
First-Principles Study of Electronic Properties of Substitutionally Doped Monolayer SnP3

Abstract
:1. Introduction
2. Computational Details
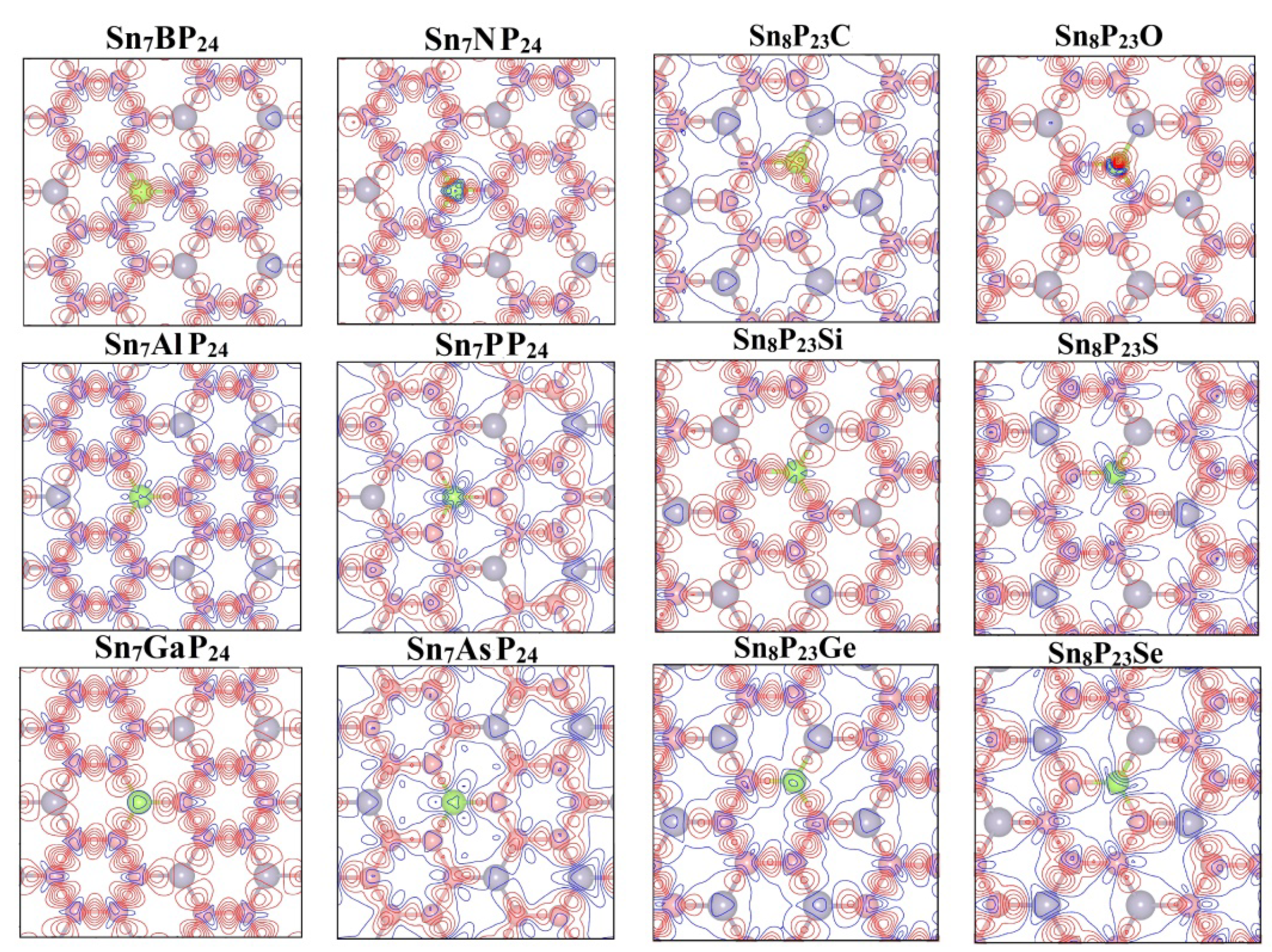
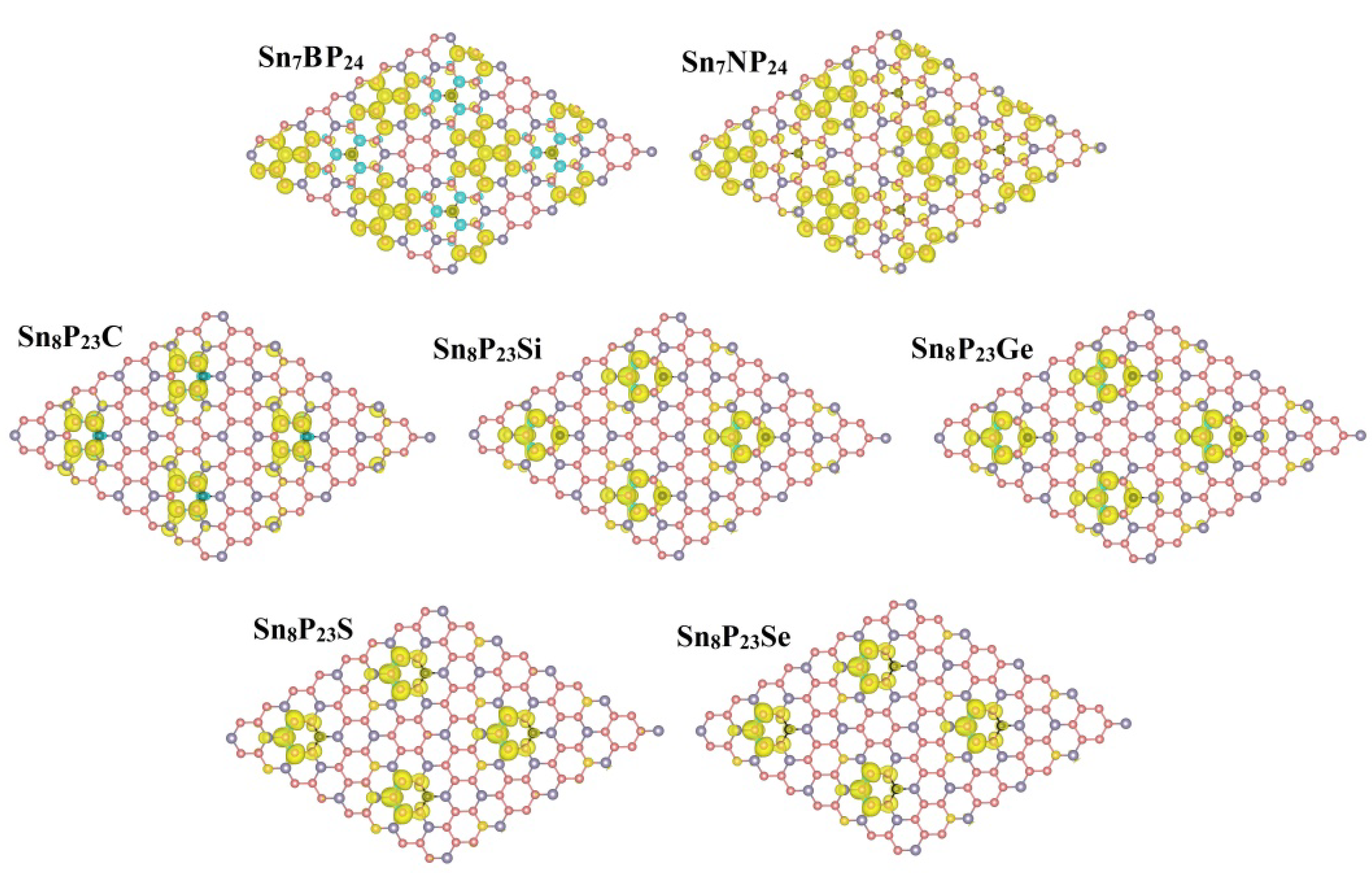
3. Result and Discussion
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Novoselov, K.S.; Geim, A.K.; Morozov, S.V.; Jiang, D.; Zhang, Y.; Dubonos, S.V.; Grigorieva, I.V.; Firsov, A.A. Electric Field Effect in Atomically Thin Carbon Films. Science 2004, 306, 666–669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geim, A.K.; Novoselov, K.S. The Rise of Graphene. Nat. Mater. 2007, 6, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Novoselov, K.S.; Geim, A.K.; Morozov, S.V.; Jiang, D.; Katsnelson, M.I.; Grigorieva, I.V.; Dubonos, S.V.; Firsov, A.A. Two-Dimensional Gas of Massless Dirac Fermions in Graphene. Nature 2005, 438, 197–200. [Google Scholar] [CrossRef]
- Kadantsev, E.S.; Hawrylak, P. Electronic Structure of a Single MoS2 Monolayer. Solid State Commun. 2012, 152, 909–913. [Google Scholar] [CrossRef]
- Dong, X.; Shi, Y.; Zhao, Y.; Chen, D.; Ye, J.; Yao, Y.; Gao, F.; Ni, Z.; Yu, T.; Shen, Z.; et al. Symmetry Breaking of Graphene Monolayers by Molecular Decoration. Phys. Rev. Lett. 2009, 102, 135501. [Google Scholar] [CrossRef] [PubMed]
- Guzmán-Arellano, R.M.; Hernández-Nieves, A.D.; Balseiro, C.A.; Usaj, G. Diffusion of Fluorine Adatoms on Doped Graphene. Appl. Phys. Lett. 2014, 105, 026805. [Google Scholar] [CrossRef] [Green Version]
