
First-Principles Study on Possible Half-Metallic Ferrimagnetism in Double Perovskites Pb2XX′O6 (X = Ti, Zr, Hf, V, Nb and Ta, X′ = Tc, Ru, Os and Rh)


Abstract
:1. Introduction
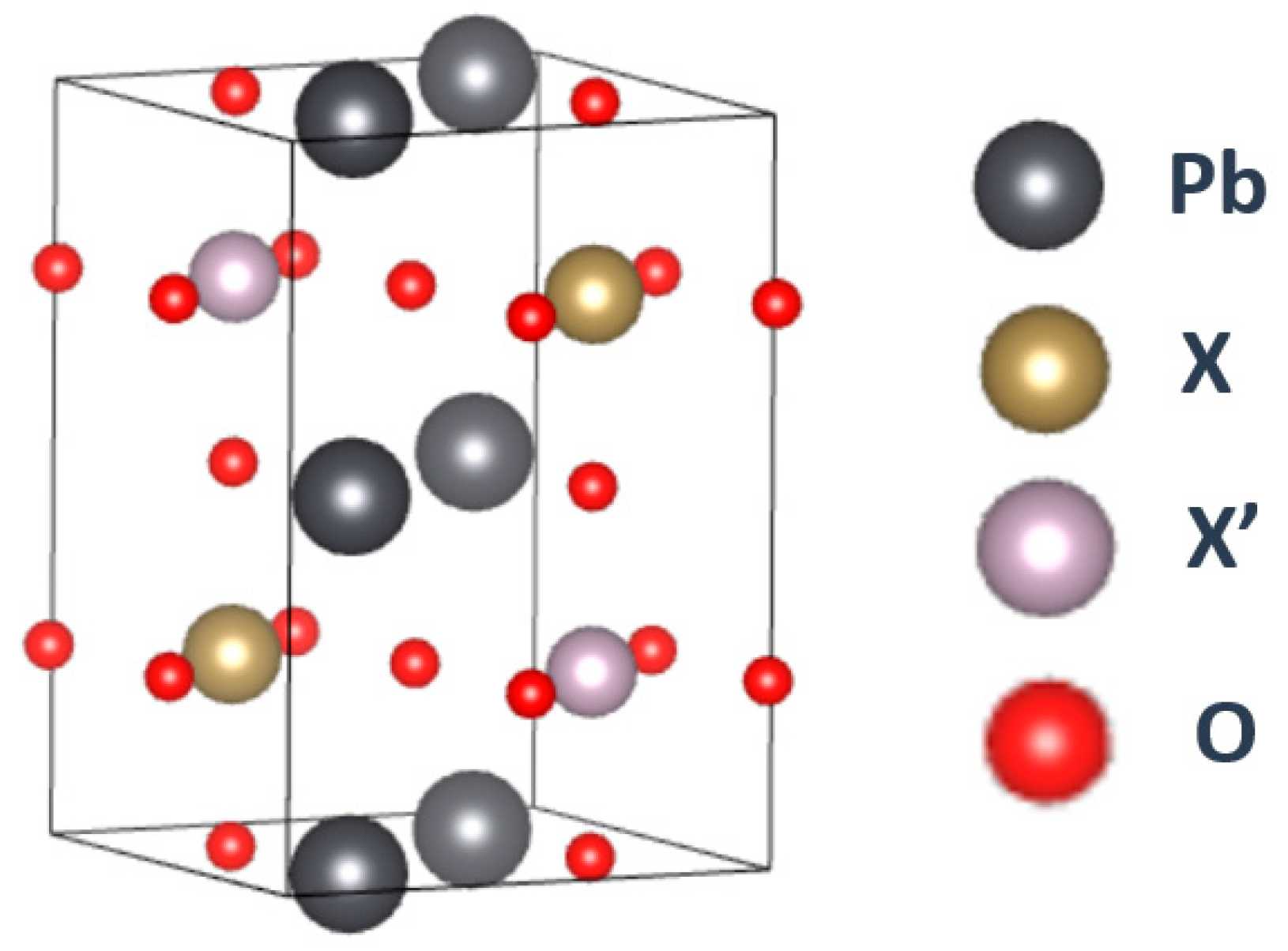
2. Materials and Methods
3. Results and Discussion
3.1. FiM-HM Compounds: Pb2XTcO6 (X = Nb and Ta)
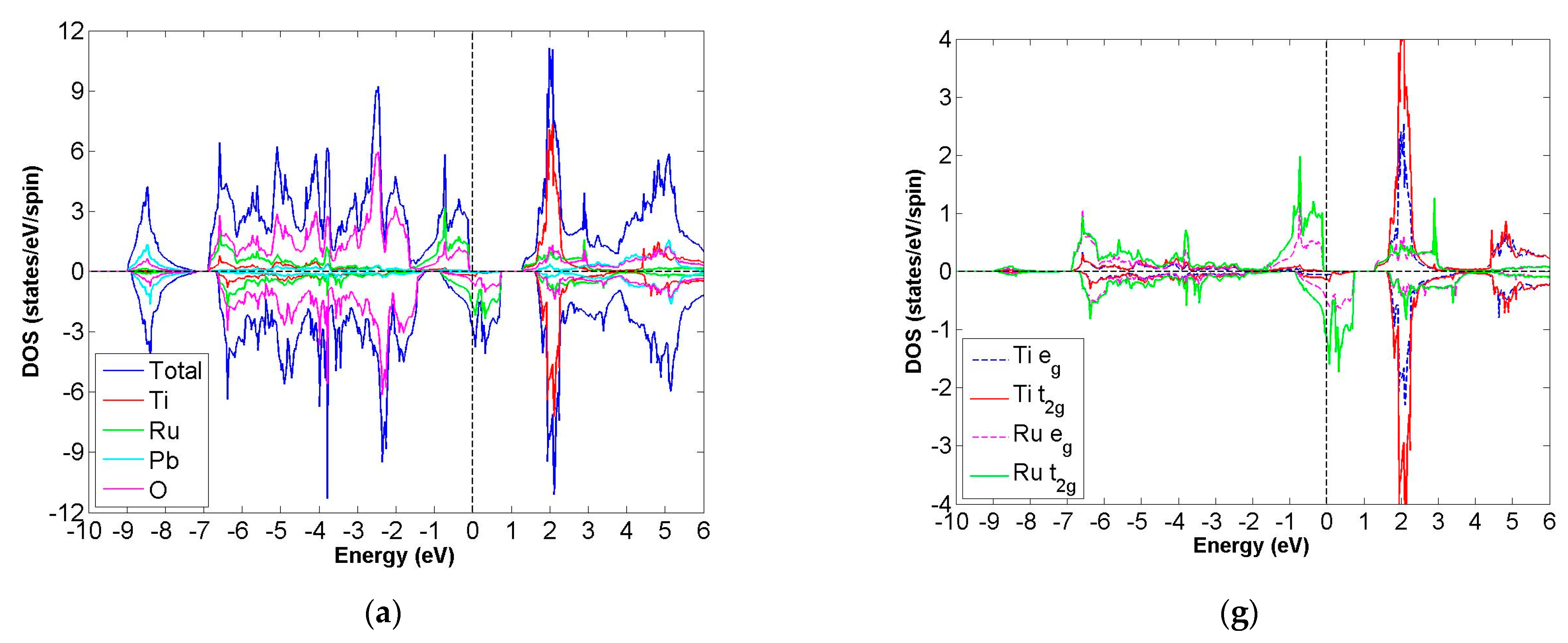
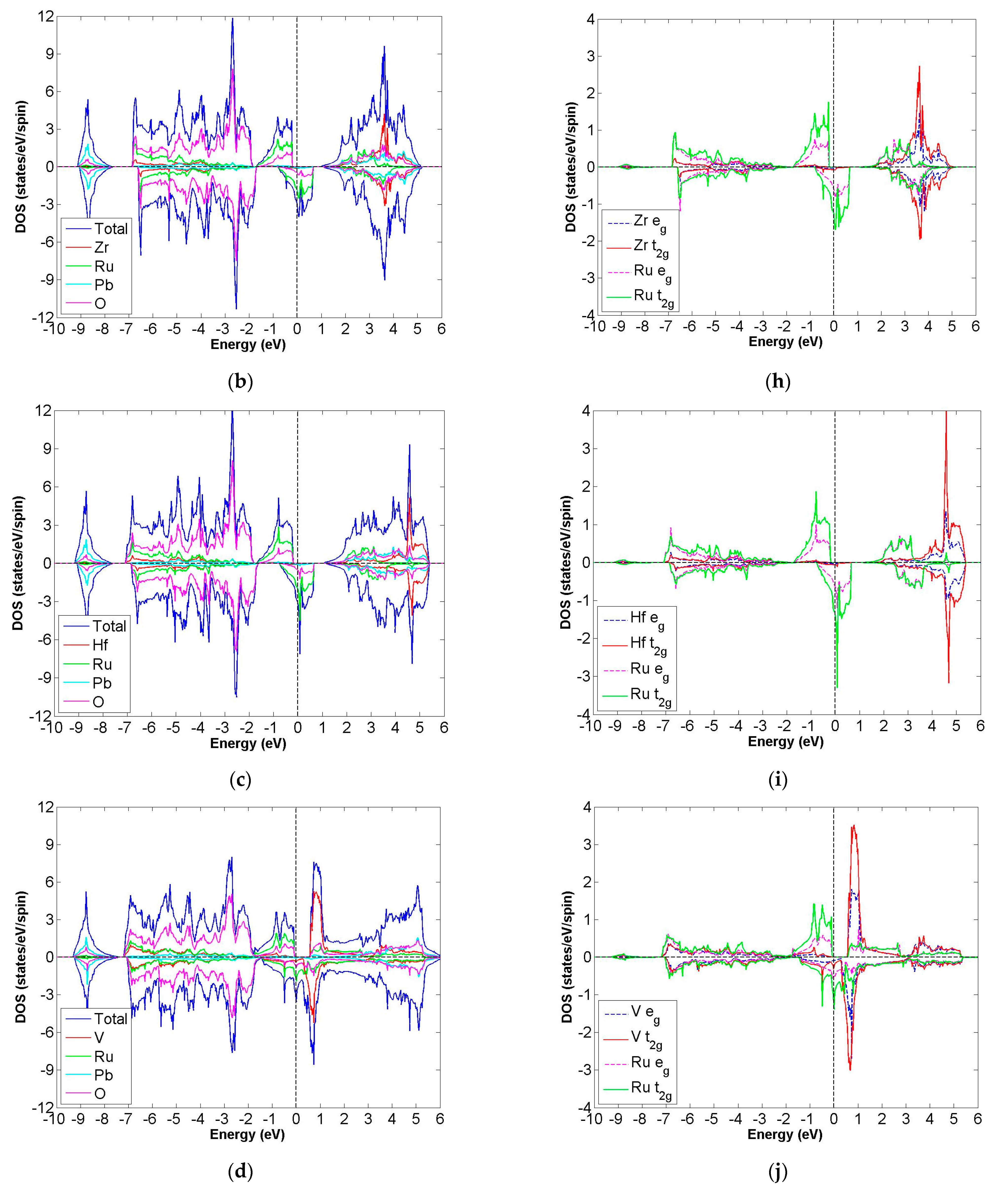
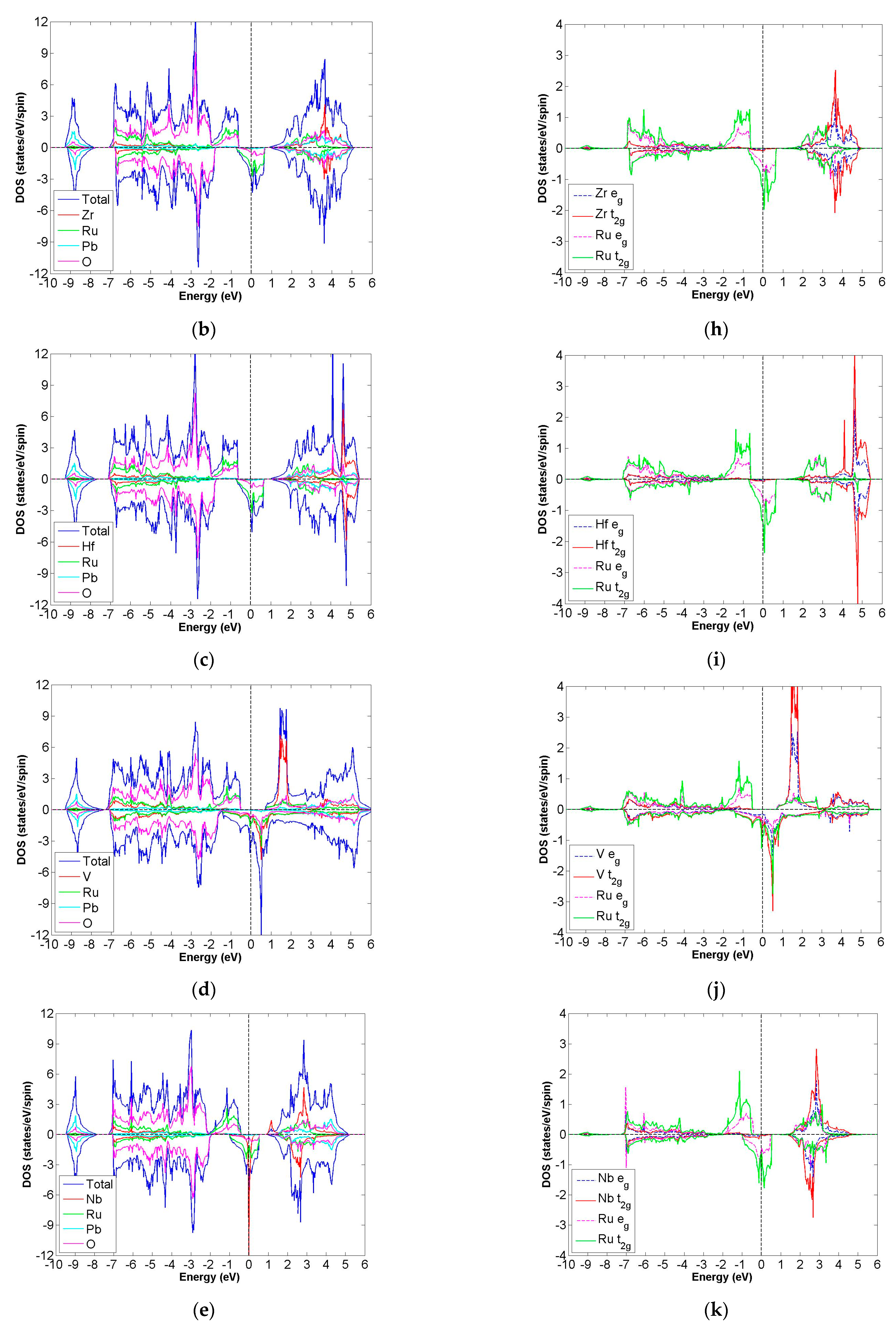
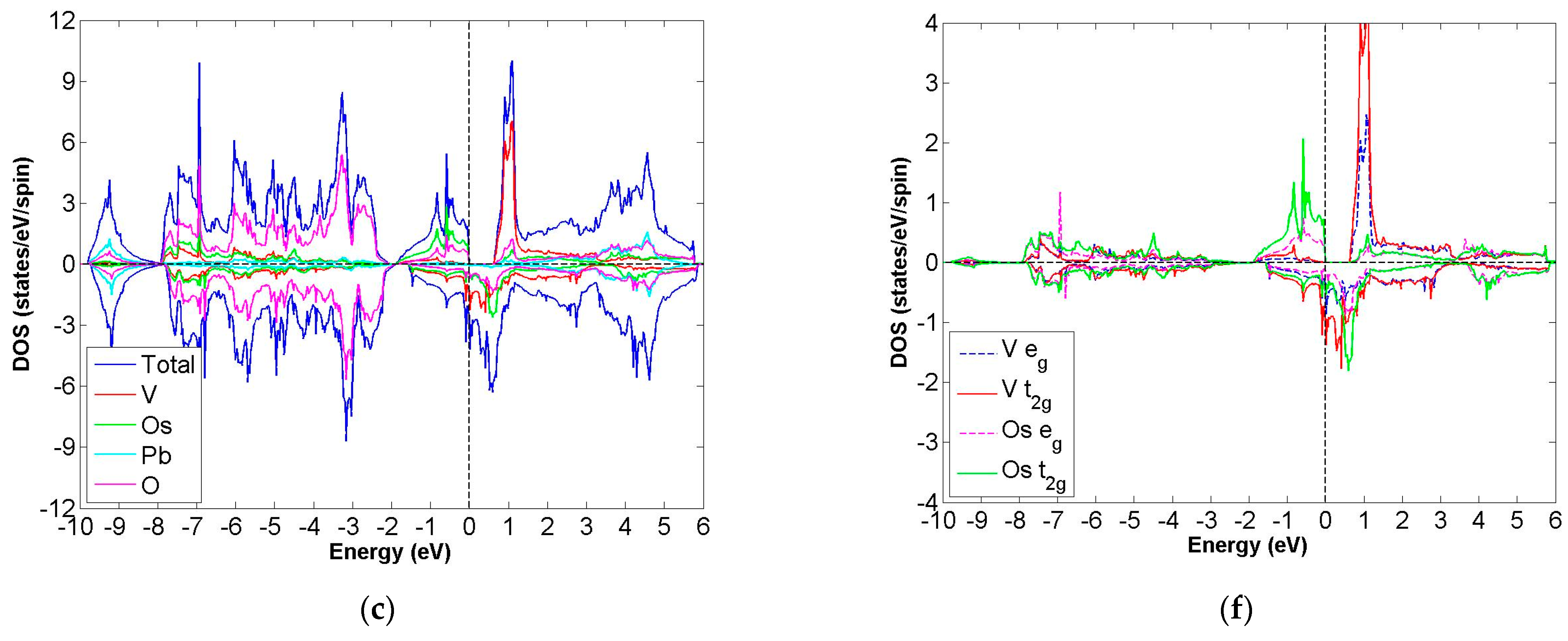
3.2. FiM-HM Compounds: Pb2XRuO6 (X = Ti, Zr, Hf, V, Nb and Ta)
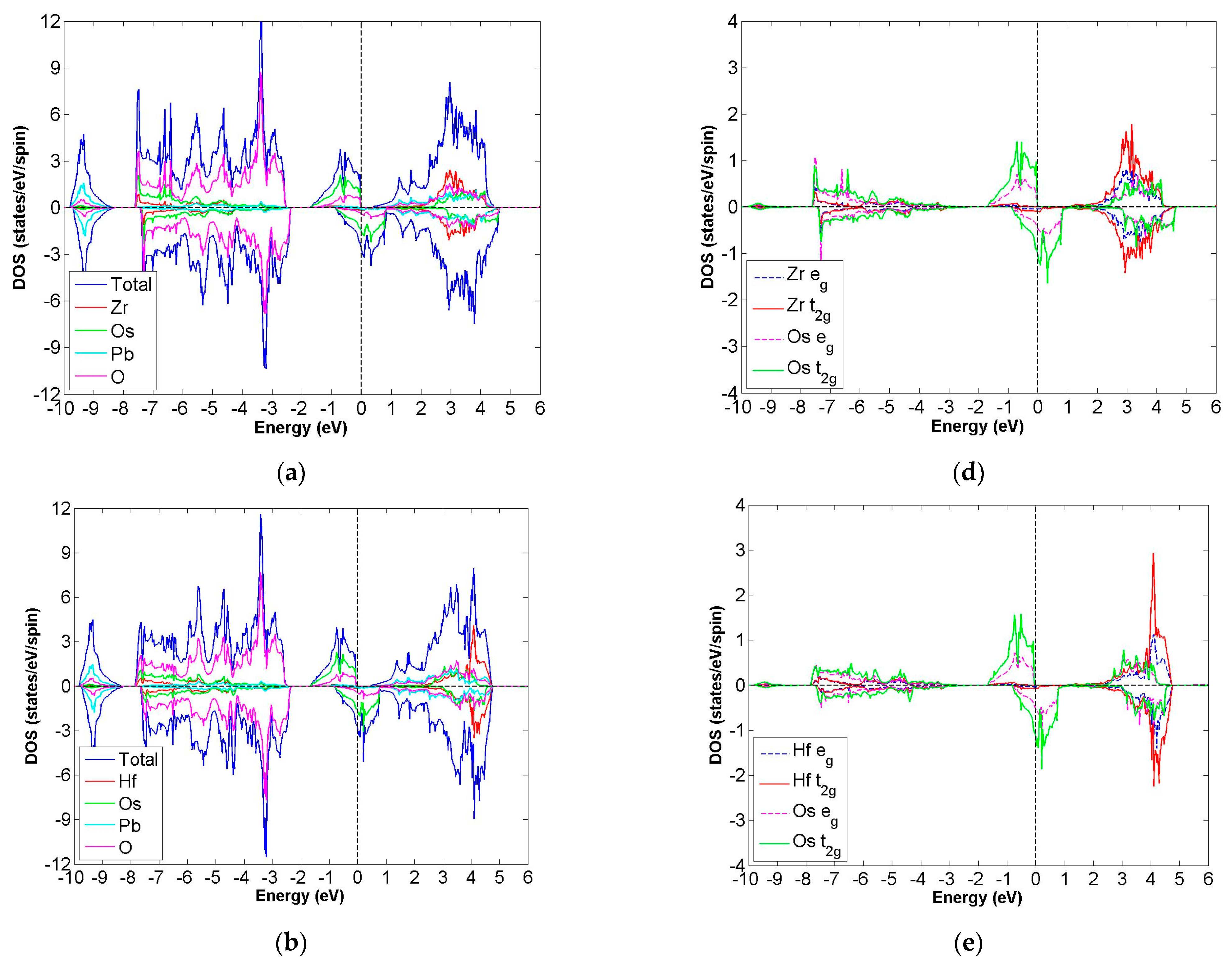
3.3. FiM-HM Compounds: Pb2XOsO6 (X = Zr, Hf and V)
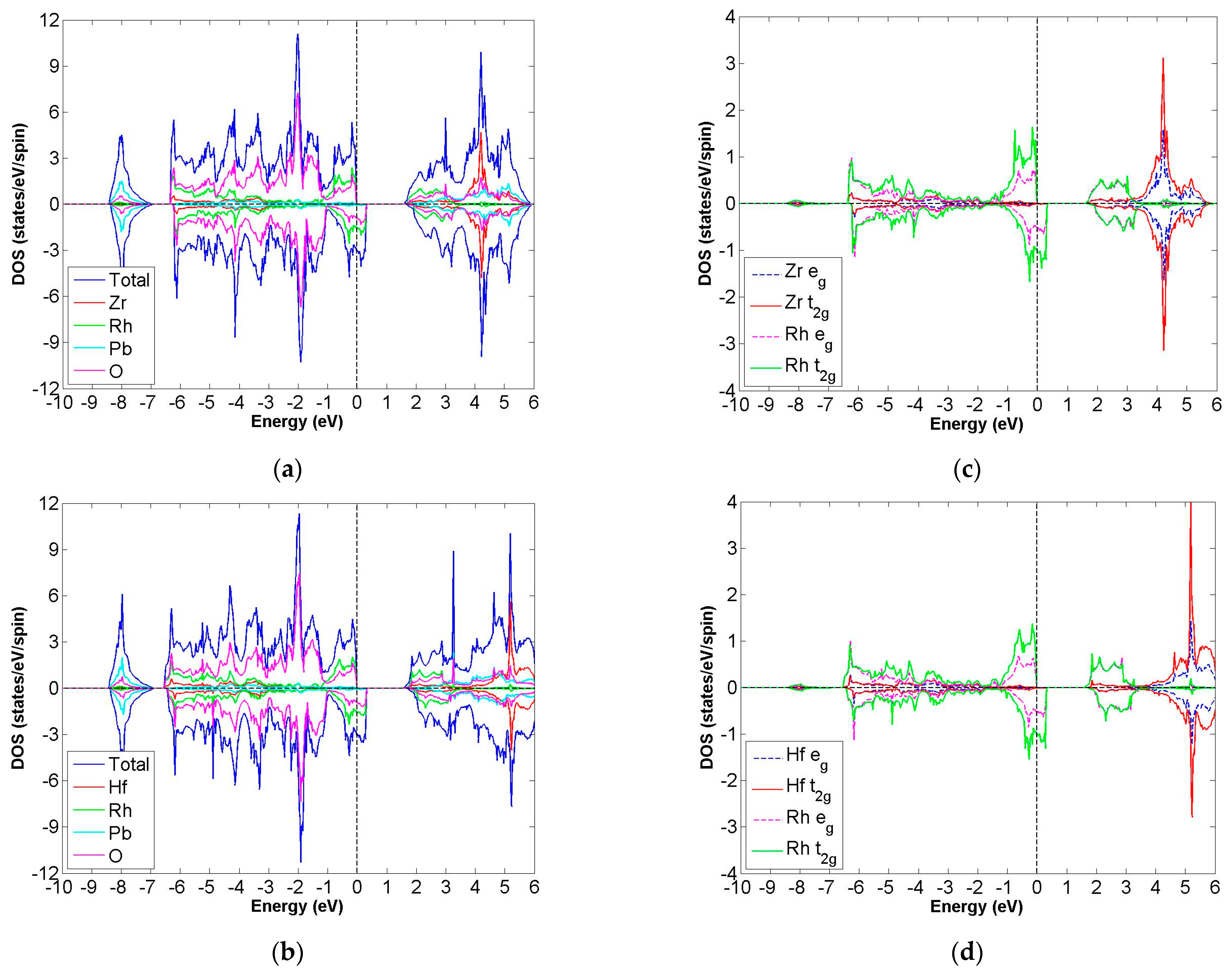
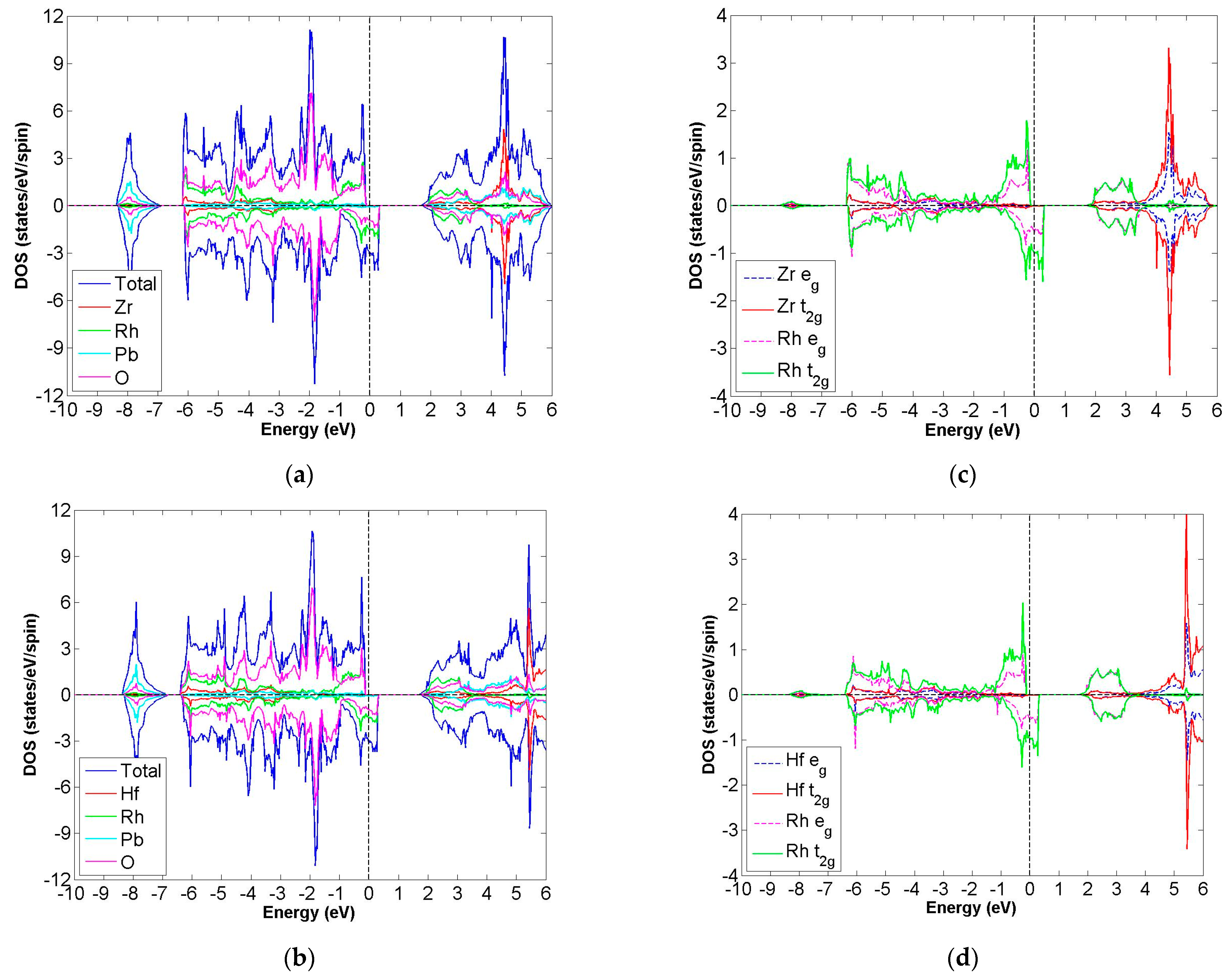
3.4. FiM-HM Compounds: Pb2XRhO6 (X = Zr and Hf)
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Prinz, G.A. Magnetoelectronics. Science 1998, 282, 1660–1663. [Google Scholar] [CrossRef] [PubMed]
- Wolf, S.A.; Awschalom, D.D.; Buhrman, R.A.; Daughton, J.M.; von Molnár, V.S.; Roukes, M.L.; Chtchelkanova, A.Y.; Treger, D.M. Spintronics: A spin-based electronics vision for the future. Science 2001, 294, 1488–1495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Žutić, I.; Fabian, J.; Sarma, S.D. Spintronics: Fundamentals and applications. Rev. Mod. Phys. 2004, 76, 323. [Google Scholar] [CrossRef] [Green Version]
- De Groot, R.A.; Mueller, F.M.; Van Engen, P.G.; Buschow, K.H.J. New class of materials: Half-metallic ferromagnets. Phys. Rev. Lett. 1983, 50, 2024. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, K.I.; Kimura, T.; Sawada, H.; Terakura, K.; Tokura, Y. Room-temperature magnetoresistance in an oxide material with an ordered double-perovskite structure. Nature 1998, 395, 677–680. [Google Scholar] [CrossRef]
- Pickett, W.E.; Moodera, J.S. Half metallic magnets. Phys. Today 2001, 54, 39–45. [Google Scholar] [CrossRef]
- Lewis, S.P.; Allen, P.B.; Sasaki, T. Band structure and transport properties of CrO2. Phys. Rev. B 1997, 55, 10253. [Google Scholar] [CrossRef] [Green Version]
- Jedema, F.J.; Filip, A.T.; Van Wees, B.J. Electrical spin injection and accumulation at room temperature in an all-metal mesoscopic spin valve. Nature 2001, 410, 345–348. [Google Scholar] [CrossRef]
