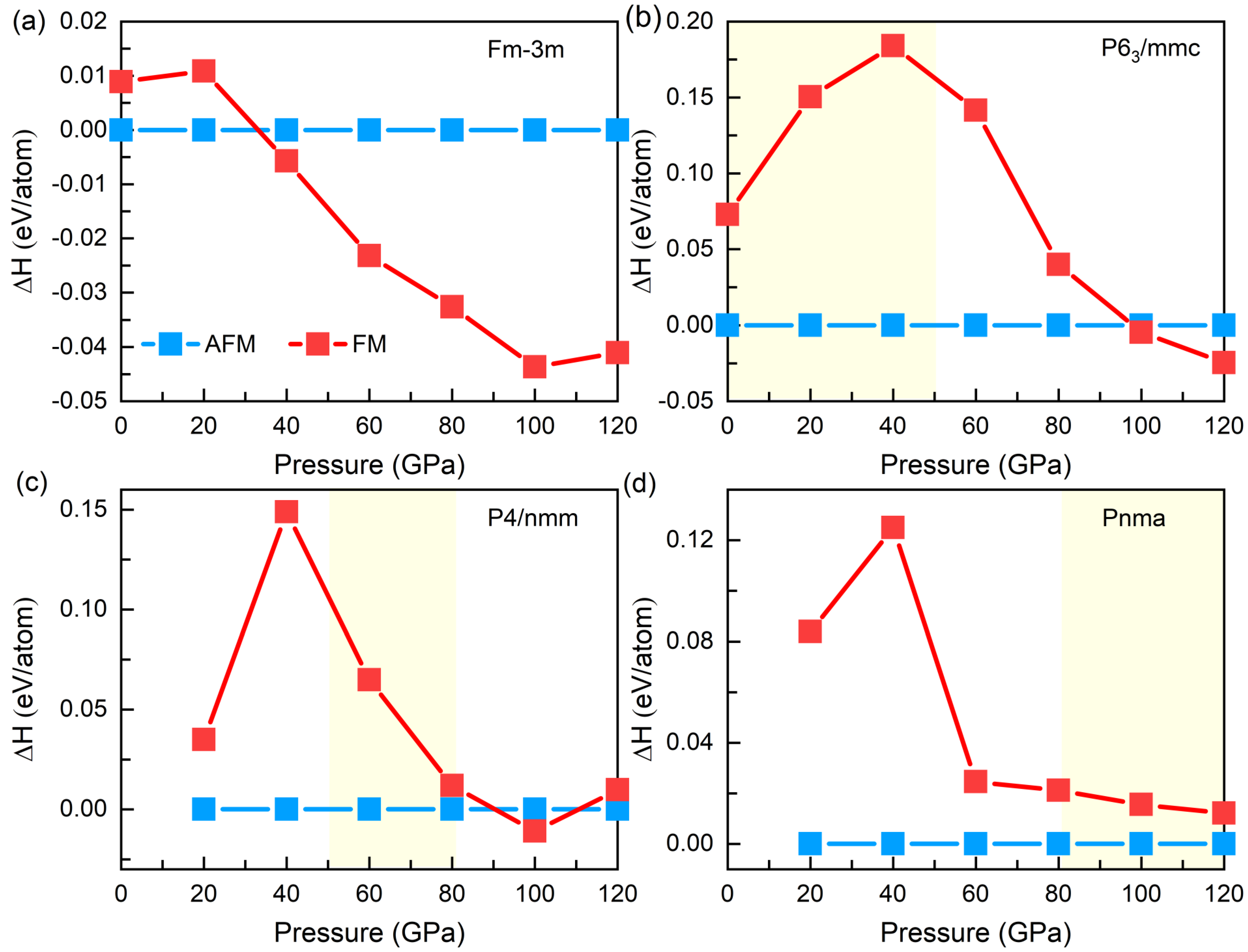
3.1. Structural Evolution under Pressure
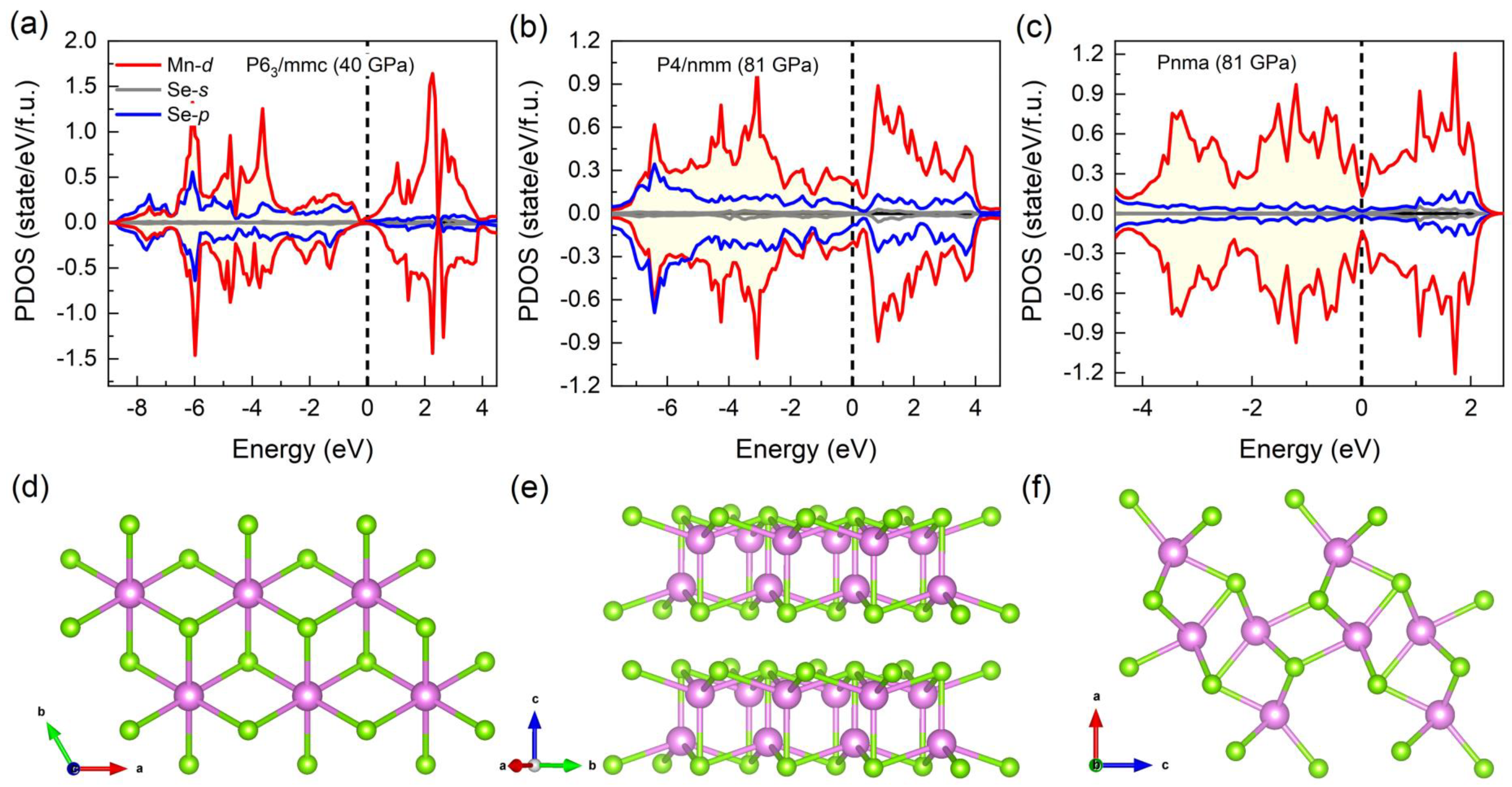
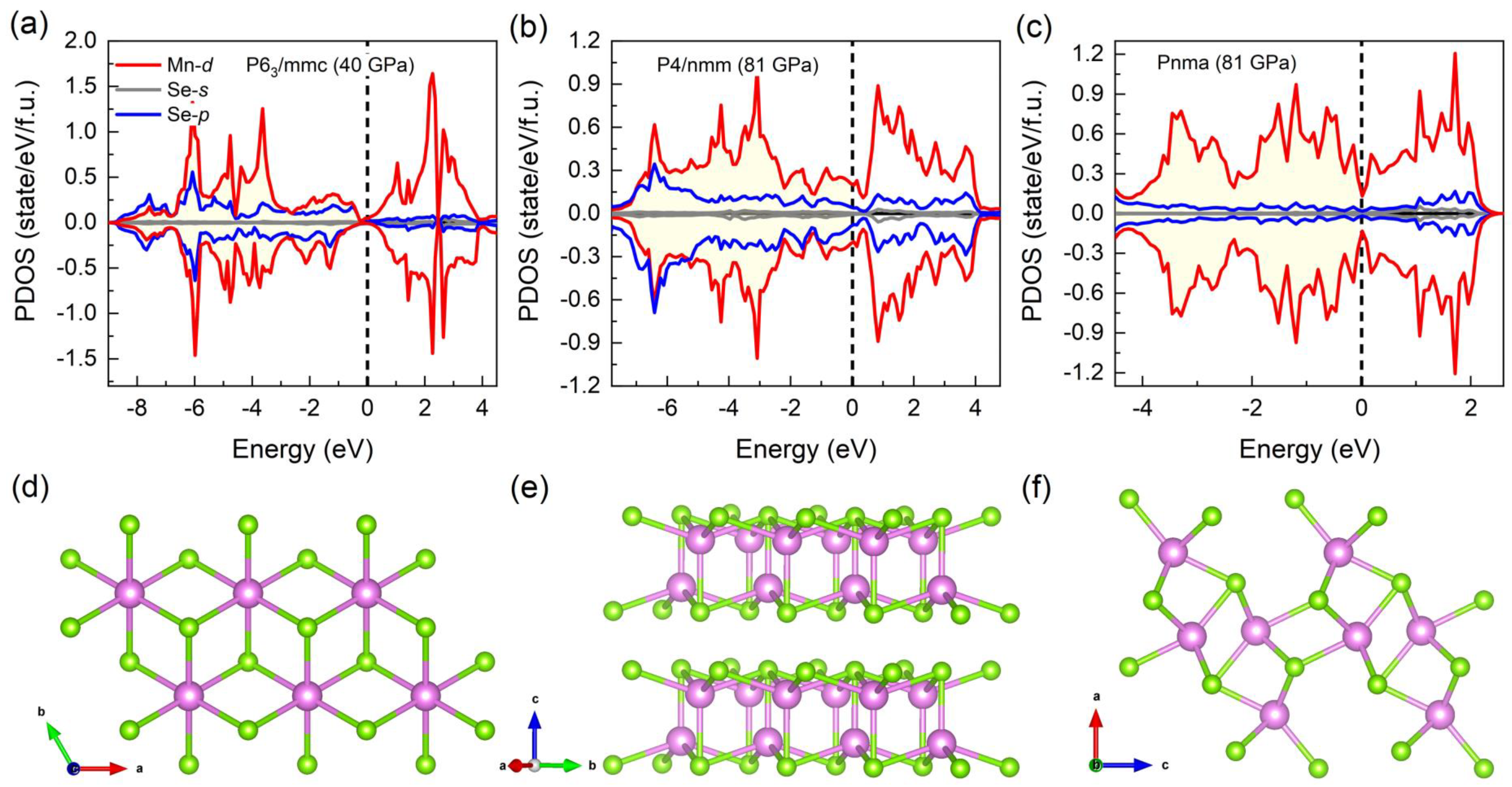
3.2. Electronic and Magnetic Properties
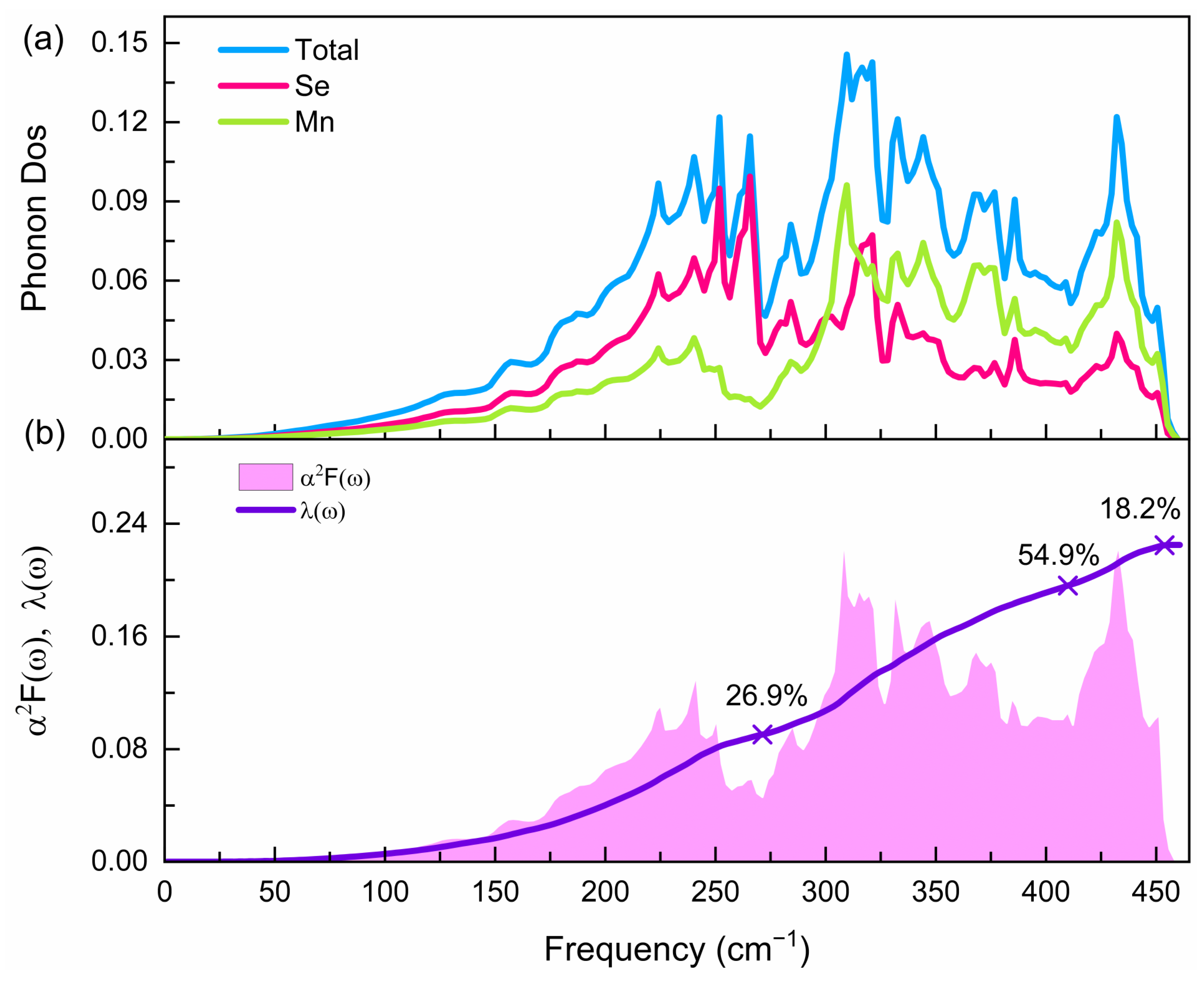
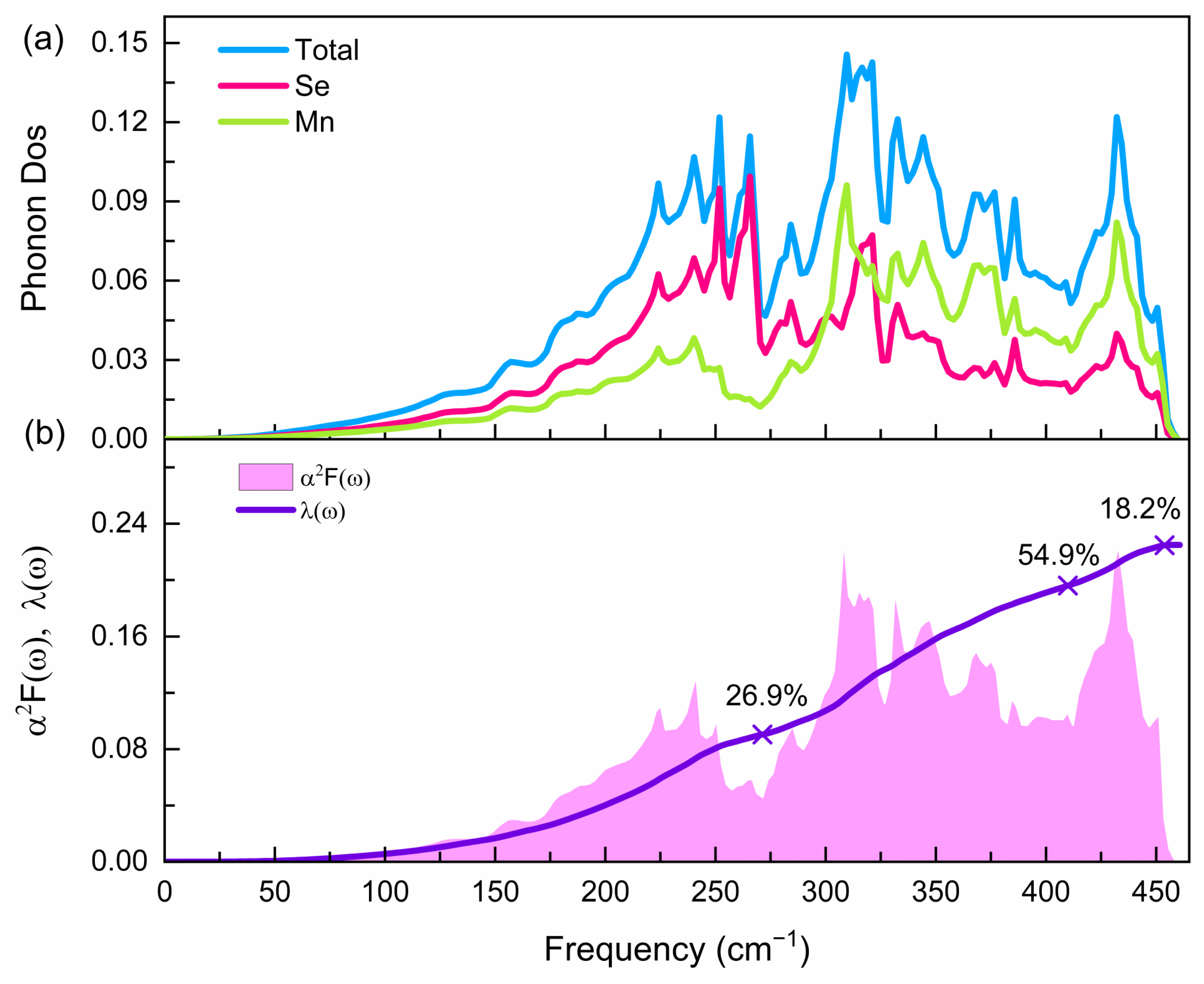
3.3. Superconducting Properties of the Pnma Phase
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Taniguchi, M.; Ley, L.; Johnson, R.L.; Ghijsen, J.; Cardona, M. Synchrotron Radiation Study of Cd1−xMnxTe (0 ≤ x ≤ 0.65). Phys. Rev. B 1986, 33, 1206–1212. [Google Scholar] [CrossRef] [PubMed]
- Ley, L.; Taniguchi, M.; Ghijsen, J.; Johnson, R.L.; Fujimori, A. Manganese-Derived Partial Density of States in Cd1−xMnxTe. Phys. Rev. B 1987, 35, 2839–2843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taniguchi, M.; Fujimori, M.; Fujisawa, M.; Mori, T.; Souma, I.; Oka, Y. Mn 3d Partial Density-of-States and p-d Hybridization in Cd1−xMnxY (Y = S, Se and Te). Solid State Commun. 1987, 62, 431–434. [Google Scholar] [CrossRef]
- Wang, Y.; Zhou, Z.; Wen, T.; Zhou, Y.; Li, N.; Han, F.; Xiao, Y.; Chow, P.; Sun, J.; Pravica, M.; et al. Pressure-Driven Cooperative Spin-Crossover, Large-Volume Collapse, and Semiconductor-to-Metal Transition in Manganese(II) Honeycomb Lattices. J. Am. Chem. Soc. 2016, 138, 15751–15757. [Google Scholar] [CrossRef] [PubMed]
