
Single Crystals of EuScCuSe3: Synthesis, Experimental and DFT Investigations

 , ,
, ,  , ,
, , 
Abstract
:1. Introduction
- -
- In the form of single crystals by the methods of reactive flux or halide flux [15],
- -
- By chemical transport reaction with I2 [9],
- -
- High-temperature alloying of elements with chalcogenides [12],
- -
- From the elements by the transport method at 1270 K,
- -
- By the interaction of binary chalcogenides at 1420 K [16],
- -
- As polycrystals by alloying the elements at 1420 K followed by annealing at 870 K for 240 h [17],
- -
- By sulfiding mixtures of oxides obtained by thermolysis of co-crystallized metal nitrates at 870–1170 K for 25 h [13].
2. Materials and Methods
2.1. Materials and Synthesis
2.2. Methods
3. Results and Discussion
3.1. Crystal Structure
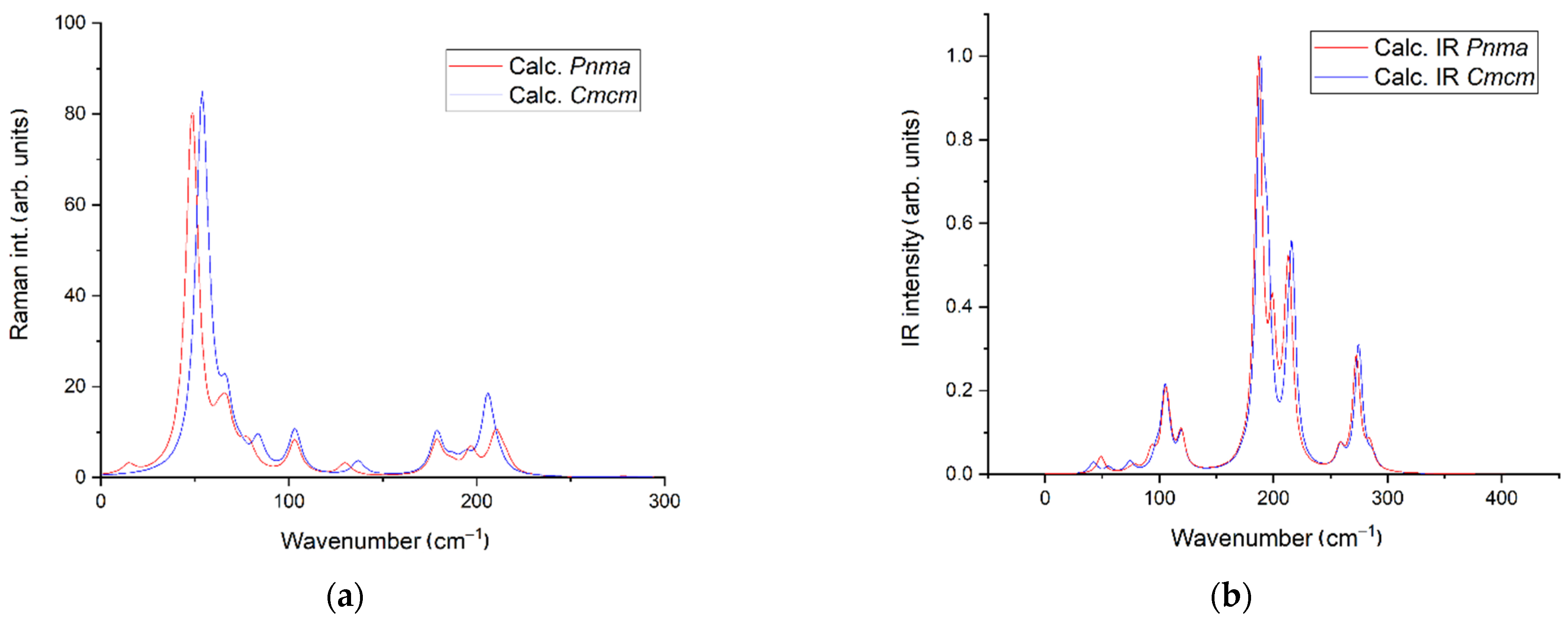
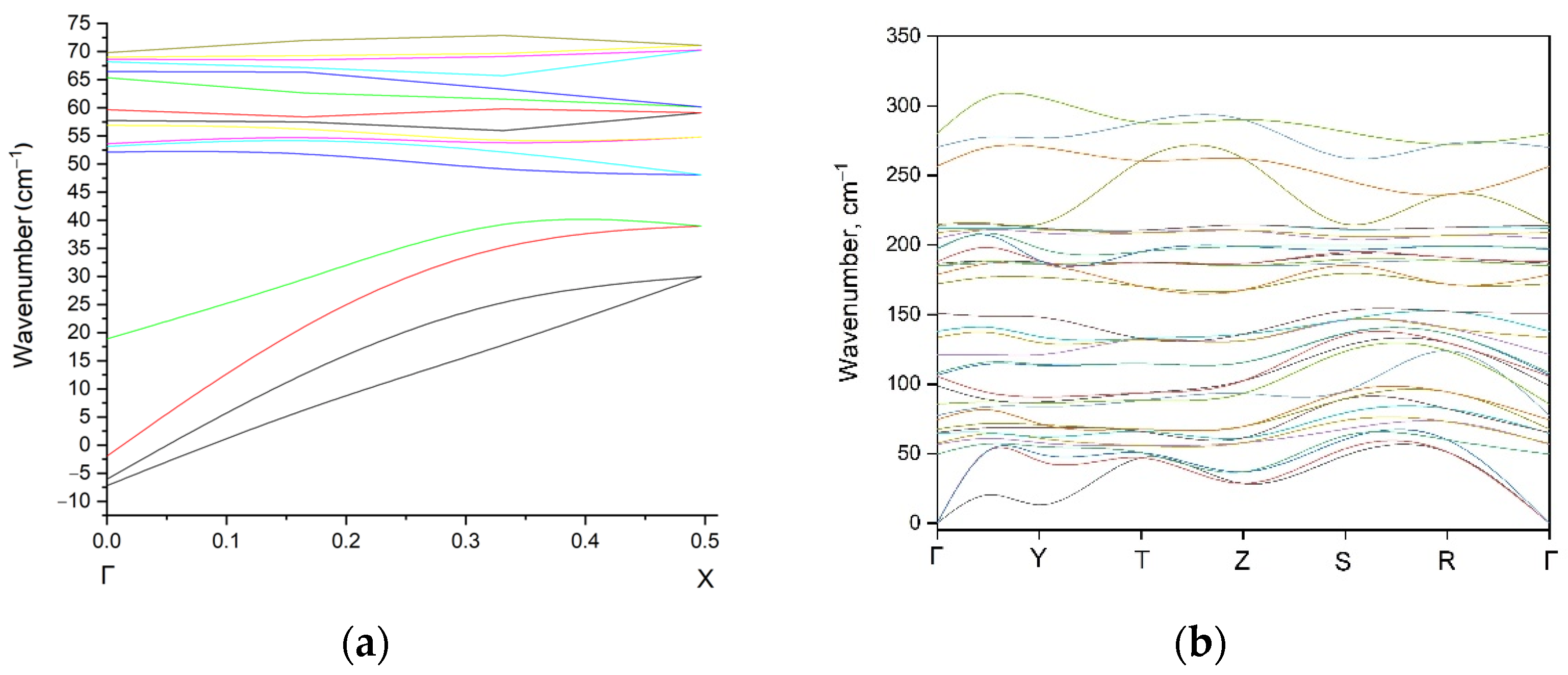
3.2. Density Functional Theory Calculations
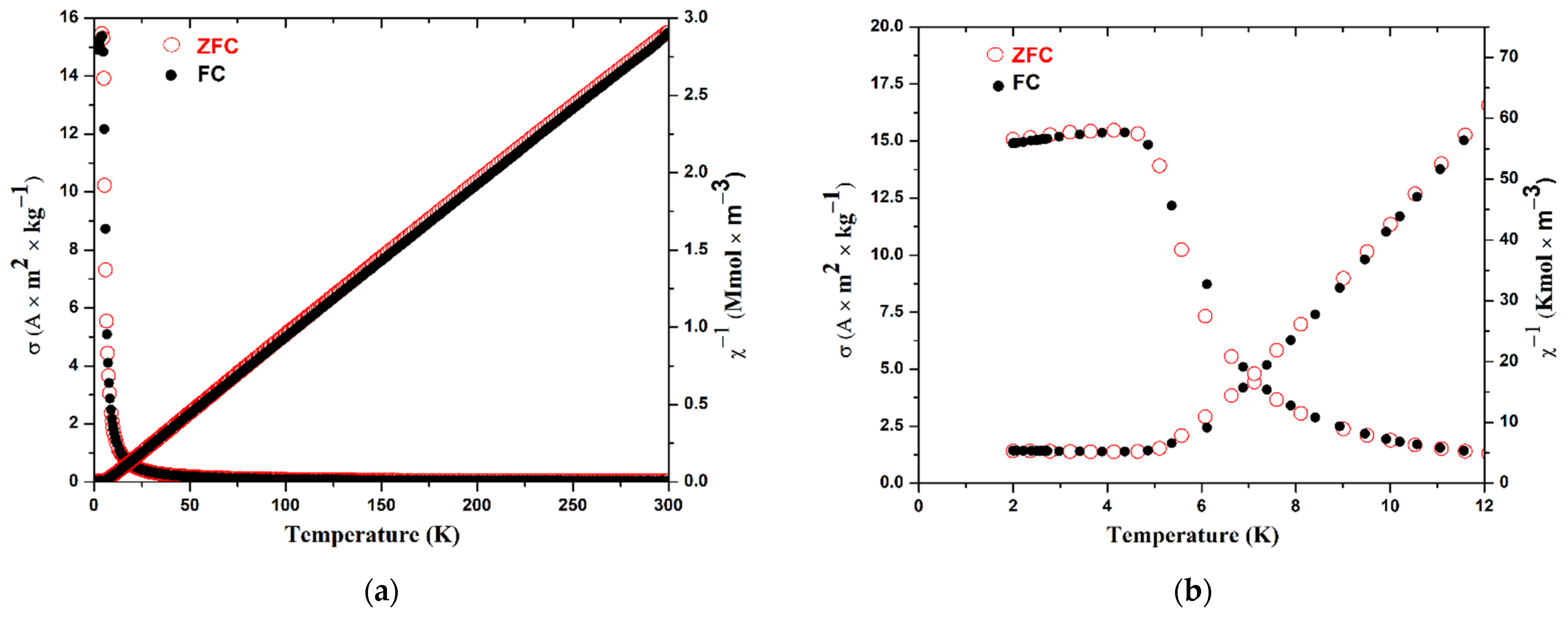
3.3. Magnetic Properties
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ishtiyak, M.; Jana, S.; Karthikeyan, R.; Ramesh, M.; Tripathy, B.; Malladi, S.K.; Niranjan, M.K.; Prakash, J. Syntheses of Five New Layered Quaternary Chalcogenides SrScCuSe3, SrScCuTe3, BaScCuSe3, BaScCuTe3, and BaScAgTe3: Crystal Structures, Thermoelectric Properties, and Electronic Structures. Inorg. Chem. Front. 2021, 8, 4086. [Google Scholar] [CrossRef]
