

Mechanistic Study of Fast Performance Decay of PtCu Alloy-based Catalyst Layers for Polymer Electrolyte Fuel Cells through Electrochemical Impedance Spectroscopy

 , , , , , , , and
, , , , , , , and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Synthesis of De-Alloyed PtCu3/KB Electrocatalyst
2.2. XRD Analysis
2.3. Scanning Electron Microscopy (SEM) Analysis
2.4. Transmission Electron Microscopy (TEM) Analysis
2.5. Thin-Film Rotating Disc Electrode (TF-RDE) Analysis
2.6. MEA Fabrication and Electrochemical Characterization
2.7. Calculations and Simulation
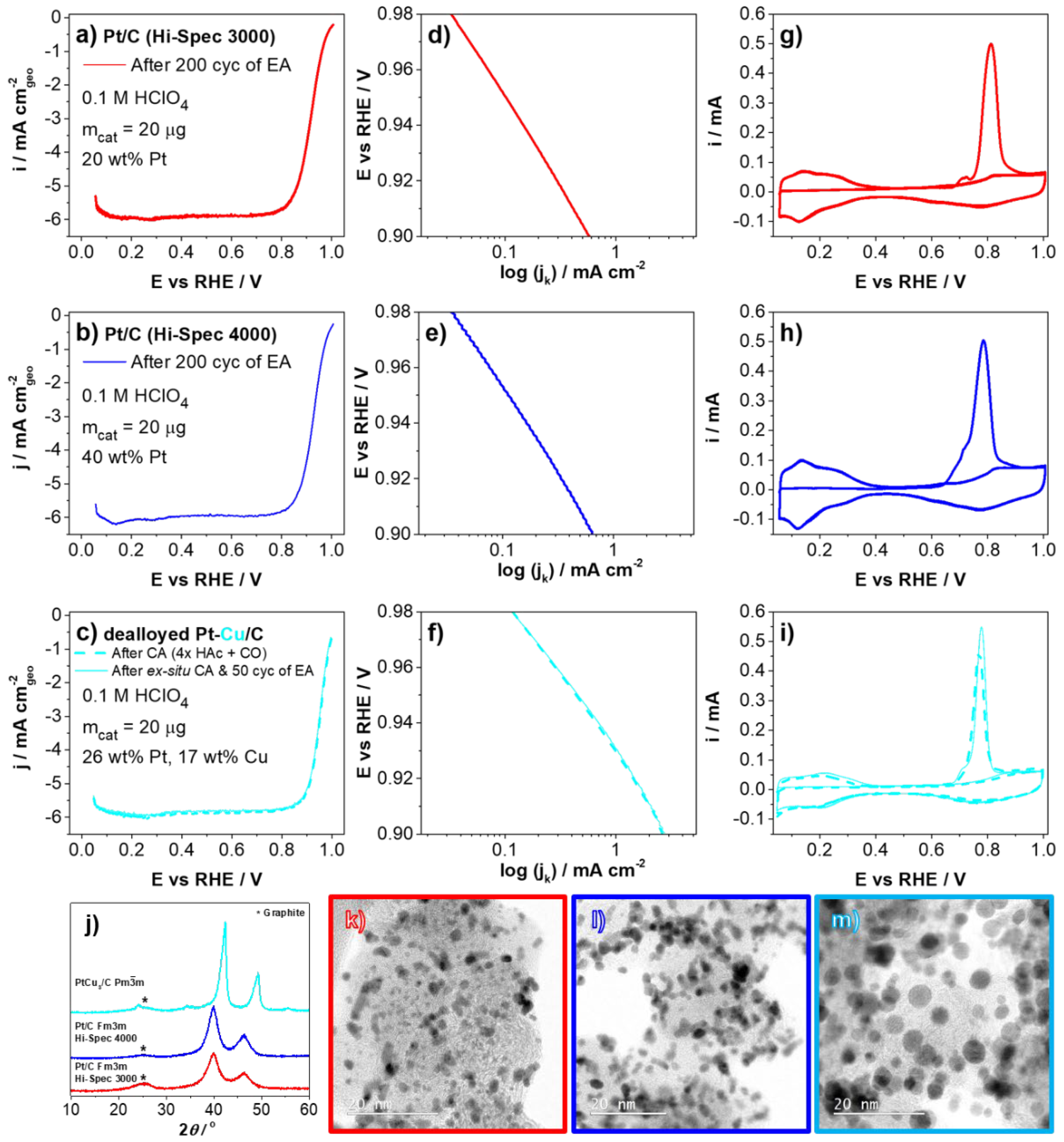
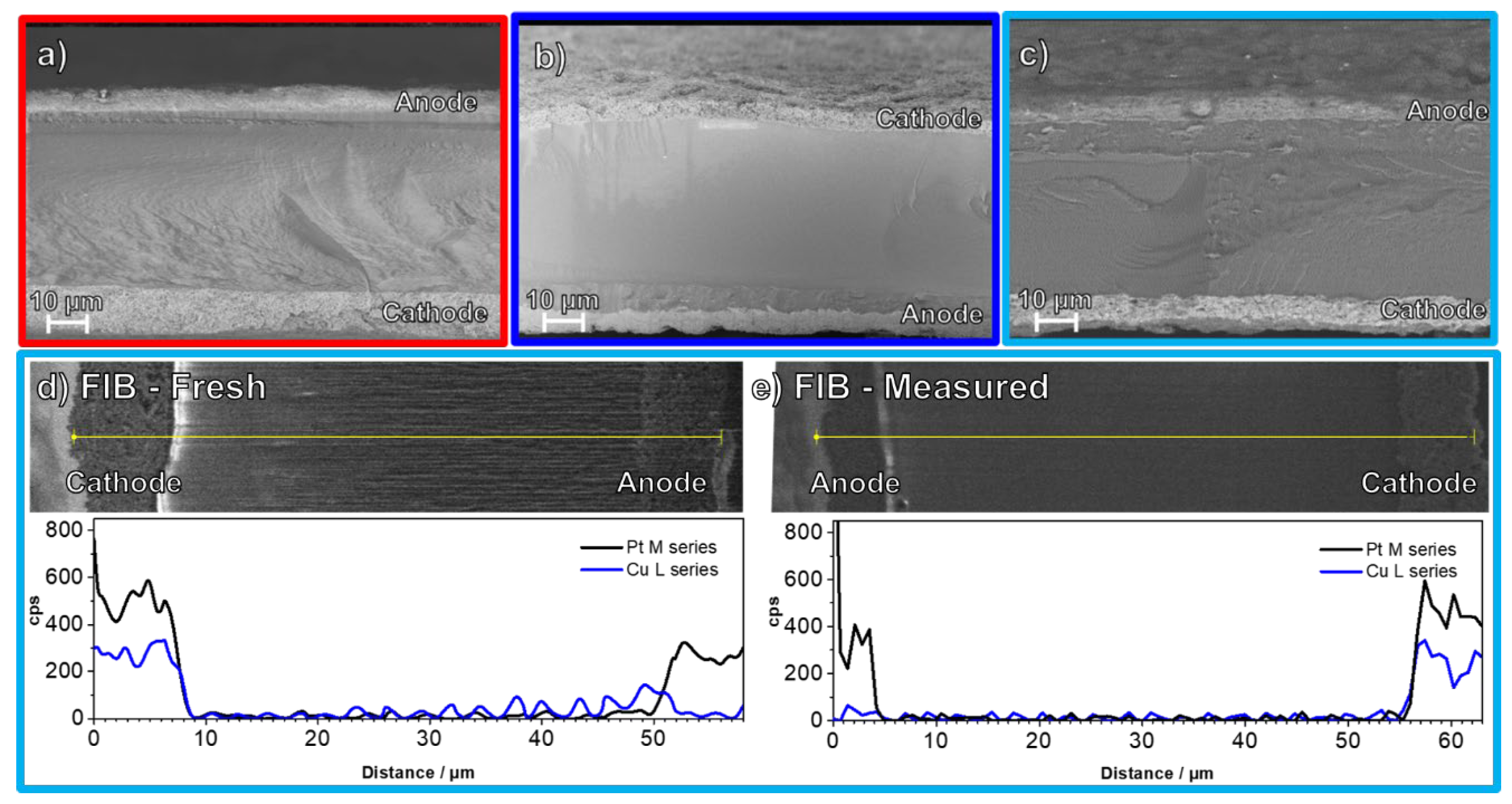
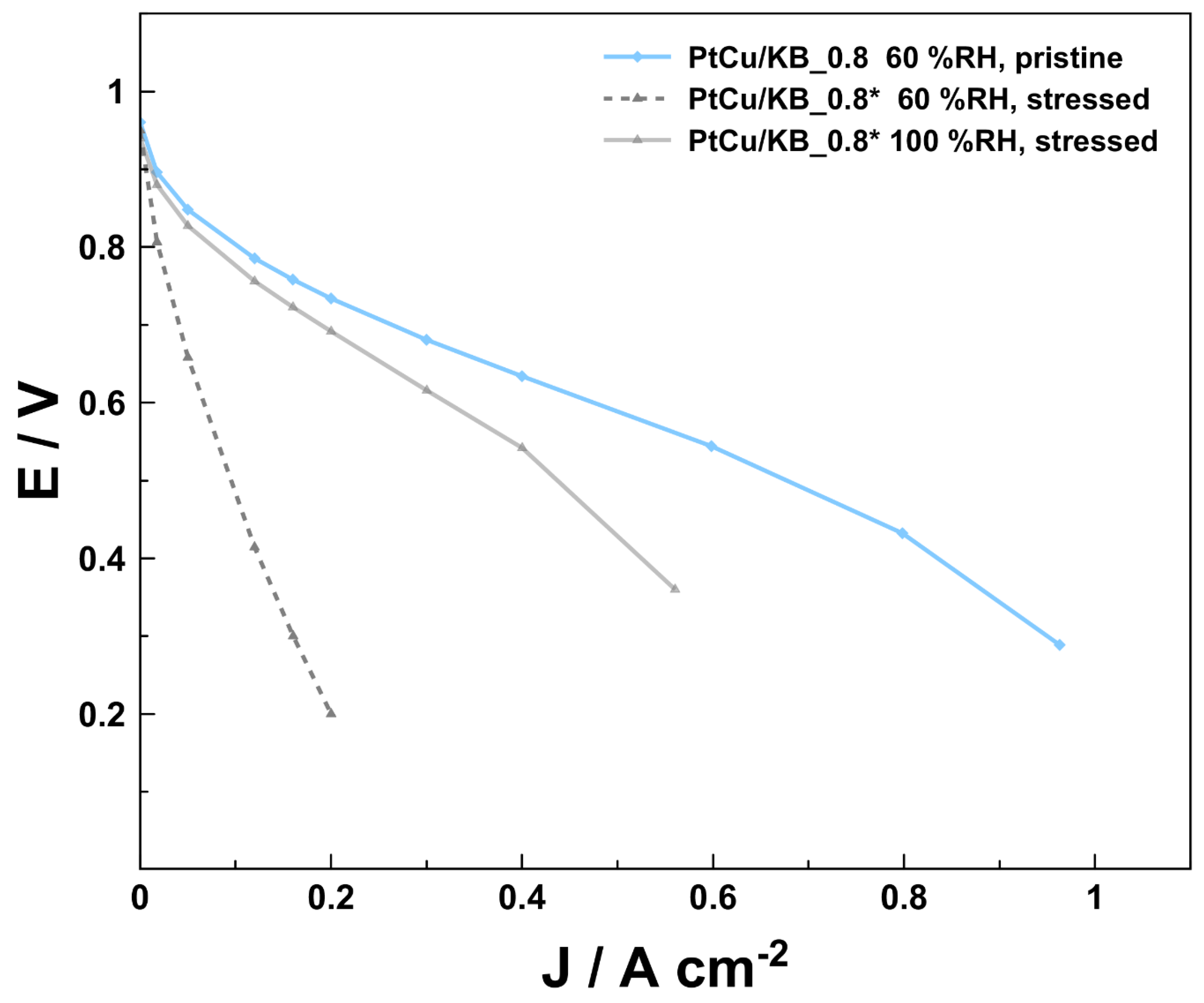
3. Results and Discussion
4. Conclusions
- (i)