- Rueff, J.-P.; Kao, C.-C.; Struzhkin, V.V.; Badro, J.; Shu, J.; Hemley, R.J.; Mao, H.K. Pressure-Induced High-Spin to Low-Spin Transition in FeS Evidenced by X-Ray Emission Spectroscopy. Phys. Rev. Lett. 1999, 82, 3284–3287. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Niu, C.; Zhang, J.; Zou, L.; Zeng, Z.; Wang, X. Spin-Crossover Induced Ferromagnetism and Layer Stacking-Order Change in Pressurized 2D Antiferromagnet MnPS3. Phys. Chem. Chem. Phys. 2021, 23, 9679–9685. [Google Scholar] [CrossRef] [PubMed]
- Wolf, S.A.; Awschalom, D.D.; Buhrman, R.A.; Daughton, J.M.; von Molnár, S.; Roukes, M.L.; Chtchelkanova, A.Y.; Treger, D.M. Spintronics: A Spin-Based Electronics Vision for the Future. Science 2001, 294, 1488–1495. [Google Scholar] [CrossRef] [Green Version]
- Ozawa, K.; Anzai, S.; Hamaguchi, Y. Effect of Pressure on the Magnetic Transition Point of Manganese Telluride. Phys. Lett. 1966, 20, 132–133. [Google Scholar] [CrossRef]
- Wang, Y.; Bai, L.; Wen, T.; Yang, L.; Gou, H.; Xiao, Y.; Chow, P.; Pravica, M.; Yang, W.; Zhao, Y. Giant Pressure-Driven Lattice Collapse Coupled with Intermetallic Bonding and Spin-State Transition in Manganese Chalcogenides. Angew. Chem. Int. Ed. 2016, 128, 10350–10353. [Google Scholar] [CrossRef]
