
Tuning the Photophysical Properties of Acceptor–Donor–Acceptor Di-2-(2-oxindolin-3-ylidene) Malononitrile Materials via Extended π–Conjugation: A Joint Experimental and Theoretical Study




Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials and Methods
2.2. Theoretical and Computational Details
3. Results
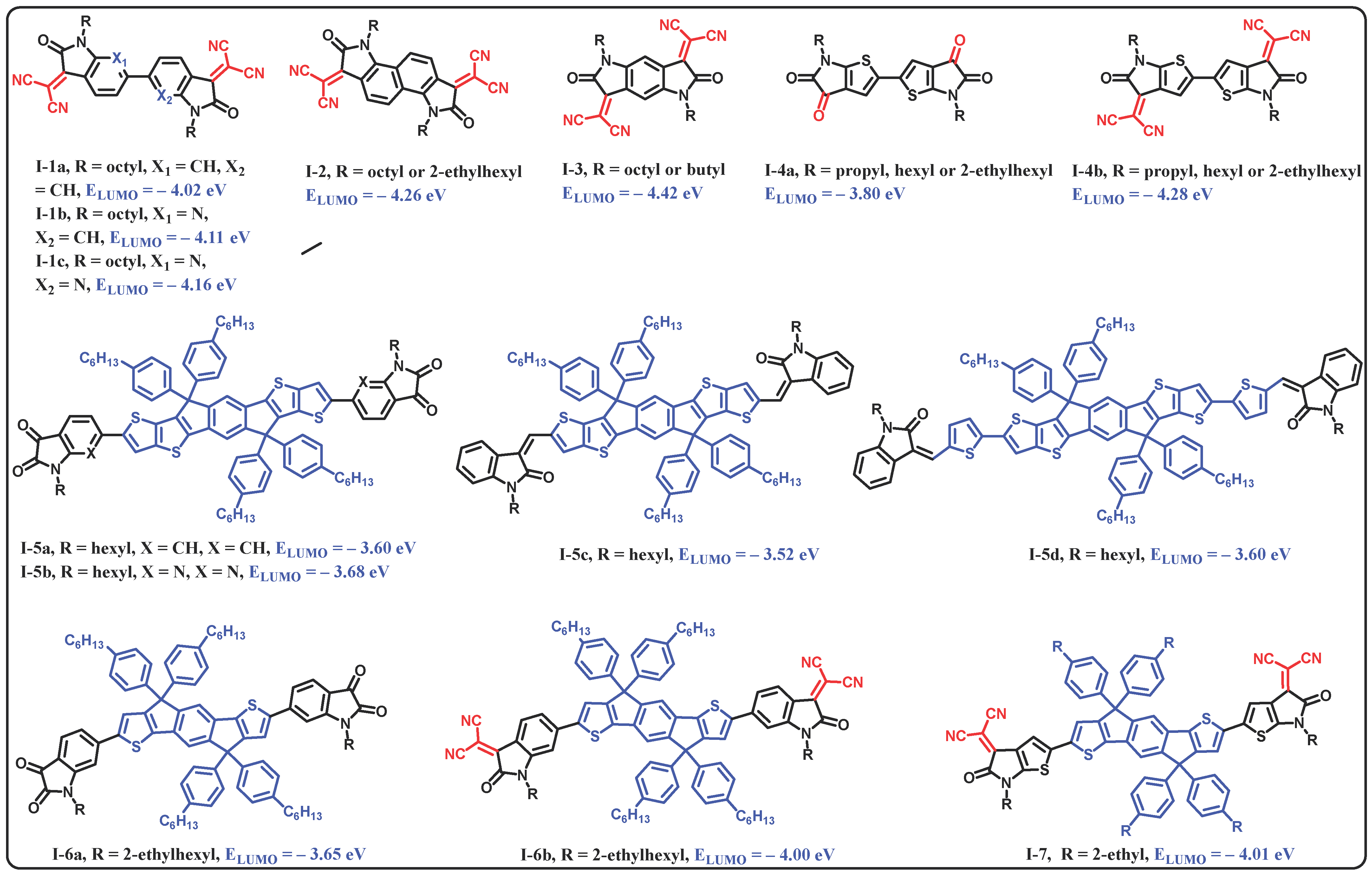
3.1. Design, Synthesis, and Characterization of A–D–A–Type OSC
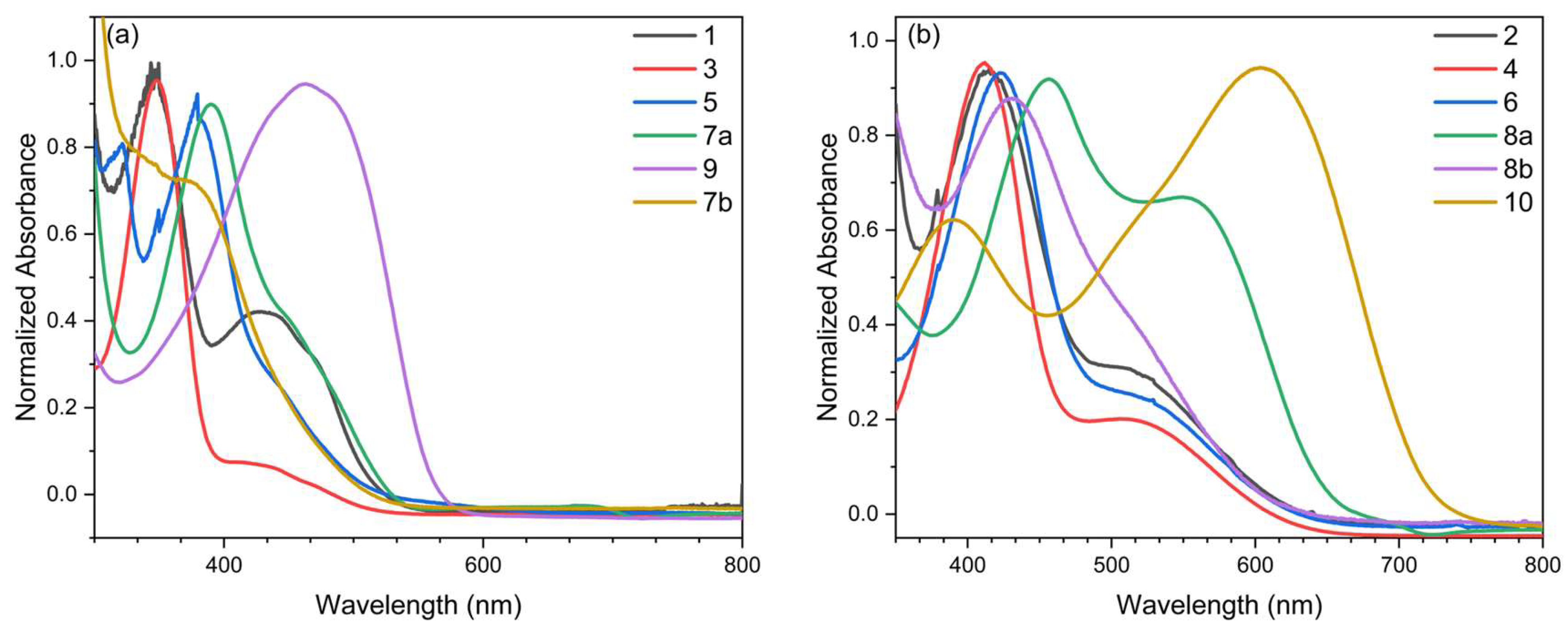
3.2. Optical Characterization
3.3. Electrochemical Study
3.4. Theoretical Studies
3.4.1. Molecular Orbitals
3.4.2. Electronic Transitions
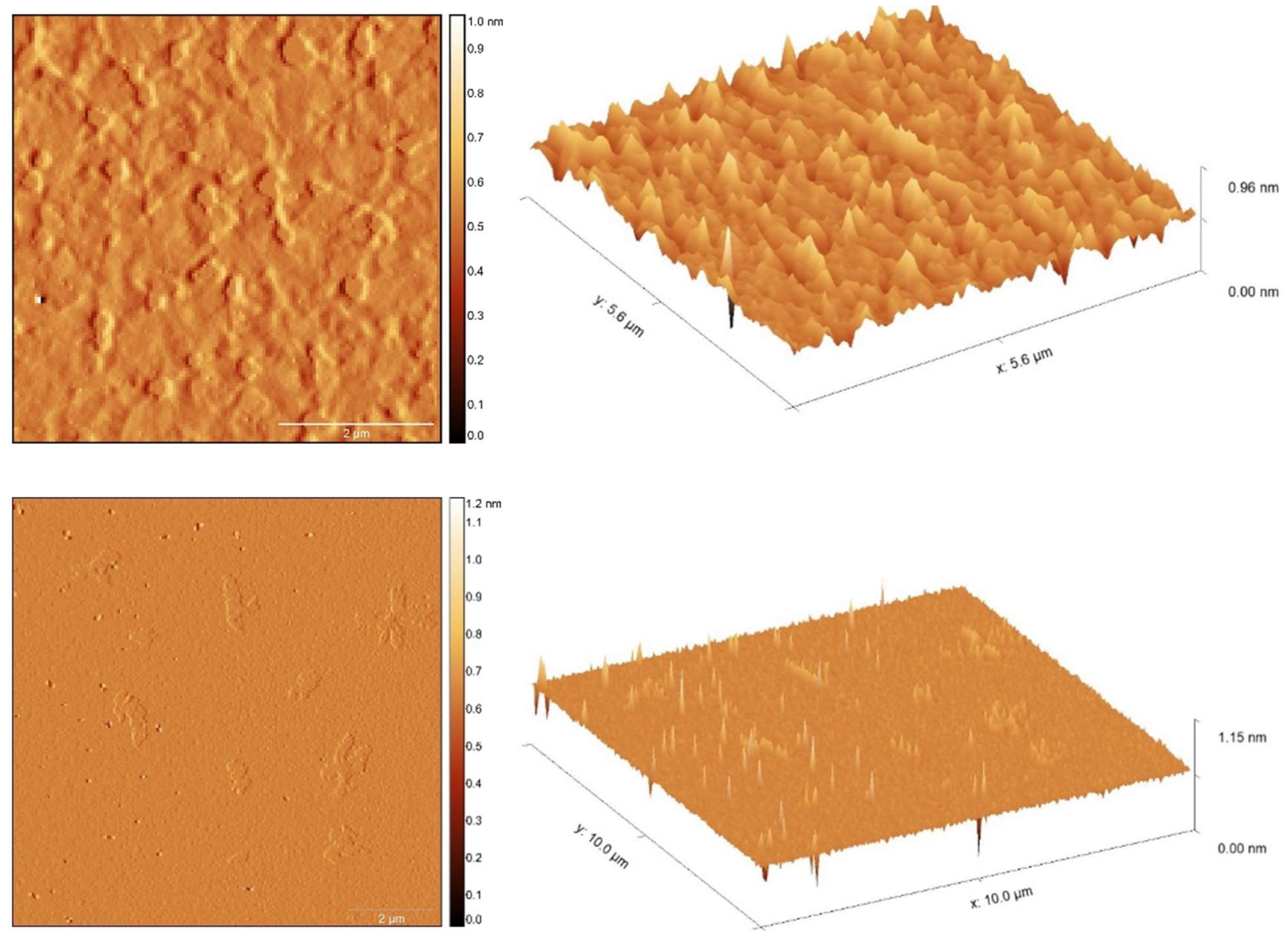
3.5. Electrical Characterization
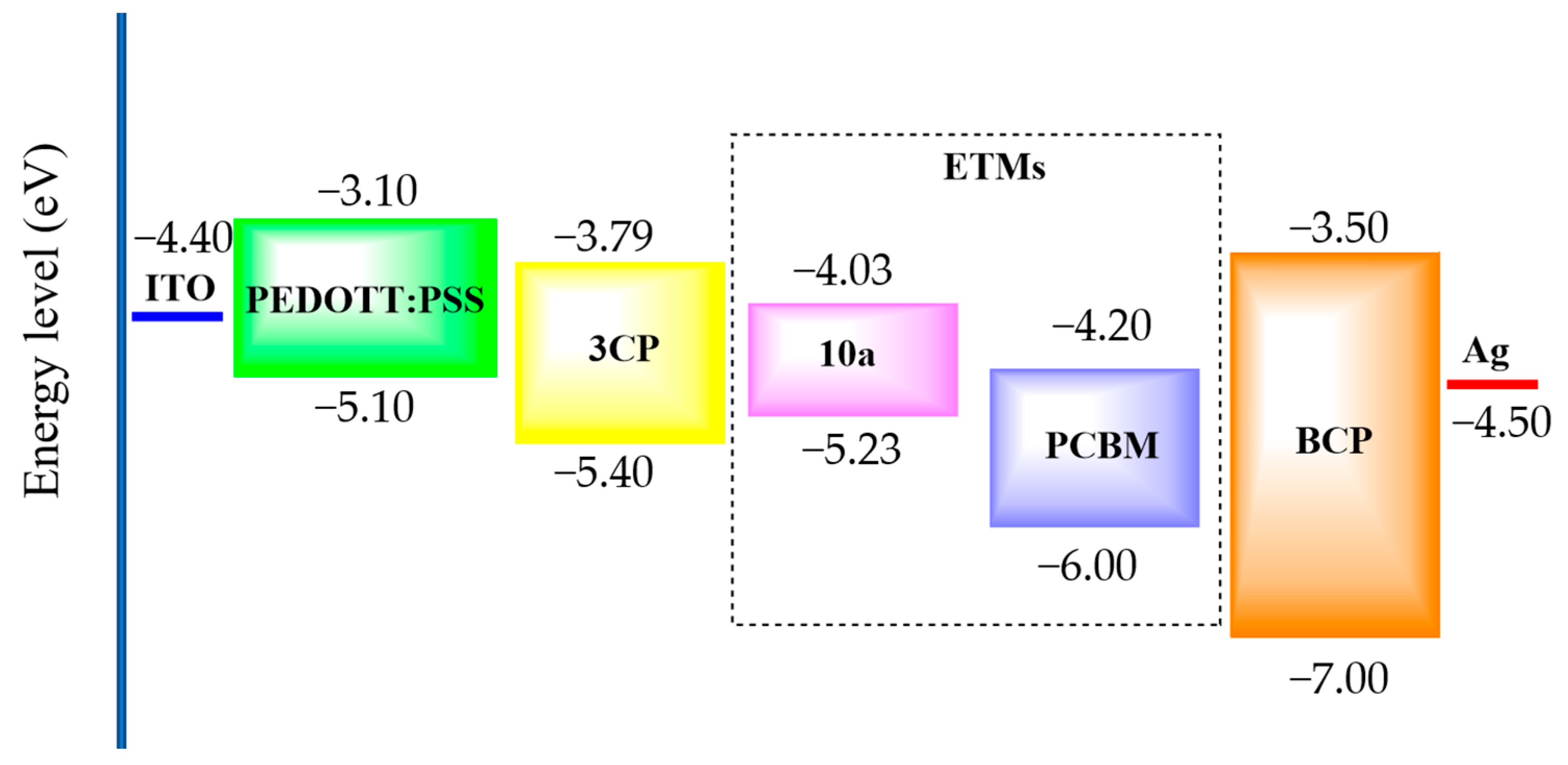
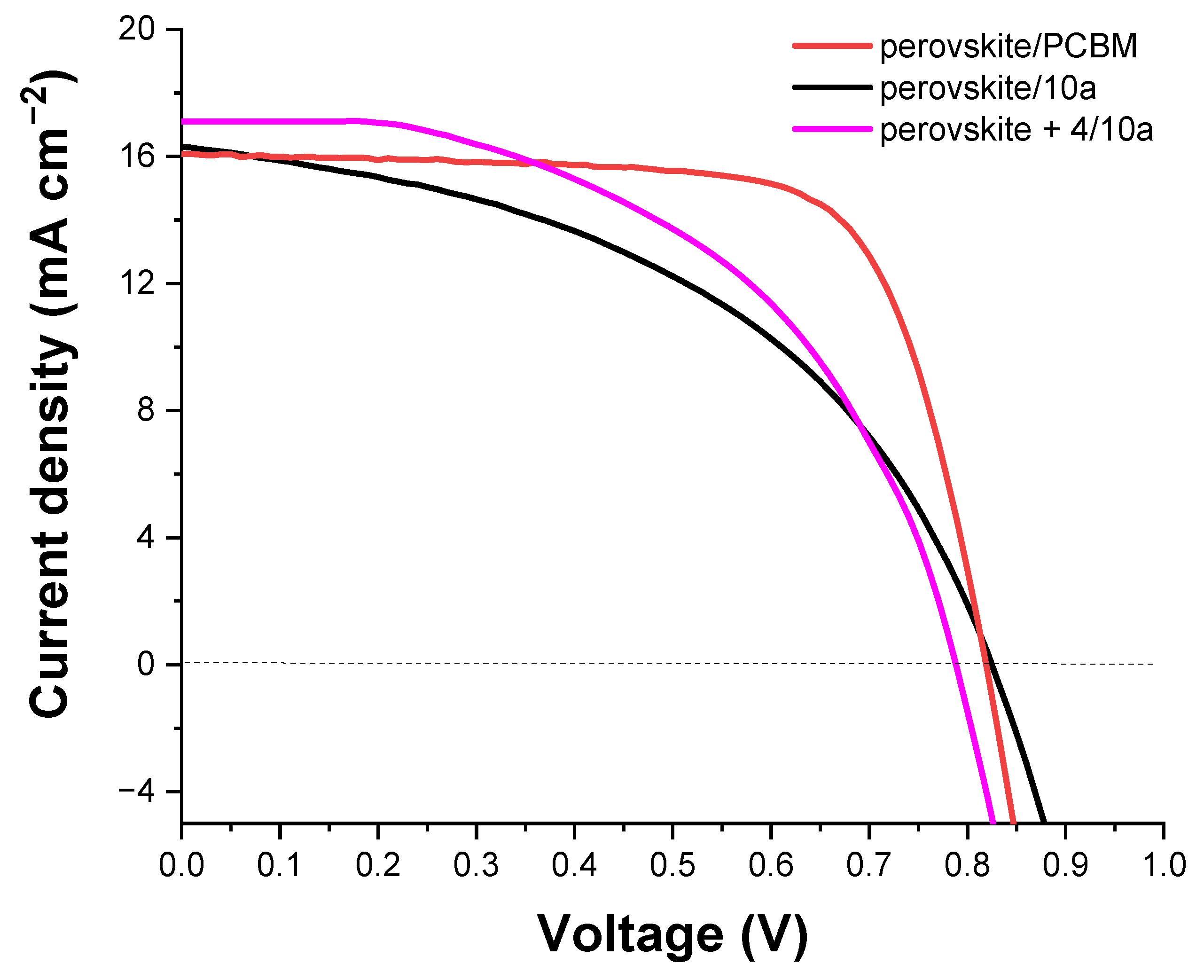
3.6. Photovoltaic Properties
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Braga, D.; Horowitz, G. High-Performance Organic Field-Effect Transistors. Adv. Mater. 2009, 21, 1473–1486. [Google Scholar] [CrossRef]
- Ren, S.; Habibi, A.; Ni, P.; Nahdi, H.; Bouanis, F.Z.; Bourcier, S.; Clavier, G.; Frigoli, M.; Yassar, A. Synthesis and characterization of solution-processed indophenine derivatives for function as a hole transport layer for perovskite solar cells. Dyes Pigments 2023, 213, 111136–111147. [Google Scholar] [CrossRef]
- Ren, S.; Ding, Y.; Zhang, W.; Wang, Z.; Wang, S.; Yi, Z. Rational Design of Novel Conjugated Terpolymers Based on Diketopyrrolopyrrole and Their Applications to Organic Thin-Film Transistors. Polymers 2023, 15, 3803. [Google Scholar] [CrossRef]
- Ren, S.; Yassar, A. Recent Research Progress in Indophenine-Based-Functional Materials: Design, Synthesis, and Optoelectronic Applications. Materials 2023, 16, 2474. [Google Scholar] [CrossRef]