- Proper chemical activation process for the PtCu alloy system that effectively removes Cu impurities from both the Pt alloy nanoparticle surface and the carbon matrix. The resulting platinum overlayer should be thick enough to present a kinetic barrier for Cu migration from the core to the surface.
- (ii)
- Completely avoid or limit operation of the cell at relative humidity above 60 %RH during all phases of operation to avoid additional dissolution of fresh Cu and consequent damage to the ionomer electrolyte near the catalyst/ionomer interface.
- (iii)
- Develop cathode ionomers resistant to Cu ions or metal ion contaminations more generally.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Marković, N.M. Electrocatalysis: Interfacing electrochemistry. Nat. Mater. 2013, 12, 101–102. [Google Scholar] [CrossRef] [PubMed]
- Gröger, O.; Gasteiger, H.A.; Suchsland, J.-P. Review—Electromobility: Batteries or Fuel Cells? J. Electrochem. Soc. 2015, 162, A2605–A2622. [Google Scholar] [CrossRef]
- Grandi, M.; Rohde, S.; Liu, D.J.; Gollas, B.; Hacker, V. Recent advancements in high performance polymer electrolyte fuel cell electrode fabrication—Novel materials and manufacturing processes. J. Power Sources 2023, 562, 232734. [Google Scholar] [CrossRef]
- Wilson, A.; Kleen, G.; Papageorgopoulos, D. DOE Hydrogen an Fuel Cells Program Record. Fuel Cell System Cost—2017. Available online: https://www.hydrogen.energy.gov/pdfs/17007_fuel_cell_system_cost_2017.pdf (accessed on 21 January 2018).
