
DFT Investigation of the Structural, Electronic, and Optical Properties of AsTi (Bi)-Phase ZnO under Pressure for Optoelectronic Applications

Abstract
:1. Introduction
2. Computational Details
3. Results and Discussion
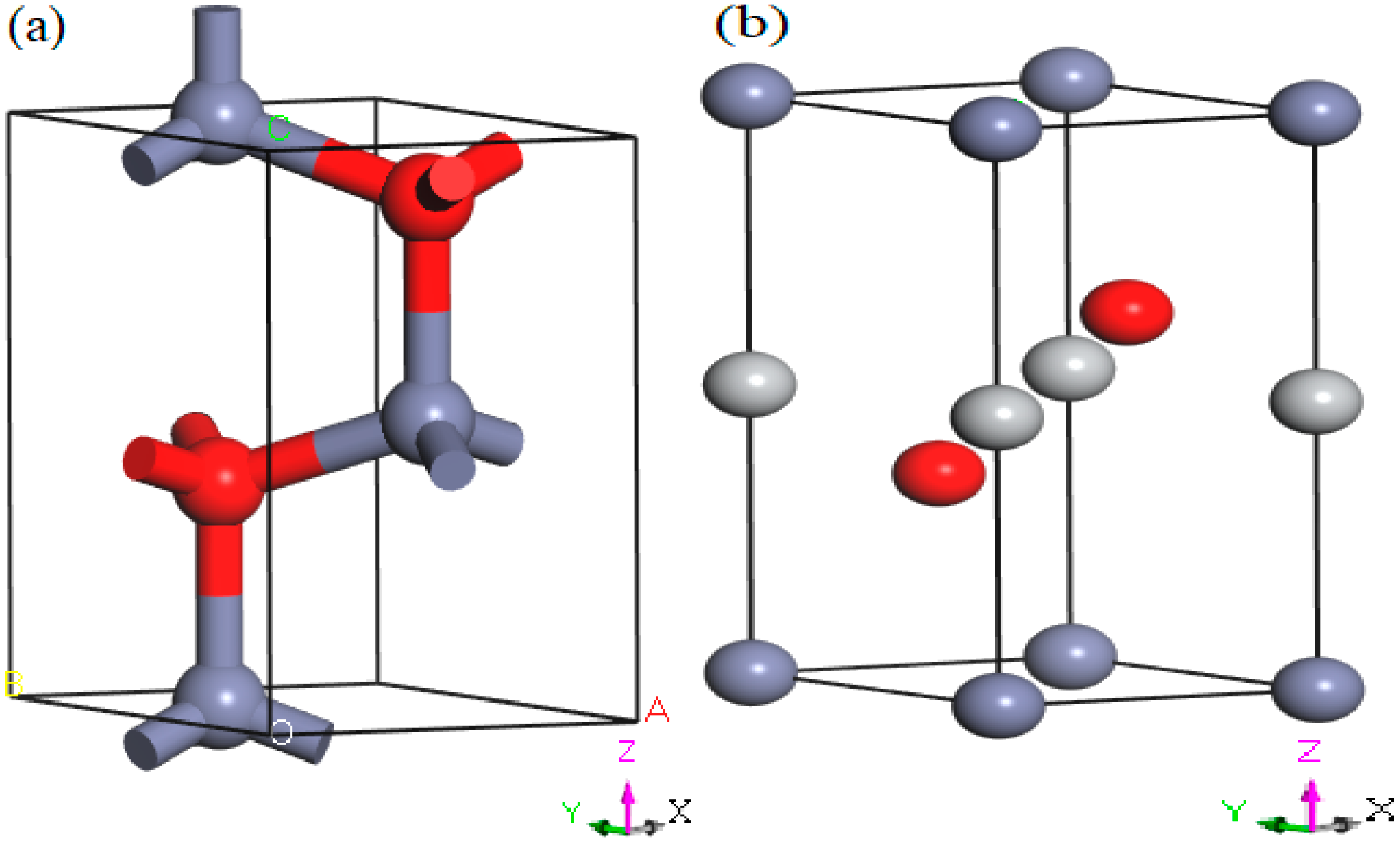
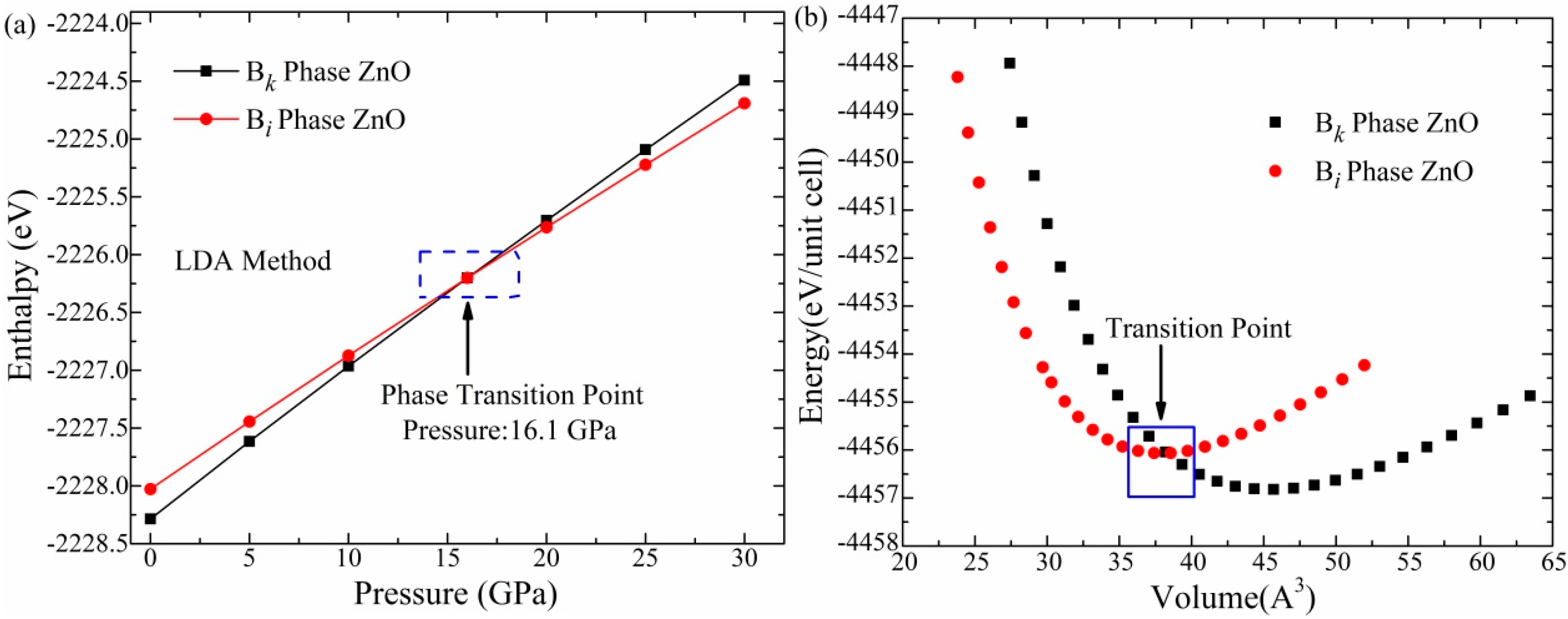
3.1. Phase Transitions and Structural Properties
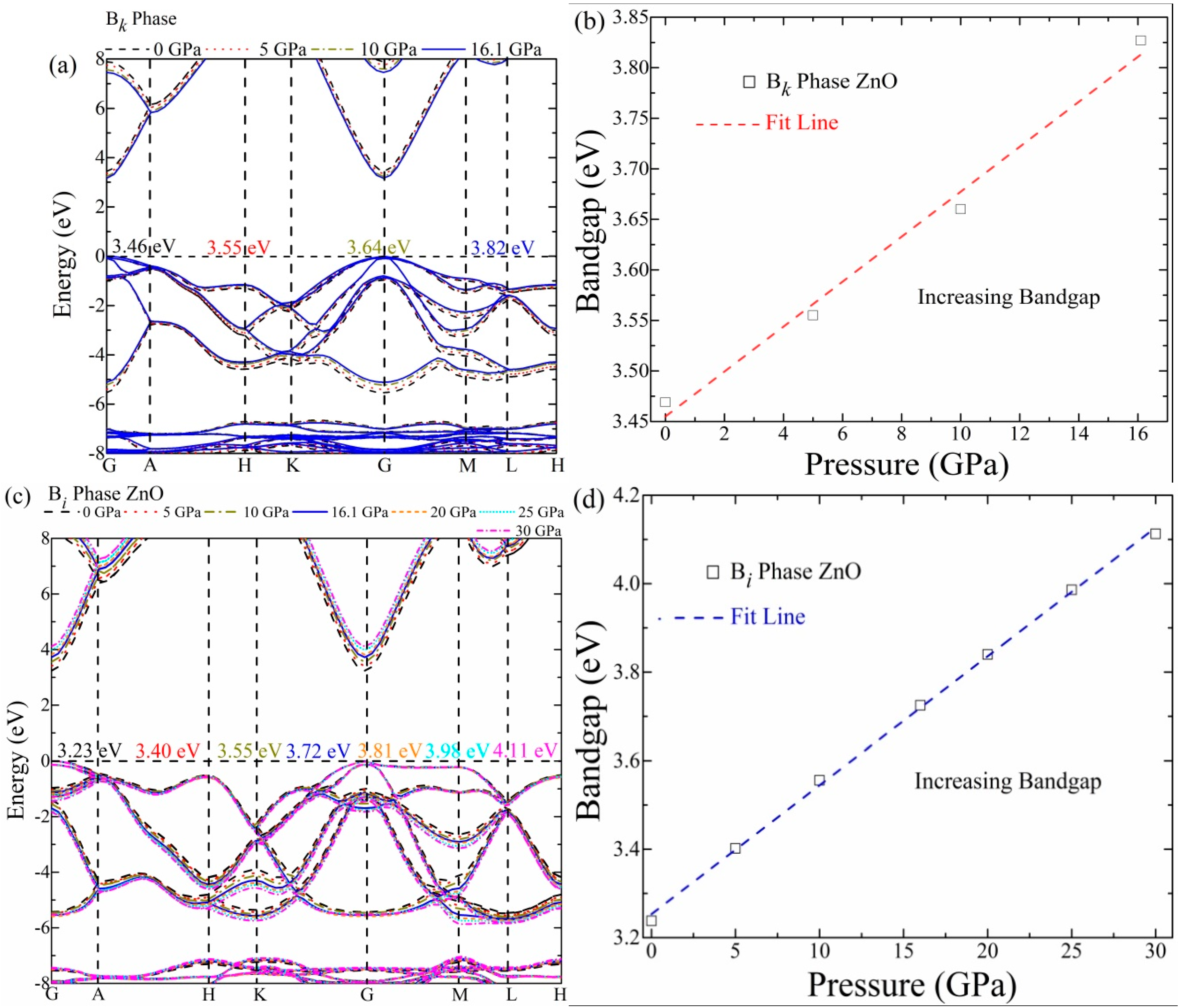
3.2. Bandgap and Structure
3.3. Electronic Density of States
3.4. Optical Properties
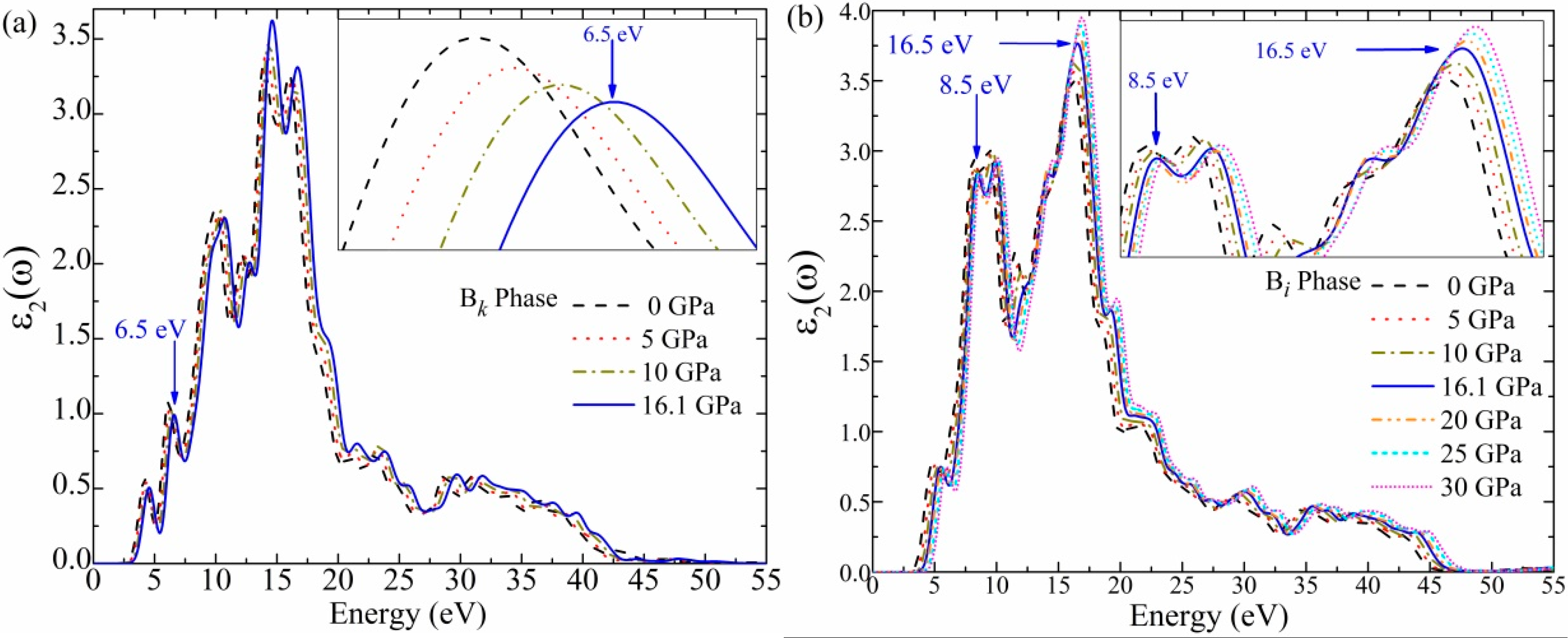
3.5. Optical Function: Dielectric Function
3.6. Optical Constants
3.7. Absorption Coefficient (α(ω))
3.7.1. Reflectivity (R(ω))
3.7.2. Refractive Index (n(ω))
3.7.3. Energy Loss Function (L(ω))
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Roy, A.; Majumdar, A. Optoelectronic and surface properties of CuO clusters: Thin film solar cell. J. Mater. Sci. Mater. Electron. 2021, 32, 27823–27836. [Google Scholar] [CrossRef]
- Yang, J.; Liu, K.; Chen, X.; Shen, D. Recent advances in optoelectronic and microelectronic devices based on ultrawide-bandgap semiconductors. Prog. Quantum Electron. 2022, 83, 100397. [Google Scholar] [CrossRef]
- Chung, J.J.; Kim, T.H.; Saeed, M.A.; Shim, J.W. Laminated indium-oxide/molybdenum-oxide nanocomposites for high-work-function electrodes in organic photovoltaics and capacitor devices. Appl. Surf. Sci. 2023, 610, 155526. [Google Scholar] [CrossRef]
- Kaminsky, F. Mineralogy of the lower mantle: A review of ‘super-deep’ mineral inclusions in diamond. Earth-Science Rev. 2012, 110, 127–147. [Google Scholar] [CrossRef]
- El Ghorfi, M.; Melcher, F.; Oberthür, T.; Boukhari, A.E.; Maacha, L.; Maddi, A.; Mhaili, M. Platinum group minerals in podiform chromitites of the Bou Azzer ophiolite, Anti Atlas, Central Morocco. Mineral. Petrol. 2008, 92, 59–80. [Google Scholar] [CrossRef]
- Wang, D.; Liu, M.; Shang, S.; Wan, P.; Shi, D.; Kan, C.; Li, B.; Jiang, M. An individual ZnO microwire homojunction LED with ultraviolet electroluminescence spectrally purified using Pt nanoparticles cladding. Opt. Laser Technol. 2023, 160, 109052. [Google Scholar] [CrossRef]