- Yousefi, M.; Faraji, M.; Asgari, R.; Moshfegh, A.Z. Effect of Boron and Phosphorus Co-doping on the Electronic and Optical Properties of Graphitic Carbon Nitride Monolayers: First-Principle Simulations. Phys. Rev. B 2018, 97, 195428. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Yu, W.; Fu, X.; Qiao, C.; Xia, C.; Jia, Y. Electronic and Magnetic Properties of SnSe Monolayers Doped by Ga, In, As, and Sb: A First-Principles Study. Phys. Chem. Chem. Phys. 2016, 18, 8158–8164. [Google Scholar] [CrossRef] [Green Version]
- Fan, X.; Shen, Z.; Liu, A.Q.; Kuo, J.L. Band gap opening of graphene by doping small boron nitride domains. Nanoscale 2012, 4, 2157–2165. [Google Scholar] [CrossRef]
- Shi, L.; Xu, A.; Zhao, T. First-Principles Investigations of the Working Mechanism of 2d h-Bn as an Interfacial Layer for the Anode of Lithium Metal Batteries. ACS Appl. Mater. Interfaces 2017, 9, 1987–1994. [Google Scholar] [CrossRef]
- Tongay, S.; Zhou, J.; Ataca, C.; Lo, K.; Matthews, T.S.; Li, J.; Grossman, J.C.; Wu, J. Thermally Driven Crossover from Indirect toward Direct Bandgap in 2D Semiconductors: MoSe2 versus MoS2. Nano Lett. 2012, 12, 5576–5580. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Liu, G.; Zhu, X.; Guo, Y. Ultrasensitive NO2 Gas Sensing Based on RGO/MoS2 Nanocomposite Film at Low Temperature. Sens. Actuators B Chem. 2017, 251, 280–290. [Google Scholar] [CrossRef]