- Jaouen, F.; Proietti, E.; Lefèvre, M.; Chenitz, R.; Dodelet, J.-P.; Wu, G.; Chung, H.T.; Johnston, C.M.; Zelenay, P. Recent advances in non-precious metal catalysis for oxygen-reduction reaction in polymer electrolyte fuel cells. Energy Environ. Sci. 2011, 4, 114–130. [Google Scholar] [CrossRef]
- Gatalo, M.; Bonastre, A.M.; Moriau, L.J.; Burdett, H.; Ruiz-Zepeda, F.; Hughes, E.; Hodgkinson, A.; Šala, M.; Pavko, L.; Bele, M.; et al. Importance of Chemical Activation and the Effect of Low Operation Voltage on the Performance of Pt-Alloy Fuel Cell Electrocatalysts. ACS Appl. Energy Mater. 2022, 5, 8862–8877. [Google Scholar] [CrossRef]
- Ramaswamy, N.; Kumaraguru, S.; Koestner, R.; Fuller, T.; Gu, W.; Kariuki, N.; Myers, D.; Dudenas, P.J.; Kusoglu, A. Editors’ Choice—Ionomer Side Chain Length and Equivalent Weight Impact on High Current Density Transport Resistances in PEMFC Cathodes. J. Electrochem. Soc. 2021, 168, 024518. [Google Scholar] [CrossRef]
- Ramaswamy, N.; Gu, W.; Ziegelbauer, J.M.; Kumaraguru, S. Carbon Support Microstructure Impact on High Current Density Transport Resistances in PEMFC Cathode. J. Electrochem. Soc. 2020, 167, 064515. [Google Scholar] [CrossRef]
- Escudero-Escribano, M.; Jensen, K.D.; Jensen, A.W. Recent Advances in Bimetallic Electrocatalysts for Oxygen Reduction: Design Principles, Structure-function Relations and Active Phase Elucidation. Curr. Opin. Electrochem. 2018, 8, 135–146. [Google Scholar] [CrossRef]
- Toda, T.; Igarashi, H.; Uchida, H.; Watanabe, M. Enhancement of the Electroreduction of Oxygen on Pt Alloys with Fe, Ni, and Co. J. Electrochem. Soc. 1999, 146, 3750–3756. [Google Scholar] [CrossRef]
- Choi, S.; Xie, S.; Shao, M.; Odell, J.H.; Lu, N.; Peng, H.-C.; Protsailo, L.; Guerrero, S.; Park, J.; Xia, X.; et al. Synthesis and Characterization of 9 nm Pt–Ni Octahedra with a Record High Activity of 3.3 A/mgPt for the Oxygen Reduction Reaction. Nano Lett. 2013, 13, 3420–3425. [Google Scholar] [CrossRef] [PubMed]
- Stamenković, V.R.; Fowler, B.; Mun, B.S.; Wang, G.; Ross, P.N.; Lucas, C.A.; Marković, N.M. Improved Oxygen Reduction Activity on Pt3Ni(111) via Increased Surface Site Availability. Science 2007, 315, 493–497. [Google Scholar] [CrossRef] [PubMed]
- Stonehart, P. Development of Advanced Noble Metal-Alloy Electrocatalysts for Phosphoric Acid Fuel Cells (PAFC). Ber. Bunsenges. Phys. Chem. 1990, 22, 913–921. [Google Scholar] [CrossRef]
- Stamenković, V.; Mun, B.S.; Mayrhofer, K.J.J.; Ross, P.N.; Marković, N.M.; Rossmeisl, J.; Greeley, J.; Nørskov, J.K. Changing the Activity of Electrocatalysts for Oxygen Reduction by Tuning the Surface Electronic Structure. Angew. Chem. 2006, 45, 2897–2901. [Google Scholar] [CrossRef]
- Strasser, P.; Koh, S.; Anniyev, T.; Greeley, J.; More, K.; Yu, C.; Liu, Z.; Kaya, S.; Nordlund, D.; Ogasawara, H.; et al. Lattice-strain Control of the Activity in Dealloyed Core-shell Fuel Cell Catalysts. Nat. Chem. 2010, 2, 454–460. [Google Scholar] [CrossRef]
- Čolić, V.; Bandarenka, A.S. Pt Alloy Electrocatalysts for the Oxygen Reduction Reaction: From Model Surfaces to Nanostructured Systems. ACS Catal. 2016, 6, 5378–5385. [Google Scholar] [CrossRef]
- Calle-Vallejo, F.; Tymoczko, J.; Colic, V.; Vu, Q.H.; Pohl, M.D.; Morgenstern, K.; Loffreda, D.; Sautet, P.; Schuhmann, W.; Bandarenka, A.S. Finding Optimal Surface Sites on Heterogeneous Catalysts by Counting Nearest Neighbors. Science 2015, 350, 185–189. [Google Scholar] [CrossRef]