- Ünal, D.; Varol, S.F.; Brault, J.; Chenot, S.; Al Khalfioui, M.; Merdan, Z. Improved performance of near UV-blue n-ZnO/p-GaN heterostructure LED with an AlN electron blocking layer. Microelectron. Eng. 2022, 262, 111830. [Google Scholar] [CrossRef]
- Khan, G.R. Optical band gap engineering of ZnO nanophosphors via Cu incorporation for ultraviolet–violet LED. Eur. Phys. J. Plus 2020, 135, 684. [Google Scholar] [CrossRef]
- Idris, M.S.; Subramani, S. Nanostructures multilayer MgO/ZnO thin film thermal interface material for LED applications: Thermal, optical, and surface temperature performance. J. Mater. Sci. Mater. Electron. 2021, 32, 16008–16023. [Google Scholar] [CrossRef]
- He, Y.; Zeng, Z.; Cao, Y.; Zhang, X.; Wu, C.; Luo, X. Ultrasenstive SERS biosensor based on Zn2+ from ZnO nanoparticle assisted DNA enzyme amplification for detection of miRNA. Anal. Chim. Acta 2022, 1228, 340340. [Google Scholar] [CrossRef]
- Sangubotla, R.; Kim, J. Fluorometric biosensor based on boronic acid-functionalized ZnO-derived nanostructures for the detection of N-acetylneuraminic acid and its in vivo bio-imaging studies. J. Taiwan Inst. Chem. Eng. 2022, 138, 104477. [Google Scholar] [CrossRef]
- RoyChaudhuri, C.; Chakraborty, B. Development of low noise FET biosensor using graphene and ZnO nanostructures on cost effective substrates for biomolecule detection with enhanced performance. CSI Trans. ICT 2020, 8, 129–136. [Google Scholar] [CrossRef]
- De Penning, R.; Monzon, N.; Padalkar, S. Flexible zinc oxide-based biosensors for detection of multiple analytes. J. Mater. Res. 2022, 37, 2942–2950. [Google Scholar] [CrossRef]
- Yin, Z.; Shan, Y.; Yu, M.; Yang, L.; Song, J.; Hu, P.; Teng, F. Enhanced performance of UV photodetector based on ZnO nanorod arrays via TiO2 as electrons trap layer. Mater. Sci. Semicond. Process. 2022, 148, 106813. [Google Scholar] [CrossRef]