- Zhou, Y.; Zou, C.; Lin, X.; Guo, Y. UV Light Activated NO2 Gas Sensing Based on Au Nanoparticles Decorated Few-Layer MoS2 Thin Film at Room Temperature. Appl. Phys. Lett. 2018, 113, 082103. [Google Scholar] [CrossRef]
- Geng, X.; Zhang, Y.; Han, Y.; Li, J.; Yang, L.; Benamara, M.; Chen, L.; Zhu, H. Two-Dimensional Water-Coupled Metallic MoS2 with Nanochannels for Ultrafast Supercapacitors. Nano Lett. 2017, 17, 1825–1832. [Google Scholar] [CrossRef]
- Dastgeer, G.; Afzal, A.M.; Nazir, G.; Sarwar, N. p-GeSe/n-ReS2 Heterojunction Rectifier Exhibiting A Fast Photoresponse with Ultra-High Frequency-Switching Applications. Adv. Mater. Interfaces 2021, 8, 2100705. [Google Scholar] [CrossRef]
- Dastgeer, G.; Afzal, A.M.; Aziz, J.; Hussain, S.; Jaffery, S.H.A.; Kim, D.K.; Imran, M.; Assiri, M.A. Flexible Memory Device Composed of Metal-Oxide and Two-Dimensional Material (Sno2/WRe2) Exhibiting Stable Resistive Switching. Materials 2021, 14, 7535. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhang, L.; Cai, X.; Li, X.; Wang, B.; Yu, W.; Zhao, R. Metal-Nonmetal Oscillations in Doped Blue Phosphorene: A First-Principles Study. Mater. Res. Express. 2018, 5, 055007. [Google Scholar] [CrossRef]
- Liu, R.; Li, Y.; Yao, B.; Ding, Z.; Jiang, Y.; Meng, L.; Deng, R.; Zhang, L.; Zhang, Z.; Zhao, H.; et al. Shallow Acceptor State in Mg-Doped CuAlO2 and Its Effect on Electrical and Optical Properties: An Experimental and First-Principles Study. ACS Appl. Mater. Interfaces 2017, 9, 12608–12616. [Google Scholar] [CrossRef]