- Rugut, E.; Joubert, D.; Jones, G. First Principle Studies on Lattice Thermal Conductivity and Thermoelectric Properties of ScCu(S,Se,Te)2. Mater. Today Commun. 2021, 26, 101905. [Google Scholar] [CrossRef]
- Pal, K.; Xia, Y.; Shen, J.; He, J.; Luo, Y.; Kanatzidis, M.G.; Wolverton, C. Accelerated Discovery of a Large Family of Quaternary Chalcogenides with Very Low Lattice Thermal Conductivity. NPJ Comput. Mater. 2021, 7, 82. [Google Scholar] [CrossRef]
- Scanlon, D.O.; Watson, G.W. Stability, Geometry, and Electronic Structure of an Alternative I-III-VI2 Material, CuScS2: A Hybrid Density Functional Theory Analysis. Appl. Phys. Lett. 2010, 97, 131904. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, J.; Wu, T.; Bu, X.; Feng, P. Three-Dimensional Open Framework Built from Cu–S Icosahedral Clusters and Its Photocatalytic Property. J. Am. Chem. Soc. 2008, 130, 15238. [Google Scholar] [CrossRef] [PubMed]
- Sagade, A.A.; Sharma, R. Copper sulphide (CuxS) as an ammonia gas sensor working at room temperature. Sens. Actuat. B Chem. 2008, 133, 135. [Google Scholar] [CrossRef]
- Shakeel, A.; Rizwan, K.; Farooq, U.; Iqbal, S.; Altaf, A.A. Advanced polymeric/inorganic nanohybrids: An integrated platform for gas sensing applications. Chemosphere 2022, 294, 133772. [Google Scholar] [CrossRef]
- Dismukes, J.P.; White, J.G. The Preparation, Properties, and Crystal Structures of Some Scandium Sulfides in the Range Sc2S3-ScS. Inorg. Chem. 1964, 3, 1220. [Google Scholar] [CrossRef]