- Li, S.; Chen, Z.; Du, Z.; He, H.; Shen, X.; Fu, Y. A self-powered, visible-blind ultraviolet photodetector based on ZnO nanorods/ZnS core–shell structure. Opt. Commun. 2022, 525, 128838. [Google Scholar] [CrossRef]
- Bakour, A.; Saadoune, A.; Bouchama, I.; Dhiabi, F.; Boudour, S.; Saeed, M.A. Effect and optimization of ZnO layer on the performance of GaInP/GaAs tandem solar cell. Micro Nanostruct. 2022, 168, 207294. [Google Scholar] [CrossRef]
- Chatterjee, A.; Ravindra, A.V.; Kumar, G.K.; Rajesh, C. Improvement in the light conversion efficiency of silicon solar cell by spin coating of CuO, ZnO nanoparticles and CuO/ZnO mixed metal nanocomposite material. J. Indian Chem. Soc. 2022, 99, 100653. [Google Scholar] [CrossRef]
- Segall, M.D.; Lindan, P.J.; Probert, M.A.; Pickard, C.J.; Hasnip, P.J.; Clark, S.J.; Payne, M.C. First-principles simulation: Ideas, illustrations and the CASTEP code. J. Phys. Condens. Matter 2002, 14, 2717–2744. [Google Scholar] [CrossRef]
- Burke, K. Perspective on density functional theory. J. Chem. Phys. 2012, 136, 150901. [Google Scholar] [CrossRef]
- Liebermann, R.C. Multi-anvil, high pressure apparatus: A half-century of development and progress. High Press. Res. 2011, 31, 493–532. [Google Scholar] [CrossRef]
- Desgreniers, S. High-density phases of ZnO: Structural and compressive parameters. Phys. Rev. B—Condens. Matter Mater. Phys. 1998, 58, 14102–14105. [Google Scholar] [CrossRef]
- Liu, H.; Tse, J.S.; Mao, H.K. Stability of rocksalt phase of zinc oxide under strong compression: Synchrotron x-ray diffraction experiments and first-principles calculation studies. J. Appl. Phys. 2006, 100, 093509. [Google Scholar] [CrossRef]
- Karzel, H.; Potzel, W.; Köfferlein, M.; Schiessl, W.; Steiner, M.; Hiller, U.; Kalvius, G.M.; Mitchell, D.W.; Das, T.P.; Blaha, P.; et al. Lattice dynamics and hyperfine interactions in ZnO and ZnSe at high external pressures. Phys. Rev. B—Condens. Matter Mater. Phys. 1996, 53, 11425–11438. [Google Scholar] [CrossRef] [PubMed]
- Özgür, Ü.; Alivov, Y.I.; Liu, C.; Teke, A.; Reshchikov, M.A.; Doğan, S.; Avrutin, V.; Cho, S.-J.; Morkoç, H. A comprehensive review of ZnO materials and devices. J. Appl. Phys. 2005, 98, 041301. [Google Scholar] [CrossRef]