- Rumer, J.W.; Schroeder, B.C.; Nielsen, C.B.; Ashraf, R.S.; Beatrup, D.; Bronstein, H.; Cryer, S.J.; Donaghey, J.E.; Holliday, S.; Hurhangee, M.; et al. Bis-lactam-based donor polymers for organic solar cells: Evolution by design. Thin Solid Films 2014, 560, 82–85. [Google Scholar] [CrossRef]
- Alsufyani, M.; Hallani, R.K.; Wang, S.; Xiao, M.; Ji, X.; Paulsen, B.D.; Xu, K.; Bristow, H.; Chen, H.; Chen, X.; et al. The effect of aromatic ring size in electron deficient semiconducting polymers for n-type organic thermoelectrics. J. Mater. Chem. C 2020, 8, 15150–15157. [Google Scholar] [CrossRef]
- Ren, S.; Zhang, W.; Wang, Z.; Yassar, A.; Liao, Z.; Yi, Z. Synergistic Use of All-Acceptor Strategies for the Preparation of an Organic Semiconductor and the Realization of High Electron Transport Properties in Organic Field-Effect Transistors. Polymers 2023, 15, 3392. [Google Scholar] [CrossRef]
- Takagi, K.; Yamamoto, S.Y.; Tsukamoto, K.; Hirano, Y.; Hara, M.; Nagano, S.; Ie, Y.; Takeuchi, D. Synthesis and Field-Effect Transistor Application of pi-Extended Lactam-Fused Conjugated Oligomers obtained by Tandem Direct Arylation. Chemistry 2018, 24, 14137–14145. [Google Scholar] [CrossRef]
- Kim, J.H.; Choi, M.W.; Kim, S.Y.; Jung, S.; Choi, Y.S.; Park, S.Y. Novel Organic Semiconductors Based on 1,5-Naphthyridine-2,6-Dione Unit for Blue-Selective Organic Phototransistor. Adv. Opt. Mater. 2020, 8, 2000695–2000704. [Google Scholar] [CrossRef]
- Yousaf, I.; Khera, R.A.; Iqbal, J.; Gul, S.; Jabeen, S.; Ihsan, A.; Ayub, K. Isatin-derived non-fullerene acceptors for efficient organic solar cells. Mater. Sci. Semicond. Process. 2021, 121, 105345–105358. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, H.; Wan, J. Recent Advances in Diversity Oriented Synthesis through Isatin-based Multicomponent Reactions. Asian J. Org. Chem. 2013, 2, 374–386. [Google Scholar] [CrossRef]