- Dismukes, J.P.; Smith, R.T.; White, J.G. Physical Properties and Crystal Structure of a New Semiconducting I-III-VI2 Compound, CuScS2. J. Phys. Chem. Solids 1971, 32, 913. [Google Scholar]
- Koscielski, L.A.; Ibers, J.A. The Structural Chemistry of Quaternary Chalcogenides of the Type AMM’Q3. Z. Anorg. Allg. Chem. 2012, 638, 2585. [Google Scholar] [CrossRef]
- Otero-Diaz, L.C.; Hiraga, K.; Sellar, J.R.; Hyde, B.G. An Electron Microscope Examination of Scandium Sesquisulphide, Sc2S3, and Its Mode of Disordering in the Electron Beam. Acta Crystallogr. Sect. B 1984, 40, 355. [Google Scholar] [CrossRef]
- Chen, L.; Corbett, J.D. Synthesis, Structure, and Bonding of Sc6MTe2 (M = Ag, Cu, Cd): Heterometal-Induced Polymerization of Metal Chains in Sc2Te. Inorg. Chem. 2002, 41, 2146. [Google Scholar] [CrossRef]
- Ruseikina, A.V.; Molokeev, M.S.; Chernyshev, V.; Aleksandrovsky, A.S.; Krylov, A.S.; Krylova, S.N.; Velikanov, D.; Grigoriev, M.V.; Maximov, N.G.; Shestakov, N.P.; et al. Synthesis, Structure, and Properties of EuScCuS3 and SrScCuS3. J. Solid State Chem. 2021, 296, 121926. [Google Scholar] [CrossRef]
- Jin, G.B.; Choi, E.S.; Albrecht-Schmitt, T.E. Syntheses, Structures, Magnetism, and Optical Properties of Gadolinium Scandium Chalcogenides. J. Solid State Chem. 2009, 182, 1075. [Google Scholar] [CrossRef]
- Babo, J.-M.; Schleid, T. CsCu2Sc3Te6 and CsCuY2Te4: Two New Quaternary Cesium Copper Rare-Earth Metal Tellurides. Solid State Sci. 2010, 12, 238. [Google Scholar] [CrossRef]