- Jaffe, J.E.; Snyder, J.A.; Lin, Z.; Hess, A.C. LDA and GGA calculations for high-pressure phase transitions in ZnO and MgO. Phys. Rev. B—Condens. Matter Mater. Phys. 2000, 62, 1660–1665. [Google Scholar] [CrossRef]
- Kuang, F.G.; Kuang, X.Y.; Kang, S.Y.; Zhong, M.M.; Mao, A.J. A first principle study of pressure-induced effects on phase transitions, band structures and elasticity of zinc oxide. Mater. Sci. Semicond. Process. 2014, 23, 63–71. [Google Scholar] [CrossRef]
- Wu, Y.; Kang, J.; Liu, F. Pressure induced wurtzite-to-zinc blende phase transition in ZnO at finite temperature. J. Mater. Res. 2008, 23, 3347–3352. [Google Scholar] [CrossRef]
- Pu, C.Y.; Tang, X.; Zhang, Q.Y. First principles study on the structural and optical properties of the high-pressure ZnO phases. Solid State Commun. 2011, 151, 1533–1536. [Google Scholar] [CrossRef]
- Wang, Q.; Wen, Y.; Chen, R.; Shan, B. A hybrid functional study of the electronic and optical properties of tetragonal PbO-type phase of ZnO under pressure. J. Alloys Compd. 2014, 586, 611–615. [Google Scholar] [CrossRef]
- Wang, Q.; Li, T.; Wang, H.; Li, H.; Miao, Y.; Chen, Q.; Wan, M.; Chen, L.; Sun, J.; He, K.; et al. The thermodynamic, electronic and optical properties of GeP type ZnO under pressure calculated by Debye model and hybrid function. Mater. Chem. Phys. 2018, 211, 206–213. [Google Scholar] [CrossRef]
- Kamboh, M.A.; Wang, H.; Wang, L.; Hao, L.; Su, Y.; Chen, L.; Wang, Q. Ab initio study of the thermodynamic, electronic and optical properties of WC (Bh) phase ZnO under pressure. Mater. Sci. Eng. B Solid-State Mater. Adv. Technol. 2021, 265, 115008. [Google Scholar] [CrossRef]
- Wang, Q.B.; Zhou, C.; Chen, L.; Wang, X.C.; He, K.H. The optical properties of NiAs phase ZnO under pressure calculated by GGA+U method. Opt. Commun. 2014, 312, 185–191. [Google Scholar] [CrossRef]