- Dyachenko, A.A.; Lukoyanov, A.V.; Shorikov, A.O.; Anisimov, V.I. Magnetically Driven Phase Transitions with a Large Volume Collapse in MnSe under Pressure: A DFT + DMFT Study. Phys. Rev. B 2018, 98, 085139. [Google Scholar] [CrossRef]
- Kimber, S.A.J.; Salamat, A.; Evans, S.R.; Jeschke, H.O.; Muthukumar, K.; Tomic, M.; Salvat-Pujol, F.; Valentí, R.; Kaisheva, M.V.; Zizak, I.; et al. Giant Pressure-Induced Volume Collapse in the Pyrite Mineral MnS2. Proc. Natl. Acad. Sci. USA 2014, 111, 5106–5110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hung, T.L.; Huang, C.H.; Deng, L.Z.; Ou, M.N.; Chen, Y.Y.; Wu, M.K.; Huyan, S.Y.; Chu, C.W.; Chen, P.J.; Lee, T.K. Pressure Induced Superconductivity in MnSe. Nat. Commun. 2021, 12, 5436. [Google Scholar] [CrossRef] [PubMed]
- Efrem D’Sa, J.B.C.; Bhobe, P.A.; Priolkar, K.R.; Das, A.; Krishna, P.S.R.; Sarode, P.R.; Prabhu, R.B. Low Temperature Magnetic Structure of MnSe. Pramana-J. Phys. 2004, 63, 227–232. [Google Scholar] [CrossRef]
- Huang, C.H.; Wang, C.W.; Chang, C.C.; Lee, Y.C.; Huang, G.T.; Wang, M.J.; Wu, M.K. Anomalous Magnetic Properties in Mn (Se, S) System. J. Magn. Magn. Mater. 2019, 483, 205–211. [Google Scholar] [CrossRef]