- Chattot, R.; Le Bacq, O.; Beermann, V.; Kühl, S.; Herranz, J.; Henning, S.; Kühn, L.; Asset, T.; Guétaz, L.; Renou, G.; et al. Surface Distortion as a Unifying Concept and Descriptor in Oxygen Reduction Reaction Electrocatalysis. Nat. Mater. 2018, 17, 827–833. [Google Scholar] [CrossRef]
- Huang, X.; Zhao, Z.; Cao, L.; Chen, Y.; Zhu, E.; Lin, Z.; Li, M.; Yan, A.; Zettl, A.; Wang, Y.M.; et al. High-performance Transition Metal-doped Pt3Ni Octahedra for Oxygen Reduction Reaction. Science 2015, 348, 1230–1234. [Google Scholar] [CrossRef]
- Chen, C.; Kang, Y.; Huo, Z.; Zhu, Z.; Huang, W.; Xin, H.L.; Snyder, J.D.; Li, D.; Herron, J.A.; Mavrikakis, M.; et al. Highly Crystalline Multimetallic Nanoframes with Three-Dimensional Electrocatalytic Surfaces. Science 2014, 343, 1339–1343. [Google Scholar] [CrossRef]
- Gatalo, M.; Moriau, L.; Petek, U.; Ruiz-Zepeda, F.; Šala, M.; Grom, M.; Galun, T.; Jovanovič, P.; Pavlišič, A.; Bele, M.; et al. CO-assisted ex-situ chemical activation of Pt-Cu/C oxygen reduction reaction electrocatalyst. Electrochim. Acta 2019, 306, 377–386. [Google Scholar] [CrossRef]
- Gatalo, M.; Bele, M.; Ruiz Zepeda, F.; Šest, E.; Šala, M.; Kamšek, A.R.; Maselj, N.; Galun, T.; Hodnik, N.; Gaberšček, M. A Double-Passivation Water-Based Galvanic Displacement Method for Reproducible Gram-Scale Production of High-Performance Platinum-Alloy Electrocatalysts. Angew. Chem. Int. Ed. 2019, 58, 13266–13270. [Google Scholar] [CrossRef]
- Logeshwaran, N.; Panneerselvam, I.R.; Ramakrishnan, S.; Kumar, R.S.; Kim, A.R.; Wang, Y.; Yoo, D.J. Quasihexagonal Platinum Nanodendrites Decorated over CoS 2-N-Doped Reduced Graphene Oxide for Electro-Oxidation of C1-, C2-, and C3-Type Alcohols. Adv. Sci. 2022, 9, 2105344. [Google Scholar] [CrossRef]
- Jia, Q.; Ramaker, D.E.; Ziegelbauer, J.M.; Ramaswamy, N.; Halder, A.; Mukerjee, S. Fundamental Aspects of Ad-Metal Dissolution and Contamination in Low and Medium Temperature Fuel Cell Electrocatalysis: A Cu Based Case Study Using In Situ Electrochemical X-ray Absorption Spectroscopy. J. Phys. Chem. C 2013, 117, 4585–4596. [Google Scholar] [CrossRef]
- Yu, Z.; Zhang, J.; Liu, Z.; Ziegelbauer, J.M.; Xin, H.; Dutta, I.; Muller, D.A.; Wagner, F.T. Comparison between Dealloyed PtCo3 and PtCu3 Cathode Catalysts for Proton Exchange Membrane Fuel Cells. J. Phys. Chem. C 2012, 116, 19877–19885. [Google Scholar] [CrossRef]
- Falina, I.; Pavlets, A.; Alekseenko, A.; Titskaya, E.; Kononenko, N. Influence of PtCu/C Catalysts Composition on Electrochemical Characteristics of Polymer Electrolyte Fuel Cell and Properties of Proton Exchange Membrane. Catalysts 2021, 11, 1063. [Google Scholar] [CrossRef]
- Bele, M.; Gatalo, M.; Jovanovič, P.; Ruiz-Zepeda, F.; Šala, M.; Šest, E.; Hodnik, N.; Hočevar, S.; Gatto, I.; Saccà, A.; et al. Insight on Single Cell Proton Exchange Membrane Fuel Cell Performance of Pt-Cu/C Cathode. Catalysts 2019, 9, 544. [Google Scholar] [CrossRef]
- Strlič, M.; Kolar, J.; Šelih, V.-S.; Kočar, D.; Pihlar, B. A Comparative Study of Several Transition Metals in Fenton-like Reaction System at Circum-neutral pH. Acta Chim. Slov. 2003, 50, 619–632. [Google Scholar]
- Frühwirt, P.; Kregar, A.; Törring, J.T.; Katrašnik, T.; Gescheidt, G. Holistic approach to chemical degradation of Nafion membranes in fuel cells: Modelling and predictions. Phys. Chem. Chem. Phys. 2020, 22, 5647–5666. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, C.Y. Modeling of Transient Platinum Degradation in a Low Pt-Loading PEFC under Current Cycling. J. Electrochem. Soc. 2017, 164, F171–F179. [Google Scholar] [CrossRef]