- van Dijk, M.; Plug, C.M. The Crystal Structure of LiScS2 and NaScS2. Mater. Res. Bull. 1980, 15, 103. [Google Scholar] [CrossRef]
- Gulay, L.D.; Shemet, V.Y.; Olekseyuk, I.D. Crystal Structures of the ScCuSe2 and Sc3CuSn3Se11 Compounds. J. Alloys Compds. 2005, 393, 174. [Google Scholar] [CrossRef]
- Perdew, J.P.; Ernzerhof, M.; Burke, K. Rationale for Mixing Exact Exchange with Density Functional Approximations. J. Chem. Phys. 1996, 105, 9982. [Google Scholar] [CrossRef]
- Pathan, H.M.; Desai, J.D.; Lokhande, C.D. Modified Chemical Deposition and Physico-Chemical Properties of Copper Sulphide (Cu2S) Thin Films. Appl. Surf. Sci. 2002, 202, 47. [Google Scholar] [CrossRef]
- Perniu, D.; Vouwzee, S.; Duta, A.; Schoonman, J. Defect Chemistry of Solar Cell Chalcopyrite Materials. J. Optoelectron. Adv. Mater. 2007, 9, 1568. [Google Scholar]
- Mansuetto, M.F.; Keane, P.M.; Ibers, J.A. Synthesis and Structures of the New Group IV Chalcogenides NaCuTiS3 and NaCuZrQ3 (Q = S, Se, Te). J. Solid State Chem. 1993, 105, 580. [Google Scholar] [CrossRef]
- Eberle, M.; Schleid, T. Expanding the SrCuRES3 Series with the Rare-Earth Metals Scandium and Yttrium. Z. Kristallogr. 2016, S36, 82. [Google Scholar]
- Eberle, M.A. Darstellung und Charakterisierung Quaternärer Seltenerdmetall-Verbindungen in Kombination mit Kupfer und Schwefel. Ph.D. Thesis, University of Stuttgart, Stuttgart, Germany, 2016. [Google Scholar]