- Anisimov, V.V.; Gunnarsson, O. Density-Functional Calculation of Effective Coulomb Interactions in Metals. Phys. Rev. B 1991, 43, 7570–7574. [Google Scholar] [CrossRef]
- Amiri, P.; Hashemifar, S.J.; Akbarzadeh, H. Density Functional Study of Narrow Cubic MnSe Nanowires: Role of MnSe Chains. Phys. Rev. B 2011, 83, 165424. [Google Scholar] [CrossRef]
- Xiao, G.; Yang, X.; Zhang, X.; Wang, K.; Huang, X.; Ding, Z.; Ma, Y.; Zou, G.; Zou, B. A Protocol to Fabricate Nanostructured New Phase: B31-Type MnS Synthesized under High Pressure. J. Am. Chem. Soc. 2015, 137, 10297–10303. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From Ultrasoft Pseudopotentials to the Projector Augmented-Wave Method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient Iterative Schemes for Ab Initio Total-Energy Calculations Using a Plane-Wave Basis Set. Phys. Rev. B 1996, 54, 11169. [Google Scholar] [CrossRef]
- Hafner, J. Ab-Initio Simulations of Materials Using Vasp: Density-Functional Theory and Beyond. J. Comput. Chem. 2008, 29, 2044–2078. [Google Scholar] [CrossRef]