- Jeng, H.T.; Guo, G.Y. First-principles investigations of orbital magnetic moments and electronic structures of the double perovskites Sr2FeMoO6, Sr2FeReO6, and Sr2CrWO6. Phys. Rev. B 2003, 67, 094438. [Google Scholar] [CrossRef]
- Chan, T.S.; Liu, R.S.; Guo, G.Y.; Hu, S.F.; Lin, J.G.; Chen, J.M.; Chang, C.R. Effects of B′-site transition metal on the properties of double perovskites Sr2FeMO6 (M = Mo, W): B′ 4d–5d system. Solid State Commun. 2005, 133, 265–270. [Google Scholar] [CrossRef]
- Wu, H. Electronic structure study of double perovskites A2FeReO6 (A = Ba, Sr, Ca) and Sr2MMoO6 (M = Cr, Mn, Fe, Co) by LSDA and LSDA+U. Phys. Rev. B 2001, 64, 125126. [Google Scholar] [CrossRef] [Green Version]
- Kato, H.; Okuda, T.; Okimoto, Y.; Tomioka, Y.; Oikawa, K.; Kamiyama, T.; Tokura, Y. Structural and electronic properties of the ordered double perovskites A2MReO6 (A = Sr, Ca; M = Mg, Sc, Cr, Mn, Fe, Co, Ni, Zn). Phys. Rev. B 2004, 69, 184412. [Google Scholar] [CrossRef]
- Wang, Y.K.; Lee, P.H.; Guo, G.Y. Half-metallic antiferromagnetic nature of La2VTcO6 and La2VCuO6 from ab initio calculations. Phys. Rev. B 2009, 80, 224418. [Google Scholar] [CrossRef]
- Chen, S.H.; Xiao, Z.R.; Liu, Y.P.; Wang, Y.K. Investigation of possible half-metallic antiferromagnets on double perovskites LaABB′O6 (A = Ca, Sr, Ba; B, B′ = transition elements). J. Appl. Phys. 2010, 108, 093908. [Google Scholar] [CrossRef]
- Weng, K.C.; Wang, Y.K. Electronic structure of half-metal antiferromagnetism in double-perovskite BiPbVRuO6 and BiPbVOsO6. J. Phys. Soc. Jpn. 2014, 83, 054715. [Google Scholar] [CrossRef]
- Lin, H.Z.; Hu, C.Y.; Lee, P.H.; Yan, A.Z.Z.; Wu, W.F.; Chen, Y.F.; Wang, Y.K. Half-Metallic Property Induced by Double Exchange Interaction in the Double Perovskite Bi2BB′O6 (B, B′ = 3d Transitional Metal) via First-Principles Calculations. Materials 2019, 12, 1844. [Google Scholar] [CrossRef] [Green Version]
- Coey, J.M.D.; Viret, M.; Von Molnár, S. Mixed-valence manganites. Adv. Phys. 1999, 48, 167–293. [Google Scholar] [CrossRef]
- Park, J.H.; Vescovo, E.; Kim, H.J.; Kwon, C.; Ramesh, R.; Venkatesan, T. Magnetic properties at surface boundary of a half-metallic ferromagnet La0.7Sr0.3MnO3. Phys. Rev. Lett. 1998, 81, 1953. [Google Scholar] [CrossRef]
- Kronik, L.; Jain, M.; Chelikowsky, J.R. Electronic structure and spin polarization of MnxGa1-xN. Phys. Rev. B 2002, 66, 041203. [Google Scholar] [CrossRef]