- Wu, P.; Christuk, A.E.; Ibers, J.A. New Quaternary Chalcogenides BaLnMQ3 (Ln = Rare Earth or Sc; M = Cu, Ag; Q= S, Se). II. Structure and Property Variation vs. Rare-Earth Element. J. Solid State Chem. 1994, 110, 337. [Google Scholar] [CrossRef]
- Shannon, R.D. Revised Effective Ionic Radii and Systematic Studies of Interatomic Distances in Halides and Chalcogenides. Acta Crystallogr. Sect. A 1976, 32, 751. [Google Scholar] [CrossRef]
- Otwinowski, Z.; Minor, W. Processing of X-Ray Diffraction Data. Methods Enzymol. 1997, 276, 307. [Google Scholar] [PubMed]
- Herrendorf, W.; Bärnighausen, H. HABITUS, A Program for the Optimization of the Crystal Shape for Numerical Absorption Correction in X-SHAPE, version 1.06; Fa. Stoe: Darmstadt, Germany, 1996. [Google Scholar]
- Sheldrick, G.M. SHELXL-97: Program for the Refinement of Crystal Structures; University of Gottingen: Göttingen, Germany, 1997. [Google Scholar]
- Sheldrick, G.M. A Short History of SHELX. Acta Crystallogr. Sect. A 2008, 64, 112. [Google Scholar] [CrossRef] [PubMed]
- Momma, K.; Izumi, F. VESTA 3 for Three-Dimensional Visualization of Crystal, Volumetric and Morphology Data. J. Appl. Crystallogr. 2011, 44, 1272. [Google Scholar] [CrossRef]
- Dovesi, R.; Saunders, V.R.; Roetti, C.; Orlando, R.; Zicovich-Wilson, C.M.; Pascale, F.; Civalleri, B.; Doll, K.; Harrison, N.M.; Bush, I.J.; et al. CRYSTAL17 User’s Manual. 2018. Available online: https://www.crystal.unito.it/ (accessed on 16 January 2023).
- Crystal Program. Available online: http://www.crystal.unito.it/index.php (accessed on 15 December 2022).