- Xie, H.-Q.; Zeng, Y.; Huang, W.-Q.; Peng, L.; Peng, P.; Wang, T.-H. First-Principles Study on Electronic and Optical Properties of La-Doped ZnS. Phys. B Condens. Matter. 2010, 405, 3899–3903. [Google Scholar] [CrossRef] [Green Version]
- Yuan, Y.; Chen, Z.; Yu, H.; Zhang, X.; Liu, T.; Xia, M.; Zheng, R.; Shui, M.; Shu, J. Heteroatom-Doped Carbon-Based Materials for Lithium and Sodium Ion Batteries. Energy Stor. Mater. 2020, 32, 65–90. [Google Scholar] [CrossRef]
- Liu, L.; Xie, Z.; Du, X.; Yu, D.; Yang, B.; Li, B.; Liu, X. Large-Scale Mechanical Preparation of Graphene Containing Nickel, Nitrogen and Oxygen Dopants as Supercapacitor Electrode Material. Chem. Eng. J. 2022, 430, 132515. [Google Scholar] [CrossRef]
- Tan, A.M.Z.; Freysoldt, C.; Hennig, R.G. First-Principles Investigation of Charged Dopants and Dopant-Vacancy Defect Complexes in Monolayer MoS2. Phys. Rev. Mater. 2020, 4, 114002. [Google Scholar] [CrossRef]
- Yu, W.; Zhu, Z.; Niu, C.Y.; Li, C.; Cho, J.H.; Jia, Y. Anomalous Doping Effect in Black Phosphorene Using First-Principles Calculations. Phys. Chem. Chem. Phys. 2015, 17, 16351–16358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, S.; Meng, F.; Wang, H.; Wang, H.; Ni, Y. Novel Two-Dimensional Semiconductor SnP3: High Stability, Tunable Bandgaps and High Carrier Mobility Explored Using First-Principles Calculations. J. Mater. Chem. A 2018, 6, 11890–11897. [Google Scholar] [CrossRef]