- Gomaa, M.A.M.; Hassan, D.K. Synthesis, characterization, and antimicrobial activity of some new N-aryl-N’-(2-oxoindolin-3-ylidene)benzohydrazonamides. Arch. Pharm. 2019, 352, 1900209–1900217. [Google Scholar] [CrossRef] [PubMed]
- Dhondge, A.P.; Chen, J.Y.; Lin, T.; Yen, F.M.; Li, K.W.; Hsieh, H.C.; Kuo, M.Y. Di-2-(2-oxindolin-3-ylidene)malononitrile Derivatives for N-Type Air-Stable Organic Field-Effect Transistors. Org. Lett. 2018, 20, 40–43. [Google Scholar] [CrossRef] [PubMed]
- Dhondge, A.P.; Tsai, P.C.; Nien, C.Y.; Xu, W.Y.; Chen, P.M.; Hsu, Y.H.; Li, K.W.; Yen, F.M.; Tseng, S.L.; Chang, Y.C.; et al. Angular-Shaped Naphthalene Bis(1,5-diamide-2,6-diylidene)malononitrile for High-Performance, Air-Stable N-Type Organic Field-Effect Transistors. Org. Lett. 2018, 20, 2538–2542. [Google Scholar] [CrossRef] [PubMed]
- Dhondge, A.P.; Huang, Y.X.; Lin, T.; Hsu, Y.H.; Tseng, S.L.; Chang, Y.C.; Chen, H.J.H.; Kuo, M.Y. Benzodipyrrole-2,6-dione-3,7-diylidenedimalononitrile Derivatives for Air-Stable n-Type Organic Field-Effect Transistors: Critical Role of N-Alkyl Substituent on Device Performance. J. Org. Chem. 2019, 84, 14061–14068. [Google Scholar] [CrossRef]
- Yoo, D.; Luo, X.; Hasegawa, T.; Ashizawa, M.; Kawamoto, T.; Masunaga, H.; Ohta, N.; Matsumoto, H.; Mei, J.; Mori, T. n-Type Organic Field-Effect Transistors Based on Bisthienoisatin Derivatives. ACS Appl. Electron. Mater. 2019, 1, 764–771. [Google Scholar] [CrossRef]
- Zhang, G.; Zhao, Y.; Kang, B.; Park, S.; Ruan, J.; Lu, H.; Qiu, L.; Ding, Y.; Cho, K. Fused Heptacyclic-Based Acceptor–Donor–Acceptor Small Molecules: N-Substitution toward High-Performance Solution-Processable Field-Effect Transistors. Chem. Mater. 2019, 31, 2027–2035. [Google Scholar] [CrossRef]