- Chen, Z.; Tan, S.; Yang, Z.; Zhang, Y. Evidence for a non-double-exchange mechanism in FeCr2S4. Phys. Rev. B 1999, 59, 11172. [Google Scholar] [CrossRef]
- Park, M.S.; Kwon, S.K.; Youn, S.J.; Min, B.I. Half-metallic electronic structures of gaint magnetoresistive spinels: Fe1-xCuxCr2S4 (x = 0.0, 0.5, 1.0). Phys. Rev. B 1999, 59, 10018. [Google Scholar] [CrossRef] [Green Version]
- Shirai, M.; Ogawa, T.; Kitagawa, I.; Suzuki, N. Band structures of zinc-blende-type MnAs and (MnAs)1(GaAs)1 superlattice. J. Magn. Magn. Mater. 1998, 177, 1383–1384. [Google Scholar] [CrossRef]
- Park, J.H.; Kwon, S.K.; Min, B.I. Electronic structures of III–V based ferromagnetic semiconductors: Half-metallic phase. Phys. B Condens. Matter 2000, 281, 703–704. [Google Scholar] [CrossRef]
- Battle, P.D.; Goodenough, J.B.; Price, R. The crystal structures and magnetic properties of Ba2LaRuO6 and Ca2LaRuO6. J. Solid State Chem. 1983, 46, 234–244. [Google Scholar] [CrossRef]
- Battle, P.D.; Jones, C.W. The crystal and magnetic structures of Sr2LuRuO6, Ba2YRuO6, and Ba2LuRuO6. J. Solid State Chem. 1989, 78, 108–116. [Google Scholar] [CrossRef]
- Ortega-San Martin, L.; Chapman, J.P.; Lezama, L.; Sánchez-Marcos, J.; Rodríguez-Fernández, J.; Arriortua, M.I.; Rojo, T. Factors determining the effect of Co (ii) in the ordered double perovskite structure: Sr2CoTeO6. J. Mater. Chem. 2005, 15, 183–193. [Google Scholar] [CrossRef]
- Izumiyama, Y.; Doi, Y.; Wakeshima, M.; Hinatsu, Y.; Shimojo, Y.; Morii, Y. Magnetic properties of the antiferromagnetic double perovskite Ba2PrRuO6. J. Phys. Condens. Matter 2001, 13, 1303–1313. [Google Scholar] [CrossRef]
- Li, M.R.; Hodges, J.P.; Retuerto, M.; Deng, Z.; Stephens, P.W.; Croft, M.C.; Deng, X.; Kotliar, G.; Sánchez-Benítez, J.; Walker, D.; et al. Mn2MnReO6: Synthesis and magnetic structure determination of a new transition-metal-only double perovskite canted antiferromagnet. Chem. Mater. 2016, 28, 3148–3158. [Google Scholar] [CrossRef]
- Zhu, W.K.; Lu, C.K.; Tong, W.; Wang, J.M.; Zhou, H.D.; Zhang, S.X. Strong ferromagnetism induced by canted antiferromagnetic order in double perovskite iridates (La1-xSrx)2ZnIrO6. Phys. Rev. B 2015, 91, 144408. [Google Scholar] [CrossRef] [Green Version]