- Jin, Y.; Li, X.; Yang, J. Single Layer of MX3 (M = Ti, Zr; X = S, Se, Te): A New Platform for Nano-Electronics and Optics. Phys. Chem. Chem. Phys. 2015, 17, 18665–18669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramzan, M.S.; Bacic, V.; Jing, Y.; Kuc, A. Electronic Properties of a New Family of Layered Materials from Groups 14 and 15: First-Principles Simulations. J. Phys. Chem. C 2019, 123, 25470–25476. [Google Scholar] [CrossRef]
- Gong, P.L.; Zhang, F.; Huang, L.F.; Zhang, H.; Li, L.; Xiao, R.C.; Deng, B.; Pan, H.; Shi, X.Q. Multifunctional Two-Dimensional Semiconductors SnP3: Universal Mechanism of Layer-Dependent Electronic Phase Transition. J. Phys. Condens. Matter 2018, 30, 475702. [Google Scholar] [CrossRef] [Green Version]
- Niu, F.; Cai, M.; Pang, J.; Li, X.; Yang, D.; Zhang, G. Gas Molecular Adsorption Effects on the Electronic and Optical Properties of Monolayer SnP3. Vacuum 2019, 168, 108823. [Google Scholar] [CrossRef]
- Du, J.; Jiang, G. First-Principle Study on Monolayer and Bilayer SnP3 Sheets as the Potential Sensors for NO2, NO, and NH3 Detection. Nanotechnology 2020, 31, 325504. [Google Scholar] [CrossRef]
- Tian, R.; Griffin, A.; McCrystall, M.; Breshears, M.; Harvey, A.; Gabbett, C.; Horváth, D.v.; Backes, C.; Jing, Y.; Heine, T.; et al. Liquid Exfoliated SnP3 Nanosheets for Very High Areal Capacity Lithium-Ion Batteries. Adv. Energy Mater. 2021, 11, 2002364. [Google Scholar] [CrossRef]