- Zhang, G.; Chen, R.; Sun, Y.; Kang, B.; Sun, M.; Lu, H.; Qiu, L.; Cho, K.; Ding, Y. Improved charge transport in fused-ring bridged hemi-isoindigo-based small molecules by incorporating a thiophene unit for solution-processed organic field-effect transistors. J. Mater. Chem. C 2020, 8, 1398–1404. [Google Scholar] [CrossRef]
- Zhao, D.; Hu, J.; Liu, Z.; Xiao, B.; Wang, X.; Zhou, E.; Zhang, Q. Isatylidene malononitrile derived acceptors for fullerene free organic solar cells. Dyes Pigments 2018, 151, 102–109. [Google Scholar] [CrossRef]
- Zhao, D.; Hu, J.; Cao, K.; Xiao, B.; Zhou, E.; Zhang, Q. Isatin-derived non-fullerene acceptors towards high open circuit voltage solar cells. Dyes Pigments 2019, 162, 898–904. [Google Scholar] [CrossRef]
- Shaikh, D.B.; Ali Said, A.; Wang, Z.; Srinivasa Rao, P.; Bhosale, R.S.; Mak, A.M.; Zhao, K.; Zhou, Y.; Liu, W.; Gao, W.; et al. Influences of Structural Modification of Naphthalenediimides with Benzothiazole on Organic Field-Effect Transistor and Non-Fullerene Perovskite Solar Cell Characteristics. ACS Appl. Mater. Interfaces 2019, 11, 44487–44500. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.; Wang, G.; Liu, T.; Lou, L.; Xiao, S.; Yang, S. Materials and structures for the electron transport layer of efficient and stable perovskite solar cells. Sci. China Chem. 2019, 62, 800–809. [Google Scholar] [CrossRef]
- Zhdankin, V.V.; Moradi, R.; Ziarani, G.M.; Lashgari, N. Recent applications of isatin in the synthesis of organic compounds. Arkivoc 2017, 1, 148–201. [Google Scholar] [CrossRef]
- Ren, S.; Habibi, A.; Wang, Y.; Yassar, A. Investigating the Effect of Cross-Conjugation Patterns on the Optoelectronic Properties of 7,7′Isoindigo-Based Materials. Electronics 2023, 12, 3313. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B Condens. Matter 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Rev. C.01; Gaussian: Wallingford, CT, USA, 2016. [Google Scholar]