- Li, M.R.; Retuerto, M.; Deng, Z.; Stephens, P.W.; Croft, M.; Huang, Q.; Wu, H.; Deng, X.; Kotliar, G.; Sánchez-Benítez, J.; et al. Giant Magnetoresistance in the Half-Metallic Double-Perovskite Ferrimagnet Mn2FeReO6. Angew. Chem. Int. Ed. 2015, 54, 12069–12073. [Google Scholar] [CrossRef]
- Arévalo-López, A.M.; McNally, G.M.; Attfield, J.P. Large Magnetization and Frustration Switching of Magnetoresistance in the Double-Perovskite Ferrimagnet Mn2FeReO6. Angew. Chem. Int. Ed. 2015, 54, 12074–12077. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhu, S.; Ou, X.; Wu, H. Ferrimagnetism in the double perovskite Ca2FeOsO6: A density functional study. Phys. Rev. B 2014, 90, 054406. [Google Scholar] [CrossRef] [Green Version]
- Demazeau, G.; Siberchicot, B.; Matar, S.; Gayet, C.; Largeteau, A. A new ferromagnetic oxide La2MnIrO6: Synthesis, characterization, and calculation of its electronic structure. J. Appl. Phys. 1994, 75, 4617–4620. [Google Scholar] [CrossRef]
- Serrate, D.; De Teresa, J.M.; Ibarra, M.R. Double perovskites with ferromagnetism above room temperature. J. Phys. Condens. Matter 2006, 19, 023201. [Google Scholar] [CrossRef]
- Attfield, J.P. Structure–property relations in doped perovskite oxides. Int. J. Inorg. Mater. 2001, 3, 1147–1152. [Google Scholar] [CrossRef]
- Karppinen, M.; Yamauchi, H. Chemistry of halfmetallic and related cation-ordered double perovskites. In Frontiers in Magnetic Materials; Narlikar, A.V., Ed.; Springer: Berlin/Heidelberg, Germany, 2005; pp. 153–184. [Google Scholar]
- King, G.; Woodward, P.M. Cation ordering in perovskites. J. Mater. Chem. 2010, 20, 5785–5796. [Google Scholar] [CrossRef]
- Kwon, O.; Kim, Y.I.; Kim, K.; Kim, J.C.; Lee, J.H.; Park, S.S.; Han, J.W.; Kim, Y.M.; Kim, G.; Jeong, H.Y. Probing One-Dimensional Oxygen Vacancy Channels Driven by Cation–Anion Double Ordering in Perovskites. Nano Lett. 2020, 20, 8353–8359. [Google Scholar] [CrossRef]
- Artner, C.; Weil, M. Lead (II) oxidotellurates (VI) with double perovskite structures. J. Solid State Chem. 2019, 276, 75–86. [Google Scholar] [CrossRef]
- Goldschmidt, V.M. Die gesetze der krystallochemie. Naturwissenschaften 1926, 14, 477–485. [Google Scholar] [CrossRef]
- Liu, Y.P.; Fuh, H.R.; Wang, Y.K. First-principles study of half-metallic materials in double-perovskite A2FeMO6 (M = Mo, Re, and W) with IVA group elements set on the A-site position. J. Phys. Chem. C 2012, 116, 18032–18037. [Google Scholar] [CrossRef]