- Huang, S.; Meng, C.; Xiao, M.; Ren, S.; Wang, S.; Han, D.; Li, Y.; Meng, Y. Multi-Shell Tin Phosphide Nanospheres as High Performance Anode Material for a Sodium Ion Battery. Sustain. Energy Fuels 2017, 1, 1944–1949. [Google Scholar] [CrossRef]
- Ghosh, B.; Puri, S.; Agarwal, A.; Bhowmick, S. SnP 3: A Previously Unexplored 2D Material. J. Phys. Chem. C 2018, 122, 18185–18191. [Google Scholar] [CrossRef]
- Zhu, X.L.; Liu, P.F.; Zhang, J.; Zhang, P.; Zhou, W.X.; Xie, G.; Wang, B.T. Monolayer SnP3: An Excellent p-Type Thermoelectric Material. Nanoscale 2019, 11, 19923–19932. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.; Wang, C.; Fan, S.; Gao, G. Strain Tunable Pudding-Mold-Type Band Structure and Thermoelectric Properties of SnP3 Monolayer. J. Phys. D 2020, 127, 155103. [Google Scholar] [CrossRef] [Green Version]
- Eivari, H.A.; Sohbatzadeh, Z.; Mele, P.; Assadi, M.H.N. Low Thermal Conductivity: Fundamentals and Theoretical Aspects in Thermoelectric Applications. Mater. Today Energy 2021, 21, 100744. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab Initio Molecular-Dynamics Simulation of the Liquid-MetalAmorphous-Semiconductor Transition in Germanium. Phys. Rev. B 1994, 49, 14251–14269. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmü, J. Efficient Iterative Schemes for Ab Initio Total-Energy Calculations Using a Plane-Wave Basis Set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [Green Version]