- Miehlich, B.; Savin, A.; Stoll, H.; Preuss, H. Results obtained with the correlation energy density functionals of becke and Lee, Yang and Parr. Chem. Phys. Lett. 1989, 157, 200–206. [Google Scholar] [CrossRef]
- Azeem, U.; Khera, R.A.; Naveed, A.; Imran, M.; Assiri, M.A.; Khalid, M.; Iqbal, J. Tuning of a A-A-D-A-A-Type Small Molecule with Benzodithiophene as a Central Core with Efficient Photovoltaic Properties for Organic Solar Cells. ACS Omega 2021, 6, 28923–28935. [Google Scholar] [CrossRef]
- Raynor, A.M.; Gupta, A.; Plummer, C.M.; Jackson, S.L.; Bilic, A.; Patil, H.; Sonar, P.; Bhosale, S.V. Significant Improvement of Optoelectronic and Photovoltaic Properties by Incorporating Thiophene in a Solution-Processable D-A-D Modular Chromophore. Molecules 2015, 20, 21787–21801. [Google Scholar] [CrossRef]
- Bondi, A.A. van der Waals Volumes and Radii. J. Phys. Chem. 1964, 68, 441–451. [Google Scholar] [CrossRef]
- Roy, J.K.; Kaur, R.; Daniel, A.; Baumann, A.; Li, Q.; Delcamp, J.H.; Leszczynski, J. Photophysical Properties of Donor–Acceptor−π Bridge–Acceptor Sensitizers with a Naphthobisthiadiazole Auxiliary Acceptor: Toward Longer-Wavelength Access in Dye-Sensitized Solar Cells. J. Phys. Chem. C 2022, 126, 11875–11888. [Google Scholar] [CrossRef]
- Zhang, C.; Chen, Z.; Zeng, W.; Yu, G.; Yang, C. Narrow band-gap copolymers with two acceptors of benzo[1,2-c;3,4-c′]bis[1,2,5]thiadiazole and Benzo[c][1,2,5] thiadiazole: Synthesis, characteristics and application in field-effect transistors. Dyes Pigments 2016, 130, 291–297. [Google Scholar] [CrossRef]
- Jean, G.N.; Nord, F.F. Studies on the chemistry of heterocyclics. XXX.1 Biaromatics in the thiophene series. III. The ultraviolet absorption spectra of biphenyl type compounds containing the thiophene ring. J. Org. Chem. 1955, 20, 1370–1378. [Google Scholar] [CrossRef]