- Zhao, J.; Zhang, H.; Niu, C.; Zhang, J.; Zeng, Z.; Wang, X. Investigations of High-Pressure Properties of MnF2 Based on the First-Principles Method. J. Phys. Chem. C 2021, 125, 21709–21717. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special Points for Brillouin-Zone Integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Pack, J.D.; Monkhorst, H.J. “Special Points for Brillouin-Zone Integrations”—A Reply. Phys. Rev. B 1977, 16, 1748–1749. [Google Scholar] [CrossRef]
- Painter, G.S. Improved Correlation Corrections to the Local-Spin-Density Approximation. Phys. Rev. B 1981, 24, 4264–4270. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dudarev, S.L.; Botton, G.A.; Savrasov, S.Y.; Humphreys, C.J.; Sutton, A.P. Electron-Energy-Loss Spectra and the Structural Stability of Nickel Oxide: An LSDA + U Study. Phys. Rev. B 1998, 57, 1505–1509. [Google Scholar] [CrossRef]
- Samarth, N.; Kłosowski, P.; Luo, H.; Giebułtowicz, T.M.; Furdyna, J.K.; Rhyne, J.J.; Larson, B.E.; Otsuka, N. Antiferromagnetic Ordering in MnSe/Znse Multilayers. J. Appl. Phys. 1991, 69, 6109. [Google Scholar] [CrossRef]
- Baroni, S.; de Gironcoli, S.; Dal Corso, A.; Giannozzi, P. Phonons and Related Crystal Properties from Density-Functional Perturbation Theory. Rev. Mod. Phys. 2001, 73, 515–562. [Google Scholar] [CrossRef] [Green Version]