- Monkhorst, H.J.; Pack, J.D. Special Points for Brillonin-Zone Integrations. Phys. Rev. B 1976, 13, 5188. [Google Scholar] [CrossRef]
- Zhang, R.; Yu, G.; Gao, Y.; Huang, X.; Chen, W. Applying Surface Strain and Coupling with Pure or N/B-Doped Graphene to Successfully Achieve High HER Catalytic Activity in 2D Layered SnP3-Based Nanomaterials: A First-Principles Investigation. Inorg. Chem. Front. 2020, 7, 647–658. [Google Scholar] [CrossRef]
- Liu, X.; Wang, C.Z.; Hupalo, M.; Lu, W.C.; Tringides, M.C.; Yao, Y.X.; Ho, K.M. Metals on graphene: Correlation between adatom adsorption behavior and growth morphology. Phys. Chem. Chem. Phys. 2012, 14, 9157. [Google Scholar] [CrossRef] [PubMed]
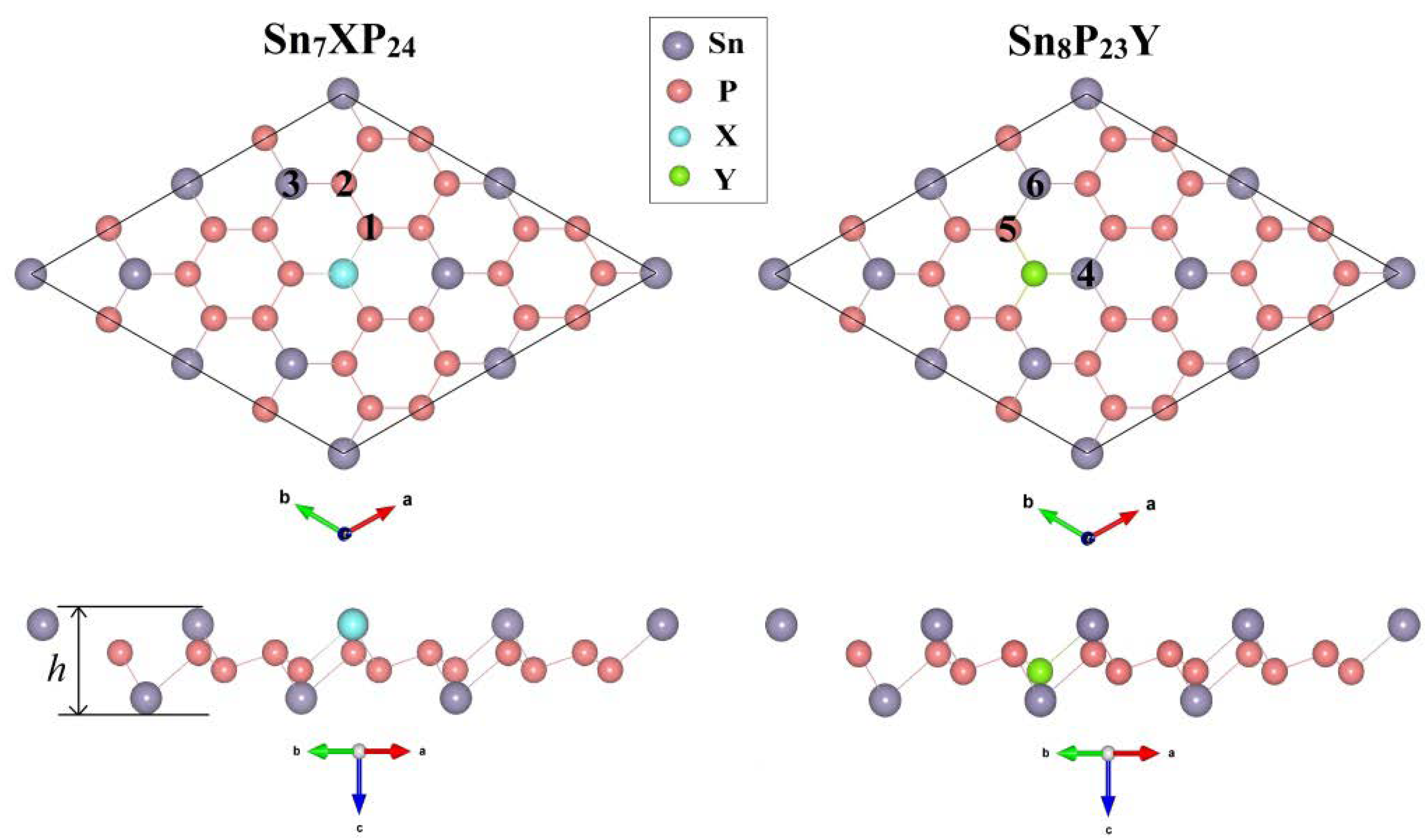
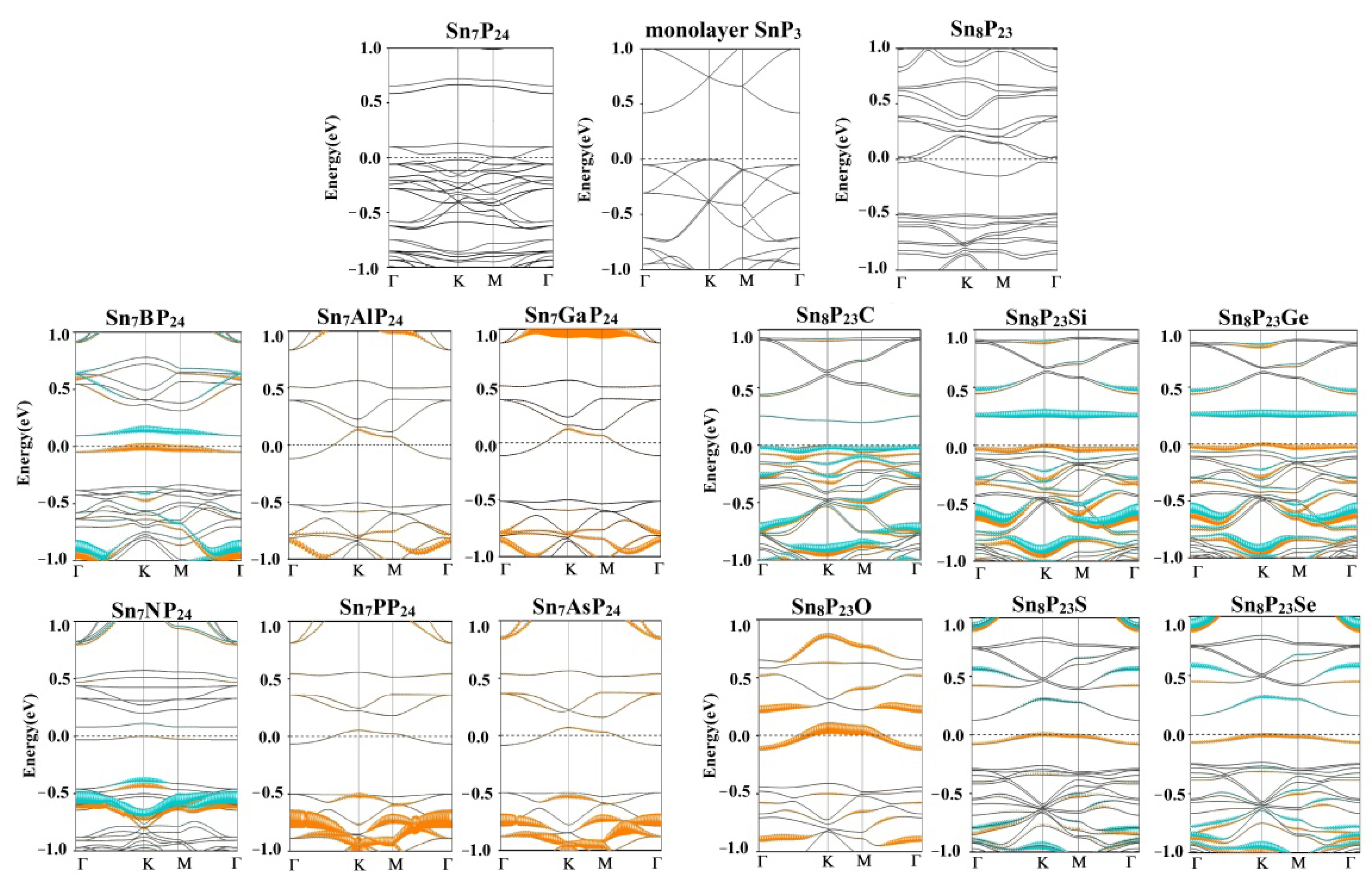
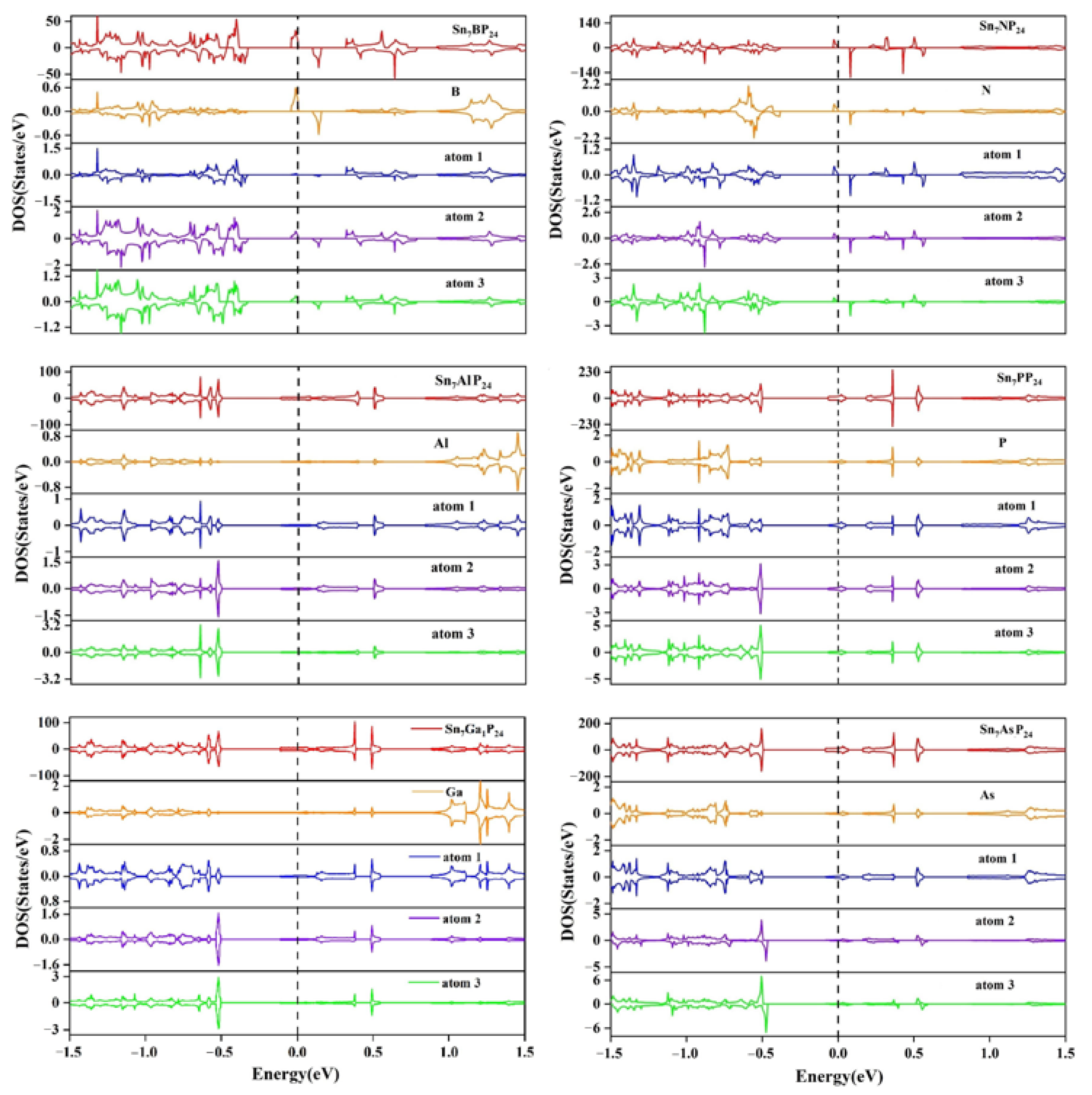
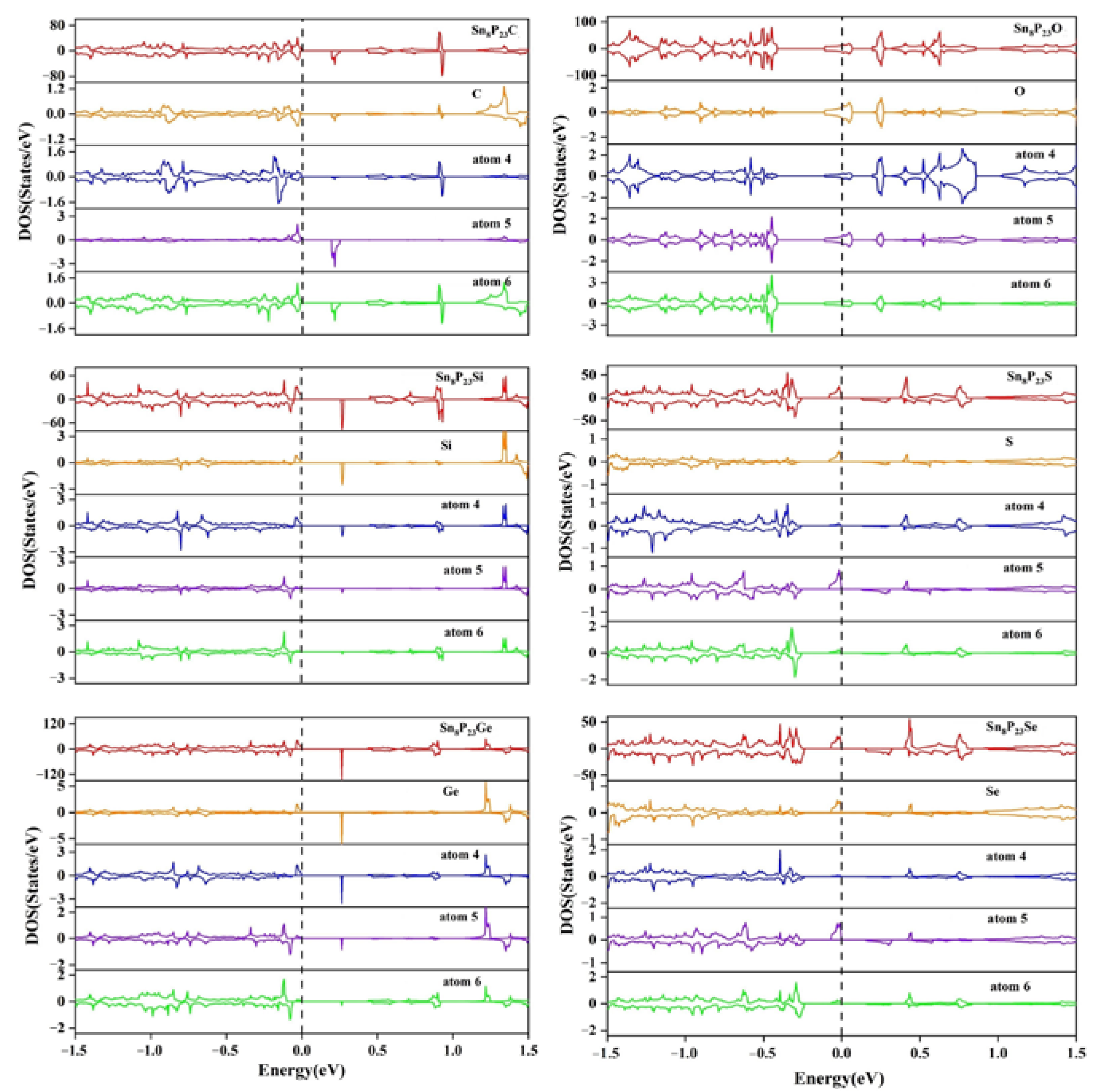

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System | a = b (Å) | Ef (eV) | d1 (Å) | d2 (Å) | h (Å) | Eg (eV) | M (μB) |
|---|---|---|---|---|---|---|---|
| SnP3 | 14.32 | / | 2.71 | 2.71 | 2.81 | 0.42 | 0.00 |
| Sn7BP24 | 14.30 | −5.662 | 1.93 | 2.71 | 2.72 | 0.09 | 1.00 |
| Sn7AlP24 | 14.32 | −3.918 | 2.32 | 2.71 | 2.85 | / | 0.00 |
| Sn7GaP24 | 14.32 | 0.592 | 2.33 | 2.71 | 2.85 | / | 0.00 |
| Sn7NP24 | 14.25 | 0.333 | 1.86 | 2.71 | 2.74 | 0.08 | 1.00 |