- Zhang, G.; Sun, M.; Wang, W.; Qiu, L. Acceptor-donor-acceptor small molecules based on fuse ring and 2-(2-oxindolin-3-ylidene)malononitrile derivatives for solution-processed n-type organic field-effect transistors. Synth. Met. 2019, 256, 116143. [Google Scholar] [CrossRef]
- Alishah, H.M.; Kazici, M.; Ongül, F.; Bozar, S.; Cantürk Rodop, M.; Kahveci, C.; Arvas, M.B.; Sahin, Y.; Gencten, M.; Kaleli, M.; et al. Effect of UV exposure of ITO/PEDOT:PSS substrates on the performance of inverted-type perovskite solar cells. J. Mater. Sci. Mater. Electron. 2020, 31, 7968–7980. [Google Scholar] [CrossRef]
- Chaturvedi, N.; Gasparini, N.; Corzo, D.; Bertrandie, J.; Wehbe, N.; Troughton, J.; Baran, D. All Slot-Die Coated Non-Fullerene Organic Solar Cells with PCE 11%. Adv. Funct. Mater. 2021, 31, 2009996–2010004. [Google Scholar] [CrossRef]
- Sharma, S.; Sakai, N.; Ray, S.; Senanayak, S.P.; Sirringhaus, H.; Snaith, H.J.; Patil, S. Inverted perovskite solar cells with air stable diketopyrrolopyrrole-based electron transport layer. Sol. Energy 2019, 186, 9–16. [Google Scholar] [CrossRef]
- Mamun, A.A.; Ava, T.T.; Zhang, K.; Baumgart, H.; Namkoong, G. New PCBM/carbon based electron transport layer for perovskite solar cells. Phys. Chem. Chem. Phys. 2017, 19, 17960–17966. [Google Scholar] [CrossRef]
- Yang, G.; Ren, Z.; Liu, K.; Qin, M.; Deng, W.; Zhang, H.; Wang, H.; Liang, J.; Ye, F.; Liang, Q.; et al. Stable and low-photovoltage-loss perovskite solar cells by multifunctional passivation. Nat. Photonics 2021, 15, 681–689. [Google Scholar] [CrossRef]