- Born, M.; Huang, K.; Lax, M. Dynamical Theory of Crystal Lattices. Am. J. Phys. 1955, 23, 837. [Google Scholar] [CrossRef]
- Mouhat, F.; Coudert, F.X. Necessary and Sufficient Elastic Stability Conditions in Various Crystal Systems. Phys. Rev. B Condens. Matter Mater. Phys. 2014, 90, 224104. [Google Scholar] [CrossRef]
- Kroumova, E.; Aroyo, M.L.; Perez-Mato, J.M.; Kirov, A.; Capillas, C.; Ivantchev, S.; Wondratschek, H. Bilbao Crystallographic Server: Useful Databases and Tools for Phase-Transition Studies. Phase Trans. 2003, 76, 155. [Google Scholar] [CrossRef]
- Sinyakova, E.F.; Vasilyeva, I.G.; Oreshonkov, A.S.; Goryanov, S.V.; Karmanov, N.S. Formation of Noble Metal Phases (Pt, Pd, Rh, Ru, Ir, Au, Ag) in the Process of Fractional Crystallization of the CuFeS2 Melt. Minerals 2022, 12, 1136. [Google Scholar] [CrossRef]
- Sukhanova, E.V.; Sagaton, N.E.; Oreshonkov, A.S.; Gavryushkin, P.N.; Popov, Z.I. Halogen-Doped Chevrel Phase Janus Monolayers for Photocatalytic Water Splitting. Nanomaterials 2023, 13, 368. [Google Scholar] [CrossRef] [PubMed]
- Oreshonkov, A.S.; Roginskii, E.M.; Atuchin, V.V. New candidate to reach Shockley-Queisser limit: The DFT study of orthorhombic silicon allotrope Si(oP32). J. Phys. Chem. Solids 2020, 137, 109219. [Google Scholar] [CrossRef]
- Grigoriev, M.V.; Solovyov, L.A.; Ruseikina, A.V.; Aleksandrovsky, A.S.; Chernyshev, V.A.; Velikanov, D.A.; Garmonov, A.A.; Molokeev, M.S.; Oreshonkov, A.S.; Shestakov, N.P.; et al. Quaternary Selenides EuLnCuSe3: Synthesis, Structures, Properties and In Silico Studies. Int. J. Mol. Sci. 2022, 23, 1503. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | EuScCuSe3 |
|---|---|
| Space group | Cmcm (no. 63) |
| Structure type | KZrCuS3 |
| a (Å) | 3.9883 (3) |
| b (Å) | 13.2776 (9) |
| c (Å) | 10.1728 (7) |
| Z | 4 |
| ρcal (g cm−3) | 6.132 |
| Vu.c. (Å3) | 538.70 (7) |
| Measuring range, ±h/±k/±l | ±5/±16/±13 |
| F(000) | 860 |
| Absorption coefficient μ (mm−1) | 36.73 |
| Measured reflections | 5112 |
| Symmetry-independent reflections | 367 |
| Rint/Rσ | 0.089/0.041 |
| R1 for n reflections with ∣Fo∣ ≥ 4σ(Fo) | 0.055 |
| n | 294 |
| R1/wR2 for all reflections | 0.042/0.091 |
| GooF | 1.087 |
| Extinction coefficient, ε | 0.0016 (4) |
| Residual electron density, ρmax/min (e− 106 pm−3) | 2.318/−2.273 |
| CSD-number | 2239558 |
| Atom | Site | Symmetry | x/a | y/b | z/c | Ueq (Å2) |
|---|---|---|---|---|---|---|
| Eu | 4c | m2m | 0 | 0.75168 (9) | 1/4 | 0.0232 (5) |
| Sc | 4a | 2/m.. | 0 | 0 | 0 | 0.0147 (7) |
| Cu | 4c | m2m | 0 | 0.4697 (2) | 1/4 | 0.0242 (7) |
| Se1 | 4c | m2m | 0 | 0.07683 (17) | 1/4 | 0.0191 (6) |
| Se2 | 8f | m.. | 0 | 0.36312 (11) | 0.05610 (16) | 0.0199 (5) |