- Wang, Q.B.; Zhou, C.; Wu, J.; Lü, T.; He, K.H. GGA+U study of the electronic and optical properties of hexagonal BN phase ZnO under pressure. Comput. Mater. Sci. 2015, 102, 196–201. [Google Scholar] [CrossRef]
- Singh, S.; Tripathi, M.N. Enhanced optoelectronic property of ZnO under negative pressure condition: A first-principles study. Mater. Res. Express 2016, 3, 086301. [Google Scholar] [CrossRef]
- Kamboh, M.A.; Hao, L.; Farhan, M.; Su, Y.; Wang, L.; Chen, Q.; Wang, H.; Zhang, J.; Wang, Q. Structural, thermodynamic, electronic, and optical properties of β-BeO phase ZnO under negative pressure: A first-principles study. J. Mater. Sci. 2022, 57, 18905–18922. [Google Scholar] [CrossRef]
- Molepo, M.P.; Joubert, D.P. Computational study of the structural phases of ZnO. Phys. Rev. B—Condens. Matter Mater. Phys. 2011, 84, 094110. [Google Scholar] [CrossRef]
- Chen, L.; Hu, H.; Kang, C.; Wang, A.; Xiong, Z.; Cui, Y.; Gao, Y. Construction of two-dimensional lateral heterostructures by graphenelike ZnO and GaN monolayers for potential optoelectronic applications. Surf. Interfaces 2023, 36, 102635. [Google Scholar] [CrossRef]
- Ghorbani, L.; Nasirian, S. Zinc oxide nanorods assisted by polyaniline network as a flexible self-powered Ultraviolet photodetector: A comprehensive study. Appl. Surf. Sci. 2020, 527, 146786. [Google Scholar] [CrossRef]
- Ferdowsi, P.; Ochoa-Martinez, E.; Alonso, S.S.; Steiner, U.; Saliba, M. Ultrathin polymeric films for interfacial passivation in wide band-gap perovskite solar cells. Sci. Rep. 2020, 10, 22260. [Google Scholar] [CrossRef]
- Mayengbam, R.; Tripathy, S.K.; Palai, G.; Dhar, S.S. First-principles study of phase transition, electronic, elastic and optical properties of defect chalcopyrite ZnGa2 Te4 semiconductor under different pressures. J. Phys. Chem. Solids 2018, 119, 193–201. [Google Scholar] [CrossRef]
- Moucherek, F.M.O.; Santos, W.O.; Novais, A.L.F.; Moreira, E.; Azevedo, D.L. Prediction of electronic and optical properties of monoclinic 1T’-phase OsSe2 monolayer using DFT principles. Mater. Today Commun. 2022, 33, 104764. [Google Scholar] [CrossRef]