- Sellin, R.C.; Mozet, K.; Ménage, A.; Dillet, J.; Didierjean, S.; Maranzana, G. Measuring electro-osmotic drag coefficients in PFSA membranes without any diffusion assumption. Int. J. Hydrogen Energy 2019, 44, 24905–24912. [Google Scholar] [CrossRef]
- Park, Y.; Caton, J. An experimental investigation of electro-osmotic drag coefficients in a polymer electrolyte membrane fuel cell. Int. J. Hydrogen Energy 2008, 33, 7513–7520. [Google Scholar] [CrossRef]
- Hinaje, M.; Raël, S.; Noiying, P.; Nguyen, D.A.; Davat, B. An Equivalent Electrical Circuit Model of Proton Exchange Membrane Fuel Cells Based on Mathematical Modelling. Energies 2012, 5, 2724–2744. [Google Scholar] [CrossRef]
- Page, S.C.; Anbuky, A.H.; Krumdieck, S.P.; Brouwer, J. Test method and equivalent circuit modeling of a PEM fuel cell in a passive state. IEEE Trans. Energy Convers. 2007, 22, 764–773. [Google Scholar] [CrossRef]
- Orazem, M.E.; Roy, S.K. Deterministic Impedance Models for Interpretation of Low-Frequency Inductive Loops in PEM Fuel Cells. ECS Trans. 2019, 3, 1031–1040. [Google Scholar] [CrossRef]
- Klotz, D. Negative capacitance or inductive loop?—A general assessment of a common low frequency impedance feature. Electrochem. Commun. 2019, 98, 58–62. [Google Scholar] [CrossRef]
- Halvorsen, I.J.; Pivac, I.; Bezmalinović, D.; Barbir, F.; Zenith, F. Electrochemical low-frequency impedance spectroscopy algorithm for diagnostics of PEM fuel cell degradation. Int. J. Hydrogen Energy 2020, 45, 1325–1334. [Google Scholar] [CrossRef]
- Roy, S.K.; Orazem, M.E.; Tribollet, B. Interpretation of Low-Frequency Inductive Loops in PEM Fuel Cells. J. Electrochem. Soc. 2007, 154, B1378. [Google Scholar] [CrossRef]
- Grandi, M.; Mayer, K.; Gatalo, M.; Kapun, G.; Ruiz-Zepeda, F.; Marius, B.; Gaberšček, M.; Hacker, V. The Influence Catalyst Layer Thickness on Resistance Contributions of PEMFC Determined by Electrochemical Impedance Spectroscopy. Energies 2021, 14, 7299. [Google Scholar] [CrossRef]
- Kuhn, H.; Andreaus, B.; Wokaun, A.; Scherer, G.G. Electrochemical impedance spectroscopy applied to polymer electrolyte fuel cells with a pseudo reference electrode arrangement. Electrochim. Acta 2006, 51, 1622–1628. [Google Scholar] [CrossRef]
- Yuan, X.; Wang, H.; Colin Sun, J.; Zhang, J. AC impedance technique in PEM fuel cell diagnosis-A review. Int. J. Hydrogen Energy 2007, 32, 4365–4380. [Google Scholar] [CrossRef]
- Lechartier, E.; Laffly, E.; Péra, M.C.; Gouriveau, R.; Hissel, D.; Zerhouni, N. Proton exchange membrane fuel cell behavioral model suitable for prognostics. Int. J. Hydrogen Energy 2015, 40, 8384–8397. [Google Scholar] [CrossRef]
- Heinzmann, M.; Weber, A.; Ivers-Tiffée, E. Advanced impedance study of polymer electrolyte membrane single cells by means of distribution of relaxation times. J. Power Sources 2018, 402, 24–33. [Google Scholar] [CrossRef]
- Bele, M.; Gaberšček, M.; Kapun, G.; Hodnik, N.; Hočevar, S. Electrocatalytic Composite(s), Associated Composition(s), and Associated Process(es). U.S. Patent US9147885B2, 29 September 2015. JP Patent JP6028027B2, 16 November 2016; EU Patent EP2735044A2, 28 May 2014; WO Patent WO2013012398A2, 24 January 2013, n.d. [Google Scholar]