- Gonze, X.; Lee, C. Dynamical Matrices, Born Effective Charges, Dielectric Permittivity Tensors, and Interatomic Force Constants from Density-Functional Perturbation Theory. Phys. Rev. B 1997, 55, 10355–10368. [Google Scholar] [CrossRef]
- Wang, Y.; Lv, J.; Zhu, L.; Ma, Y. Crystal Structure Prediction Via Particle Swarm Optimization. Phys. Rev. B 2010, 82, 094116. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Jian, L.; Zhu, L.; Ma, Y. Calypso: A Method for Crystal Structure Prediction. Comput. Phys. Commun. 2012, 183, 2063–2070. [Google Scholar] [CrossRef] [Green Version]
- Giannozzi, P.; Baroni, S.; Bonini, N.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Chiarotti, G.L.; Cococcioni, M.; Dabo, I.; et al. Quantum Espresso: A Modular and Open-Source Software Project for Quantum Simulations of Materials. J. Phys. Condens. Matter 2009, 21, 395502. [Google Scholar] [CrossRef] [PubMed]
- Bardeen, J.; Cooper, L.N.; Schrieffer, J.R. Theory of Superconductivity. Phys. Rev. 1957, 108, 1175–1204. [Google Scholar] [CrossRef] [Green Version]
- Prandini, G.; Marrazzo, A.; Castelli, I.E.; Mounet, N.; Marzari, N. Precision and Efficiency in Solid-State Pseudopotential Calculations. NPJ Comput. Mater. 2018, 4, 72. [Google Scholar] [CrossRef]