- Sharma, S.B.; Qattan, I.A.; Jaoude, M.A.; Abedrabbo, S. First-principles DFT study of structural, electronic and optical properties of Cu-doped TiO (112) surface for enhanced visible-light photocatalysis. Comput. Mater. Sci. 2023, 218, 111952. [Google Scholar] [CrossRef]
- Nesa, M.; Momin, M.A.; Sharmin, M.; Bhuiyan, A.H. Structural, optical and electronic properties of CuO and Zn doped CuO: DFT based First-principles calculations. Chem. Phys. 2020, 528, 110536. [Google Scholar] [CrossRef]
- Sun, L.; Liu, Y.; Wu, P.; Zhou, W. Band structure and optical properties of MoS2/SnO2 hetero-bilayer from hybrid functional calculations. Mater. Chem. Phys. 2020, 239, 122071. [Google Scholar] [CrossRef]
- Garwood, T.; Modine, N.A.; Krishna, S. Electronic structure modeling of InAs/GaSb superlattices with hybrid density functional theory. Infrared Phys. Technol. 2017, 81, 27–31. [Google Scholar] [CrossRef]
- Lee, H.; Cheong, S.W.; Kim, B.G. Hybrid functional band gap calculation of SnO6 containing perovskites and their derived structures. J. Solid State Chem. 2015, 228, 214–220. [Google Scholar] [CrossRef]
- Mesquita, W.D.; de Jesus, S.R.; Oliveira, M.C.; Ribeiro, R.A.P.; de Cássia Santos, M.R.; Junior, M.G.; Longo, E.; do Carmo Gurgel, M.F. Barium strontium titanate-based perovskite materials from DFT perspective: Assessing the structural, electronic, vibrational, dielectric and energetic properties. Theor. Chem. Acc. 2021, 140, 27. [Google Scholar] [CrossRef]
- Pham, A.; Assadi, M.H.N.; Yu, A.B.; Li, S. Critical role of Fock exchange in characterizing dopant geometry and magnetic interaction in magnetic semiconductors. Phys. Rev. B—Condens. Matter Mater. Phys. 2014, 89, 155110. [Google Scholar] [CrossRef]
- Ceperley, D.M.; Alder, B.J. Ground State of the Electron Gas by a Stochastic Method. Phys. Rev. Lett. 1980, 45, 566–569. [Google Scholar] [CrossRef]