- Daskeviciute-Geguziene, S.; Zhang, Y.; Rakstys, K.; Xiao, C.; Xia, J.; Qiu, Z.; Daskeviciene, M.; Paskevicius, T.; Jankauskas, V.; Asiri, A.M.; et al. Passivating Defects of Perovskite Solar Cells with Functional Donor-Acceptor–Donor Type Hole Transporting Materials. Adv. Funct. Mater. 2022, 33, 2208317–2208326. [Google Scholar] [CrossRef]
- Byranvand, M.M.; Saliba, M. Defect Passivation of Perovskite Films for Highly Efficient and Stable Solar Cells. Sol. RRL 2021, 5, 2100295–2100330. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | λmax sol (nm) a | λonset sol (nm) a | Eg opt (eV) b |
|---|---|---|---|
| 1 | 430, 347 | 530 | 2.34 |
| 2 | 514, 415 | 628 | 1.97 |
| 3 | 434, 349 | 532 | 2.33 |
| 4 | 515, 414 | 630 | 1.97 |
| 5 | 440, 380 | 546 | 2.27 |
| 6 | 533, 423 | 651 | 1.90 |
| 7a | 440, 393 | 553 | 2.24 |
| 7b | 375 | 526 | 2.35 |
| 8a | 550, 457 | 670 | 1.85 |
| 8b | 432, 520 | 642 | 1.93 |
| 9a,b | 462 | 586 | 2.11 |
| 10a,b | 605, 383 | 758 | 1.63 |
| Ered | Eredonset | ELUMO | EHOMO | Egcv | |
|---|---|---|---|---|---|
| (V) 1 | (V) | (eV) 2 | (eV) 2 | (eV) 3 | |
| 1 | −1.11 | −0.87 | −3.40 | −5.46 | 2.06 |
| 2 | −0.61 | −0.36 | −3.91 | −5.92 | 2.01 |
| 3 | −1.03 | −0.81 | −3.46 | −5.52 | 2.06 |
| 4 | −0.58 | −0.34 | −3.93 | −5.87 | 1.94 |
| 5 | −0.93 | −0.74 | −3.53 | −5.53 | 2.00 |
| 6 | −0.52 | −0.29 | −3.98 | −5.82 | 1.84 |
| 7a | −0.94 | −0.73 | −3.54 | −5.35 | 1.81 |
| 8a | −0.46 | −0.19 | −4.08 | −5.80 | 1.88 |
| 8b | −0.30 | −0.04 | −4.23 | −5.96 | 1.73 |
| 9a,b | −0.95 | −0.71 | −3.56 | −5.11 | 1.55 |
| 10a,b | −0.47 | −0.24 | −4.03 | −5.23 | 1.20 |
| Eg-optDFT | ELUMODFT | EHOMODFT | EgDFT | |
|---|---|---|---|---|
| (eV) | (eV) | (eV) | (eV) | |
| 1 | 2.30 | −3.37 | −5.60 | 2.23 |
| 2 | 1.83 | −4.01 | −5.78 | 1.77 |
| 3 | 2.35 | −3.48 | −5.68 | 2.20 |
| 4 | 1.78 | −4.13 | −5.76 | 1.63 |
| 5 | 2.17 | −3.67 | −5.69 | 2.02 |
| 6 | 1.72 | −4.20 | −5.76 | 1.56 |