- Vanderbilt, D. Soft Self-Consistent Pseudopotentials in a Generalized Eigenvalue Formalism. Phys. Rev. B 1990, 41, 7892–7895. [Google Scholar] [CrossRef]
- Perdew, J.P.; Ruzsinszky, A.; Csonka, G.I.; Vydrov, O.A.; Scuseria, G.E.; Constantin, L.A.; Zhou, X.; Burke, K. Restoring the Density-Gradient Expansion for Exchange in Solids and Surfaces. Phys. Rev. Lett. 2008, 100, 136406. [Google Scholar] [CrossRef] [Green Version]
- Mouhat, F.; Coudert, F.X. Necessary and Sufficient Elastic Stability Conditions in Various Crystal Systems. Phys. Rev. B 2014, 90, 224104. [Google Scholar] [CrossRef] [Green Version]
- Pickart, S.J.; Nathans, R.; Shirane, G. Magnetic Structure Transitions in LixMn1−xSe. Phys. Rev. 1961, 121, 707–714. [Google Scholar] [CrossRef]
- McMillan, W.L. Transition Temperature of Strong-Coupled Superconductors. Phys. Rev. 1968, 167, 331–344. [Google Scholar] [CrossRef]
- Dynes, R.C. Mcmillan’s Equation and the Tc of Superconductors. Solid State Commun. 1972, 10, 615–618. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| P (GPa) | SG | Z | a (Å) | b (Å) | c (Å) | WP | x | y | z |
|---|---|---|---|---|---|---|---|---|---|
| 0 | P63/mmc | 2 | 3.761 | 3.761 | 6.188 | Mn(2a) | 0.0000 | 0.0000 | 0.0000 |
| Se(2c) | 0.3333 | 0.6667 | 0.2500 | ||||||