- Vanderbilt, D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism. Phys. Rev. B 1990, 41, 7892–7895. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Pfrommer, B.G.; Côté, M.; Louie, S.G.; Cohen, M.L. Relaxation of Crystals with the Quasi-Newton Method. J. Comput. Phys. 1997, 131, 233–240. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Crystal Structure and Pair Potentials: A Molecular-Dynamics Study. Phys. Rev. Lett. 1980, 45, 1196–1199. [Google Scholar] [CrossRef]
- Bauernschmitt, R.; Ahlrichs, R. Treatment of electronic excitations within the adiabatic approximation of time dependent density functional theory. Chem. Phys. Lett. 1996, 256, 454–464. [Google Scholar] [CrossRef]
- Huang, Y.; Yang, L.; Liu, C.; Liu, X.; Liu, J.; Huang, X.; Zhu, P.; Cui, T.; Sun, C.; Bao, Y. Lasing-Mode Switch of a Hexagonal ZnO Pyramid Driven by Pressure within a Diamond Anvil Cell. J. Phys. Chem. Lett. 2019, 10, 610–616. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.D.; Guo, M.L.; Li, W.X.; Liu, C.L. First-principles study of electronic and optical properties in wurtzite Zn1−xCdxO. J. Appl. Phys. 2008, 103, 063721. [Google Scholar] [CrossRef]
- Zhang, X.; Le, M.-Q.; Zahhaf, O.; Capsal, J.-F.; Cottinet, P.-J.; Petit, L. Enhancing dielectric and piezoelectric properties of micro-ZnO/PDMS composite-based dielectrophoresis. Mater. Des. 2020, 192, 108783. [Google Scholar] [CrossRef]
- Cheng, P.; Chen, M.; Shen, X.; Chen, S.; Liu, Y.; Cao, D.; Wang, Q. Heterojunction structure for suppressing dark current toward high-performance ZnO microrod metal-semiconductor-metal ultraviolet photodetectors. Micro Nanostruct. 2022, 171, 207422. [Google Scholar] [CrossRef]
- Sarmah, S.; Das, M.; Sarkar, D. UV–Visible photo sensing properties of CdS-porous silicon (PS): P-Si and ZnO-PS: P-Si heterostructures metal-semiconductor-metal (MSM) based devices. Mater. Today Proc. 2021, 46, 6234–6238. [Google Scholar] [CrossRef]
- Reinosa, J.J.; Leret, P.; Álvarez-Docio, C.M.; del Campo, A.; Fernández, J.F. Enhancement of UV absorption behavior in ZnO–TiO2 composites. Boletín Soc. Española Cerámica Vidr. 2016, 55, 55–62. [Google Scholar] [CrossRef]