| Compound | Space Group | Structure Type | Lattice Constants (Å) | V (Å3) | ρ (g cm−3) | |||
|---|---|---|---|---|---|---|---|---|
| EuScCuSe3 | Calc. | Pnma | Eu2CuS3 | 4.01674 | 13.38926 | 10.03507 | 539.698 | 6.160 |
| EuScCuSe3 | Calc. | Cmcm | KZrCuS3 | 4.02028 | 13.38869 | 10.02436 | 539.574 | 6.162 |
| EuScCuSe3 | Exp. | Cmcm | KZrCuS3 | 3.9883(3) | 13.2776(9) | 10.1728(7) | 538.70(7) | 6.132 |
| Compound | Space Group | Structure Type | C11 | C12 | C13 | C22 | C23 | C33 | C44 | C55 | C66 | B | HVcal |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| EuScCuSe3 | Pnma | Eu2CuS3 | 122 | 33 | 38 | 150 | 44 | 99 | 46 | 21 | 33 | 66 | 5.7 |
| EuScCuSe3 | Cmcm | KZrCuS3 | 151 | 43 | 27 | 98 | 37 | 128 | 13 | 33 | 45 | 65 | 4.8 |
| Compound | Space Group | Structure Type | Averaging Scheme | B | G | Young’s Modulus | Poisson Ratio |
|---|---|---|---|---|---|---|---|
| EuScCuSe3 | Pnma | Eu2CuS3 | Voigt | 67 | 37 | 94 | 0.263 |
| Reuss | 65 | 34 | 86 | 0.278 | |||
| Hill | 66 | 36 | 90 | 0.271 | |||
| EuScCuSe3 | Cmcm | KZrCuS3 | Voigt | 66 | 36 | 92 | 0.267 |
| Reuss | 64 | 28 | 74 | 0.309 | |||
| Hill | 65 | 32 | 82 | 0.287 |
| Magnetic Characteristics | Calculated | M(H) at 300 K | M(T) at 500 kOe |
|---|---|---|---|
| C (K m3 kmol−1) | 0.098999 | 0.0995 | 0.0977 |
| μ (μB) | 7.9373 | 7.96 | 7.89 |
| θW (K) | - | - | 6.0 |
| Tc (K) | - | - | 4.5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grigoriev, M.V.; Ruseikina, A.V.; Chernyshev, V.A.; Oreshonkov, A.S.; Garmonov, A.A.; Molokeev, M.S.; Locke, R.J.C.; Elyshev, A.V.; Schleid, T. Single Crystals of EuScCuSe3: Synthesis, Experimental and DFT Investigations. Materials 2023, 16, 1555. https://doi.org/10.3390/ma16041555
Grigoriev MV, Ruseikina AV, Chernyshev VA, Oreshonkov AS, Garmonov AA, Molokeev MS, Locke RJC, Elyshev AV, Schleid T. Single Crystals of EuScCuSe3: Synthesis, Experimental and DFT Investigations. Materials. 2023; 16(4):1555. https://doi.org/10.3390/ma16041555
Chicago/Turabian StyleGrigoriev, Maxim V., Anna V. Ruseikina, Vladimir A. Chernyshev, Aleksandr S. Oreshonkov, Alexander A. Garmonov, Maxim S. Molokeev, Ralf J. C. Locke, Andrey V. Elyshev, and Thomas Schleid. 2023. "Single Crystals of EuScCuSe3: Synthesis, Experimental and DFT Investigations" Materials 16, no. 4: 1555. https://doi.org/10.3390/ma16041555
APA StyleGrigoriev, M. V., Ruseikina, A. V., Chernyshev, V. A., Oreshonkov, A. S., Garmonov, A. A., Molokeev, M. S., Locke, R. J. C., Elyshev, A. V., & Schleid, T. (2023). Single Crystals of EuScCuSe3: Synthesis, Experimental and DFT Investigations. Materials, 16(4), 1555. https://doi.org/10.3390/ma16041555