- Bele, M.; Jovanovič, P.; Pavlišič, A.; Jozinović, B.; Zorko, M.; Rečnik, A.; Chernyshova, E.; Hočevar, S.; Hodnik, N.; Gaberšček, M. A Highly Active PtCu3 Intermetallic Core-shell, Multilayered Pt-skin, Carbon Embedded Electrocatalyst Produced by a Scale-up sol-gel Synthesis. Chem. Commun. 2014, 50, 13124–13126. [Google Scholar] [CrossRef] [PubMed]
- Hodnik, N.; Bele, M.; Hočevar, S. New Pt-skin Electrocatalysts for Oxygen Reduction and Methanol Oxidation Reactions. Electrochem. Commun. 2012, 23, 125–128. [Google Scholar] [CrossRef]
- Hodnik, N.; Jeyabharathi, C.; Meier, J.C.; Kostka, A.; Phani, K.L.; Rečnik, A.; Bele, M.; Hočevar, S.; Gaberšček, M.; Mayrhofer, K.J.J. Effect of Ordering of PtCu3 Nanoparticle Structure on the Activity and Stability for the Oxygen Reduction Reaction. Phys. Chem. Chem. Phys. 2014, 16, 13610–13615. [Google Scholar] [CrossRef]
- Gatalo, M.; Hodnik, N.; Gaberšček, M.; Bele, M. Method for Preparation of a Supported Noble Metal-Metal Alloy Composite, and the Obtained Supported Noble Metal-Metal Alloy Composite. LU101157, 17 March 2020. [Google Scholar]
- Gatalo, M.; Hodnik, N.; Bele, M.; Jovanovič, P.; Gaberšček, M.; Grom, M. Method of Treating a Platinum-Alloy Catalyst, and Device for Carrying Out the Method of Treating a Platinum-Alloy Catalyst. EP18196870.2, 2 April 2020. [Google Scholar]
- van der Vliet, D.; Strmčnik, D.; Wang, C.; Stamenković, V.R.; Marković, N.M.; Koper, M.T.M. On the Importance of Correcting for the Uncompensated Ohmic Resistance in Model Experiments of the Oxygen Reduction Reaction. J. Electroanal. Chem. 2010, 647, 29–34. [Google Scholar] [CrossRef]
- Bard, A.J. Electrochemical Methods: Fundamentals and Applications; Bard, A.J., Faulkner, L.R., Eds.; Wiley: New York, NY, USA, 1980; ISBN 0471055425. [Google Scholar]
- Mayrhofer, K.J.J.; Strmčnik, D.; Blizanac, B.B.; Stamenković, V.R.; Arenz, M.; Marković, N.M. Measurement of Oxygen Reduction Activities via the Rotating Disc Electrode Method: From Pt Model Surfaces to Carbon-supported High Surface Area Catalysts. Electrochim. Acta 2008, 53, 3181–3188. [Google Scholar] [CrossRef]
- Afsahi, F.; Mathieu-Potvin, F.; Kaliaguine, S. Impact of Ionomer Content on Proton Exchange Membrane Fuel Cell Performance. Fuel Cells 2016, 16, 107–125. [Google Scholar] [CrossRef]
- Kim, K.H.; Lee, K.Y.; Kim, H.J.; Cho, E.A.; Lee, S.Y.; Lim, T.H.; Yoon, S.P.; Hwang, I.C.; Jang, J.H. The effects of Nafion®ionomer content in PEMFC MEAs prepared by a catalyst-coated membrane (CCM) spraying method. Int. J. Hydrogen Energy 2010, 35, 2119–2126. [Google Scholar] [CrossRef]
- Bodner, M. Degradation Phenomena in Polymer Electrolyte Fuel Cells; Graz University of Technology: Graz, Austria, 2017. [Google Scholar]
- Ahluwalia, R.K.; Arisetty, S.; Peng, J.-K.; Subbaraman, R.; Wang, X.; Kariuki, N.; Myers, D.J.; Mukundan, R.; Borup, R.; Polevaya, O. Dynamics of Particle Growth and Electrochemical Surface Area Loss due to Platinum Dissolution. J. Electrochem. Soc. 2014, 161, F291–F304. [Google Scholar] [CrossRef]
- Wan, T.H.; Saccoccio, M.; Chen, C.; Ciucci, F. Influence of the Discretization Methods on the Distribution of Relaxation Times Deconvolution: Implementing Radial Basis Functions with DRTtools. Electrochim. Acta 2015, 184, 483–499. [Google Scholar] [CrossRef]
- Cruz-Manzo, S.; Chen, R. Electrochemical impedance study on estimating the mass transport resistance in the polymer electrolyte fuel cell cathode catalyst layer. J. Electroanal. Chem. 2013, 702, 45–48. [Google Scholar] [CrossRef]