- Fuh, H.R.; Weng, K.C.; Liu, Y.P.; Wang, Y.K. Theoretical prediction of a new type of half-metallic materials in double derovskite Pb2BB′O6 (B, B′= 3d Transition Metal) via first-principle calculations. Spin 2014, 4, 1450001. [Google Scholar] [CrossRef]
- Shimakawa, Y.; Azuma, M.; Ichikawa, N. Multiferroic compounds with double-perovskite structures. Materials 2011, 4, 153–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuh, H.R.; Liu, Y.P.; Wang, Y.K. First principle research of possible HM-AFM in double perovskites A2MoOsO6 and A2TcReO6 (A = Si, Ge, Sn, and Pb) with group IVA elements set on the A-site position. Solid State Sci. 2013, 19, 94–98. [Google Scholar] [CrossRef]
- Rached, H.; Bendaoudia, S.; Rached, D. Investigation of Iron-based double perovskite oxides on the magnetic phase stability, mechanical, electronic and optical properties via first-principles calculation. Mater. Chem. Phys. 2017, 193, 453–469. [Google Scholar] [CrossRef]
- Fuh, H.R.; Liu, Y.P.; Chen, S.H.; Wang, Y.K. Electronic structures of compensated magnetism in double perovskites A2CrRu(Os)O6 (A = Si, Ge, Sn, and Pb) from ab initio calculations. J. Alloys Compd. 2013, 547, 126–131. [Google Scholar] [CrossRef]
- Anderson, M.T.; Greenwood, K.B.; Taylor, G.A.; Poeppelmeier, K.R. B-cation arrangements in double perovskites. Prog. Solid State Chem. 1993, 22, 197–233. [Google Scholar] [CrossRef]
- Deng, Z.Q.; Smit, J.P.; Niu, H.J.; Evans, G.; Li, M.R.; Xu, Z.L.; Claridge, J.B.; Rosseinsky, M.J. B cation ordered double perovskite Ba2CoMo0.5Nb0.5O6-δ as a potential SOFC cathode. Chem. Mater. 2009, 21, 5154–5162. [Google Scholar] [CrossRef]
- Glazer, A.M. Simple ways of determining perovskite structures. Acta Crystallogr. A 1975, 31, 756–762. [Google Scholar] [CrossRef] [Green Version]
- Dowben, P.A.; Skomski, R. Are half metallic ferromagnets half metals? J. Appl. Phys. 2004, 95, 7453. [Google Scholar] [CrossRef] [Green Version]
- Kohn, W.; Sham, L.J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 1965, 140, A1133. [Google Scholar] [CrossRef] [Green Version]
- Fuoco, L.; Rodriguez, D.; Peppel, T.; Maggard, P.A. Molten-Salt-Mediated Syntheses of Sr2FeReO6, Ba2FeReO6, and Sr2CrReO6: Particle Sizes, B/B′ Site Disorder, and Magnetic Properties. Chem. Mater. 2011, 23, 5409–5414. [Google Scholar] [CrossRef]