- Bouhjar, F.; Derbali, L.; Marí, B. High performance novel flexible perovskite solar cell based on a low-cost-processed ZnO:Co electron transport layer. Nano Res. 2020, 13, 2546–2555. [Google Scholar] [CrossRef]
- Chen, Z.; Wang, J.; Wu, H.; Yang, J.; Wang, Y.; Zhang, J.; Bao, Q.; Wang, M.; Ma, Z.; Tress, W.; et al. A Transparent Electrode Based on Solution-Processed ZnO for Organic Optoelectronic Devices. Nat. Commun. 2022, 13, 4387. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pressure (GPa) | Phase | Space Group | Reference | a = b (Å) | c (Å) | Bond Angle α = β | γ | Bond Length (Å) | Volume (Å3) |
|---|---|---|---|---|---|---|---|---|---|
| 0 | Bk | P63MC | Theory a | 3.397 | 5.132 | 90° | 120° | - | - |
| Theory b | 3.161 | 5.165 | 90° | 120° | - | 22.36 | |||
| Present | 3.357 | 4.360 | 90° | 120° | 2.180 | 21.287 | |||
| Bi | P-3M1 | Theory b | 2.93 | 4.97 | 90° | 120° | - | 18.76 | |
| Present | 2.920 | 5.024 | 90° | 120° | 2.102 | 18.557 | |||
| 16.1 | Bk | P63MC | Present | 3.290 | 4.220 | 90° | 120° | 2.11 | 19.785 |
| Bi | P-3M1 | Present | 2.851 | 4.949 | 90° | 120° | 2.059 | 17.721 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adnan, M.; Wang, Q.; Sohu, N.; Du, S.; He, H.; Peng, Z.; Liu, Z.; Zhang, X.; Bai, C. DFT Investigation of the Structural, Electronic, and Optical Properties of AsTi (Bi)-Phase ZnO under Pressure for Optoelectronic Applications. Materials 2023, 16, 6981. https://doi.org/10.3390/ma16216981
Adnan M, Wang Q, Sohu N, Du S, He H, Peng Z, Liu Z, Zhang X, Bai C. DFT Investigation of the Structural, Electronic, and Optical Properties of AsTi (Bi)-Phase ZnO under Pressure for Optoelectronic Applications. Materials. 2023; 16(21):6981. https://doi.org/10.3390/ma16216981
Chicago/Turabian StyleAdnan, Muhammad, Qingbo Wang, Najamuddin Sohu, Shiyu Du, Heming He, Zhenbo Peng, Zhen Liu, Xiaohong Zhang, and Chengying Bai. 2023. "DFT Investigation of the Structural, Electronic, and Optical Properties of AsTi (Bi)-Phase ZnO under Pressure for Optoelectronic Applications" Materials 16, no. 21: 6981. https://doi.org/10.3390/ma16216981