| Sn7PP24 | 14.25 | −1.184 | 2.32 | 2.71 | 2.76 | / | 0.00 |
| Sn7AsP24 | 14.26 | −0.699 | 2.44 | 2.71 | 2.77 | / | 0.00 |
| Sn8P23C | 14.22 | −6.609 | 1.74 | 2.35 | 2.72 | 0.21 | 1.00 |
| Sn8P23Si | 14.37 | −0.206 | 2.19 | 2.67 | 2.69 | 0.26 | 1.00 |
| Sn8P23Ge | 14.41 | 0.579 | 2.28 | 2.72 | 2.65 | 0.27 | 1.00 |
| Sn8P23O | 14.20 | −0.636 | 1.75 | 2.69 | 2.80 | / | 0.00 |
| Sn8P23S | 14.29 | 0.756 | 2.20 | 2.74 | 2.75 | 0.12 | 1.00 |
| Sn8P23Se | 14.37 | 0.661 | 2.35 | 2.83 | 2.72 | 0.16 | 1.00 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, N.; Li, X.; Ruan, S.; Chen, X.; Li, S.; Hu, T. First-Principles Study of Electronic Properties of Substitutionally Doped Monolayer SnP3. Materials 2022, 15, 2462. https://doi.org/10.3390/ma15072462
Zhang N, Li X, Ruan S, Chen X, Li S, Hu T. First-Principles Study of Electronic Properties of Substitutionally Doped Monolayer SnP3. Materials. 2022; 15(7):2462. https://doi.org/10.3390/ma15072462
Chicago/Turabian StyleZhang, Ningxia, Xiaodan Li, Shihao Ruan, Xiong Chen, Shenghao Li, and Taotao Hu. 2022. "First-Principles Study of Electronic Properties of Substitutionally Doped Monolayer SnP3" Materials 15, no. 7: 2462. https://doi.org/10.3390/ma15072462
APA StyleZhang, N., Li, X., Ruan, S., Chen, X., Li, S., & Hu, T. (2022). First-Principles Study of Electronic Properties of Substitutionally Doped Monolayer SnP3. Materials, 15(7), 2462. https://doi.org/10.3390/ma15072462