- Rai, D.P.; Shankar, A.; Ghimire, M.P.; Sandeep; Thapa, R.K. The electronic, magnetic and optical properties of double perovskite A2FeReO6 (A = Sr, Ba) from first principles approach. Comput. Mater. Sci. 2015, 101, 313–320. [Google Scholar] [CrossRef]
- Ivanov, S.A.; Bush, A.A.; Stash, A.I.; Kamentsev, K.E.; Shkuratov, V.Y.; Kvashnin, Y.O.; Autieri, C.; Marco, I.D.; Sanyal, B.; Eriksson, O.; et al. Polar Order and Frustrated Antiferromagnetism in Perovskite Pb2MnWO6 Single Crystals. Inorg. Chem. 2016, 55, 2791–2805. [Google Scholar] [CrossRef] [PubMed]
- Galasso, F.; Pyle, J. Ordering in compounds of the A(B’0.33Ta0.67) O3 type. Inorg. Chem. 1963, 2, 482–484. [Google Scholar] [CrossRef]
- Nakamura, T.; Choy, J.H. Determination of ionic valency pairs via lattice constants in ordered perovskites (ALa)(Mn2+ Mo5+) O6 (A= Ba, Sr, Ca) with applications to (ALa)(Fe3+Mo4+) O6, Ba2(Bi3+Bi5+) O6 and Ba2(Bi3+Sb5+) O6. J. Solid State Chem. 1977, 20, 233–244. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anisimov, V.I.; Zaanen, J.; Andersen, O.K. Band theory and Mott insulators: Hubbard U instead of Stoner, I. Phys. Rev. B 1991, 44, 943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solovyev, I.V.; Dederichs, P.H.; Anisimov, V.I. Corrected atomic limit in the local-density approximation and the electronic structure of d impurities in Rb. Phys. Rev. B 1994, 50, 16861. [Google Scholar] [CrossRef]
- Zener, C. Interaction between the d-shells in the transition metals. II. Ferromagnetic compounds of manganese with perovskite structure. Phys. Rev. 1951, 82, 403. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953. [Google Scholar] [CrossRef] [Green Version]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for open-shell transition metals. Phys. Rev. B 1993, 48, 13115. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Materials Pb2XX′O6 | Spin Magnetic Moment (μB/f.u.) | d Orbital Electrons ↑/↓ | Band Gap (eV) | ΔE (meV/f.u.) FiM-AF | ||||
|---|---|---|---|---|---|---|---|---|
| mX | mX′ | mtot | X | X′ | ||||
| NbTc | (0,0) | −1.527 | 1.846 | 2.000 | 1.016/1.112 | 3.200/1.375 | 0.47/0.00 | −21 |
| (2,2) | −1.886 | 1.980 | 2.000 | 0.913/1.034 | 3.251/1.292 | 0.95/0.00 | −26 | |
| TaTc | (0,0) | −0.072 | 1.588 | 2.000 | 1.054/1.119 | 3.099/1.527 | 0.30/0.00 | −96 |