| 60 | P4/nmm | 2 | 3.160 | 3.160 | 4.898 | Mn(2c) | 0.0000 | 0.5000 | 0.1553 |
| Se(2c) | 0.5000 | −0.000 | 0.3134 | ||||||
| 100 | Pnma | 4 | 5.265 | 2.891 | 5.280 | Mn(4c) | −0.015 | 0.7500 | 0.3065 |
| Se(4c) | −0.197 | 0.7500 | 0.9281 |
| Space Group | P63/mmc | P4/nmm | Pnma |
|---|---|---|---|
| (0 GPa) | (50.5 GPa) | (81 GPa) | |
| C11 | 122.8 | 178.0 | 361.7 |
| C12 | 54.1 | 97.9 | 261.2 |
| C13 | 49.7 | 89.3 | 180.3 |
| C22 | 122.8 | 178.0 | 426.6 |
| C23 | 49.7 | 89.3 | 200.1 |
| C33 | 142.4 | 385.5 | 449.1 |
| C44 | 36.9 | 72.8 | 171.5 |
| C55 | 36.9 | 72.8 | 234.7 |
| C66 | 34.4 | 169.4 | 271.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, J.; Zhang, H.; Niu, C.; Wang, X. Investigations of Structural, Electronic and Magnetic Properties of MnSe under High Pressure. Materials 2022, 15, 1109. https://doi.org/10.3390/ma15031109
Zhao J, Zhang H, Niu C, Wang X. Investigations of Structural, Electronic and Magnetic Properties of MnSe under High Pressure. Materials. 2022; 15(3):1109. https://doi.org/10.3390/ma15031109
Chicago/Turabian StyleZhao, Jing, Hanxing Zhang, Caoping Niu, and Xianlong Wang. 2022. "Investigations of Structural, Electronic and Magnetic Properties of MnSe under High Pressure" Materials 15, no. 3: 1109. https://doi.org/10.3390/ma15031109