| 7a | 2.17 | −3.60 | −5.54 | 1.94 |
| 8a | 1.62 | −4.21 | −5.62 | 1.41 |
| 8b | 1.78 | −4.46 | −6.06 | 1.60 |
| 9a,b | 1.68 | −3.48 | −5.00 | 1.52 |
| 10a,b | 1.30 | −4.05 | −5.12 | 1.07 |
| ETM | Jsc [mA cm−2] | Voc [V] | FF [%] | PCE [%] |
|---|---|---|---|---|
| PCBM | 16.70 | 0.81 | 73 | 9.44 |
| (15.85) | (0.80) | (72) | (9.05) | |
| 10a | 16.30 | 0.82 | 47 | 6.24 |
| (14.50) | (0.78) | (46) | (5.34) | |
| 4/10a | 17.11 | 0.79 | 51 | 6.94 |
| (17.53) | (0.81) | (50) | (6.78) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ren, S.; Habibi, A.; Ni, P.; Zhang, Y.; Yassar, A. Tuning the Photophysical Properties of Acceptor–Donor–Acceptor Di-2-(2-oxindolin-3-ylidene) Malononitrile Materials via Extended π–Conjugation: A Joint Experimental and Theoretical Study. Materials 2023, 16, 6410. https://doi.org/10.3390/ma16196410
Ren S, Habibi A, Ni P, Zhang Y, Yassar A. Tuning the Photophysical Properties of Acceptor–Donor–Acceptor Di-2-(2-oxindolin-3-ylidene) Malononitrile Materials via Extended π–Conjugation: A Joint Experimental and Theoretical Study. Materials. 2023; 16(19):6410. https://doi.org/10.3390/ma16196410
Chicago/Turabian StyleRen, Shiwei, Amirhossein Habibi, Pingping Ni, Yuexing Zhang, and Abderrahim Yassar. 2023. "Tuning the Photophysical Properties of Acceptor–Donor–Acceptor Di-2-(2-oxindolin-3-ylidene) Malononitrile Materials via Extended π–Conjugation: A Joint Experimental and Theoretical Study" Materials 16, no. 19: 6410. https://doi.org/10.3390/ma16196410
APA StyleRen, S., Habibi, A., Ni, P., Zhang, Y., & Yassar, A. (2023). Tuning the Photophysical Properties of Acceptor–Donor–Acceptor Di-2-(2-oxindolin-3-ylidene) Malononitrile Materials via Extended π–Conjugation: A Joint Experimental and Theoretical Study. Materials, 16(19), 6410. https://doi.org/10.3390/ma16196410