| (2,2) | −0.116 | 1.691 | 2.011 | 1.004/1.115 | 3.141/1.467 | 0.80/0.00 | −178 | |
| TiRu | (0,0) | −0.072 | 1.348 | 2.000 | 0.955/1.019 | 3.675/2.341 | 1.35/0.00 | −43 |
| (2,2) | −0.098 | 1.378 | 2.000 | 0.916/1.008 | 3.691/2.329 | 1.80/0.00 | −24 | |
| ZrRu | (0,0) | −0.031 | 1.386 | 2.000 | 0.747/0.771 | 3.696/2.325 | 1.18/0.00 | −55 |
| (2,2) | −0.036 | 1.406 | 2.000 | 0.720/0.750 | 3.708/2.317 | 1.52/0.00 | −106 | |
| HfRu | (0,0) | −0.028 | 1.379 | 2.000 | 0.784/0.802 | 3.690/2.326 | 1.25/0.00 | −50 |
| (2,2) | −0.029 | 1.394 | 2.000 | 0.758/0.780 | 3.699/2.321 | 1.60/0.00 | −92 | |
| VRu | (0,0) | −0.364 | 0.906 | 1.000 | 1.614/1.964 | 3.492/2.598 | 0.62/0.00 | −18 |
| (3,2) | −0.875 | 1.174 | 1.000 | 1.341/2.193 | 3.613/2.452 | 1.23/0.00 | −139 | |
| NbRu | (0,0) | −1.283 | 0.999 | 1.000 | 1.046/1.150 | 3.549/2.562 | 1.00/0.00 | −49 |
| (2,2) | −1.531 | 1.052 | 1.000 | 0.960/1.081 | 3.578/2.538 | 1.30/0.00 | −115 | |
| TaRu | (0,0) | −0.041 | 0.761 | 1.000 | 1.062/1.099 | 3.453/2.700 | 0.85/0.00 | −22 |
| (2,2) | −0.051 | 0.767 | 1.000 | 1.029/1.077 | 3.466/2.706 | 1.12/0.00 | −60 | |
| ZrOs | (0,0) | −0.030 | 1.372 | 2.000 | 0.746/0.772 | 3.386/2.032 | 0.38/0.00 | −27 |
| (2,2) | −0.044 | 1.462 | 2.000 | 0.714/0.756 | 3.442/1.998 | 0.75/0.00 | −112 | |
| HfOs | (0,0) | −0.025 | 1.372 | 2.000 | 0.782/0.800 | 3.386/2.032 | 0.47/0.00 | −28 |
| (2,2) | −0.032 | 1.452 | 2.000 | 0.752/0.779 | 3.436/2.002 | 0.82/0.00 | −97 | |
| VOs | (0,0) | −0.837 | 1.218 | 1.000 | 1.366/2.182 | 3.310/2.109 | 0.65/0.00 | −30 |
| (3,2) | −1.457 | 1.629 | 1.000 | 1.050/2.475 | 3.492/1.884 | 1.65/0.00 | −309 | |
| ZrRh | (0,0) | −0.021 | 0.557 | 1.000 | 0.752/0.769 | 3.700/3.147 | 1.60/0.00 | −10 |
| (2,2) | −0.021 | 0.529 | 1.000 | 0.728/0.746 | 3.968/3.172 | 1.82/0.00 | −36 | |
| HfRh | (0,0) | −0.020 | 0.548 | 1.000 | 0.788/0.802 | 3.695/3.150 | 1.57/0.00 | −8 |
| (2,2) | −0.019 | 0.517 | 1.000 | 0.764/0.779 | 3.690/3.175 | 1.82/0.00 | −34 | |
| Materials Pb2XX′O6 | Final States | E (eV/f.u.) | Materials Pb2XX′O6 | Final States | E (eV/f.u.) | ||
|---|---|---|---|---|---|---|---|
| NbTc | (0,0) | AF | −74.269 | TaRu | (0,0) | AF | −74.610 |
| (2,2) | AF | −72.512 | (2,2) | AF | −72.606 | ||
| (0,0) | FiM | −74.290 | (0,0) | FiM | −74.632 | ||
| (2,2) | FiM | −72.538 | (2,2) | FiM | −72.666 | ||
| TaTc | (0,0) | AF | −76.443 | ZrOs | (0,0) | AF | −73.595 |
| (2,2) | AF | −74.486 | (2,2) | AF | −71.755 | ||
| (0,0) | FiM | −76.539 | (0,0) | FiM | −73.622 | ||
| (2,2) | FiM | −74.664 | (2,2) | FiM | −71.867 | ||
| TiRu | (0,0) | AF | −70.600 | HfOs | (0,0) | AF | −75.677 |
| (2,2) | AF | −68.628 | (2,2) | AF | −73.851 | ||
| (0,0) | FiM | −70.643 | (0,0) | FiM | −75.705 | ||
| (2,2) | FiM | −68.752 | (2,2) | FiM | −73.948 | ||
| ZrRu | (0,0) | AF | −71.763 | VOs | (0,0) | AF | −71.377 |
| (2,2) | AF | −69.993 | (3,2) | AF | −67.913 | ||
| (0,0) | FiM | −71.818 | (0,0) | FiM | −71.407 | ||
| (2,2) | FiM | −70.099 | (3,2) | FiM | −68.222 | ||
| HfRu | (0,0) | AF | −73.897 | ZrRh | (0,0) | AF | −69.142 |
| (2,2) | AF | −72.119 | (2,2) | AF | −67.422 | ||
| (0,0) | FiM | −73.947 | (0,0) | FiM | −69.152 | ||
| (2,2) | FiM | −72.211 | (2,2) | FiM | −67.458 | ||
| VRu | (0,0) | AF | −69.729 | HfRh | (0,0) | AF | −71.277 |
| (3,2) | AF | −66.201 | (2,2) | AF | −69.542 | ||
| (0,0) | FiM | −69.747 | (0,0) | FiM | −71.285 | ||
| (3,2) | FiM | −66.340 | (2,2) | FiM | −69.576 | ||
| NbRu | (0,0) | AF | −72.123 | ||||
| (2,2) | AF | −70.084 | |||||
| (0,0) | FiM | −72.172 | |||||
| (2,2) | FiM | −70.199 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, B.-Y.; Lee, P.-H.; Wang, Y.-K. First-Principles Study on Possible Half-Metallic Ferrimagnetism in Double Perovskites Pb2XX′O6 (X = Ti, Zr, Hf, V, Nb and Ta, X′ = Tc, Ru, Os and Rh). Materials 2022, 15, 3311. https://doi.org/10.3390/ma15093311
Chen B-Y, Lee P-H, Wang Y-K. First-Principles Study on Possible Half-Metallic Ferrimagnetism in Double Perovskites Pb2XX′O6 (X = Ti, Zr, Hf, V, Nb and Ta, X′ = Tc, Ru, Os and Rh). Materials. 2022; 15(9):3311. https://doi.org/10.3390/ma15093311
Chicago/Turabian StyleChen, Bo-Yu, Po-Han Lee, and Yin-Kuo Wang. 2022. "First-Principles Study on Possible Half-Metallic Ferrimagnetism in Double Perovskites Pb2XX′O6 (X = Ti, Zr, Hf, V, Nb and Ta, X′ = Tc, Ru, Os and Rh)" Materials 15, no. 9: 3311. https://doi.org/10.3390/ma15093311
APA StyleChen, B.-Y., Lee, P.-H., & Wang, Y.-K. (2022). First-Principles Study on Possible Half-Metallic Ferrimagnetism in Double Perovskites Pb2XX′O6 (X = Ti, Zr, Hf, V, Nb and Ta, X′ = Tc, Ru, Os and Rh). Materials, 15(9), 3311. https://doi.org/10.3390/ma15093311