- Hirschorn, B.; Orazem, M.E.; Tribollet, B.; Vivier, V.; Frateur, I.; Musiani, M. Determination of effective capacitance and film thickness from constant-phase-element parameters. Electrochim. Acta 2010, 55, 6218–6227. [Google Scholar] [CrossRef]
- Shoar Abouzari, M.R.; Berkemeier, F.; Schmitz, G.; Wilmer, D. On the physical interpretation of constant phase elements. Solid State Ion. 2009, 180, 922–927. [Google Scholar] [CrossRef]
- McNealy, B.E.; Hertz, J.L. On the use of the constant phase element to understand variation in grain boundary properties. Solid State Ion. 2014, 256, 52–60. [Google Scholar] [CrossRef]
- Huang, J.; Li, Z.; Liaw, B.Y.; Zhang, J. Graphical analysis of electrochemical impedance spectroscopy data in Bode and Nyquist representations. J. Power Sources 2016, 309, 82–98. [Google Scholar] [CrossRef]
- Pavko, L.; Gatalo, M.; Križan, G.; Križan, J.; Ehelebe, K.; Ruiz-Zepeda, F.; Šala, M.; Dražić, G.; Geuß, M.; Kaiser, P.; et al. Toward the Continuous Production of Multigram Quantities of Highly Uniform Supported Metallic Nanoparticles and Their Application for Synthesis of Superior Intermetallic Pt-Alloy ORR Electrocatalysts. ACS Appl. Energy Mater. 2021, 4, 13819–13829. [Google Scholar] [CrossRef]
- Molaeimanesh, G.R.; Nazemian, M. Investigation of GDL compression effects on the performance of a PEM fuel cell cathode by lattice Boltzmann method. J. Power Sources 2017, 359, 494–506. [Google Scholar] [CrossRef]
- Yoshida, T.; Kojima, K. Toyota MIRAI Fuel Cell Vehicle and Progress Toward a Future Hydrogen Society. Interface Mag. 2015, 24, 45–49. [Google Scholar] [CrossRef]
- Darling, R.M.; Meyers, J.P. Kinetic model of platinum dissolution in PEMFCs. J. Electrochem. Soc. 2003, 150, 1523–1527. [Google Scholar] [CrossRef]
- US Department of Energy (DOE). Multi-Year Research, Development, and Demonstration Plan: 3.4 Fuel Cells; Fuel Cell Technologies Office: Washington, DC, USA, 2012. [CrossRef]
- Padgett, E.; Yarlagadda, V.; Holtz, M.E.; Ko, M.; Levin, B.D.A.; Kukreja, R.S.; Ziegelbauer, J.M.; Andrews, R.N.; Ilavsky, J.; Kongkanand, A.; et al. Mitigation of PEM Fuel Cell Catalyst Degradation with Porous Carbon Supports. J. Electrochem. Soc. 2019, 166, F198–F207. [Google Scholar] [CrossRef]
- Yarlagadda, V.; Carpenter, M.K.; Moylan, T.E.; Kukreja, R.S.; Koestner, R.; Gu, W.; Thompson, L.; Kongkanand, A. Boosting Fuel Cell Performance with Accessible Carbon Mesopores. ACS Energy Lett. 2018, 3, 618–621. [Google Scholar] [CrossRef]
- Padgett, E.; Andrejevic, N.; Liu, Z.; Kongkanand, A.; Gu, W.; Moriyama, K.; Jiang, Y.; Kumaraguru, S.; Moylan, T.E.; Kukreja, R.; et al. Editors’ Choice—Connecting Fuel Cell Catalyst Nanostructure and Accessibility Using Quantitative Cryo-STEM Tomography. J. Electrochem. Soc. 2018, 165, F173–F180. [Google Scholar] [CrossRef]
- Kreitmeier, S.; Wokaun, A.; Büchi, F.N. Local Catalyst Support Degradation during Polymer Electrolyte Fuel Cell Start-Up and Shutdown. J. Electrochem. Soc. 2012, 159, F787–F793. [Google Scholar] [CrossRef]
- Owejan, J.P.; Soc, J.E.; Owejan, J.P.; Owejan, J.E.; Gu, W. Impact of Platinum Loading and Catalyst Layer Structure on PEMFC Performance Impact of Platinum Loading and Catalyst Layer Structure on PEMFC Performance. J. Electrochem. Soc. 2013, 160, F824–F833. [Google Scholar] [CrossRef]


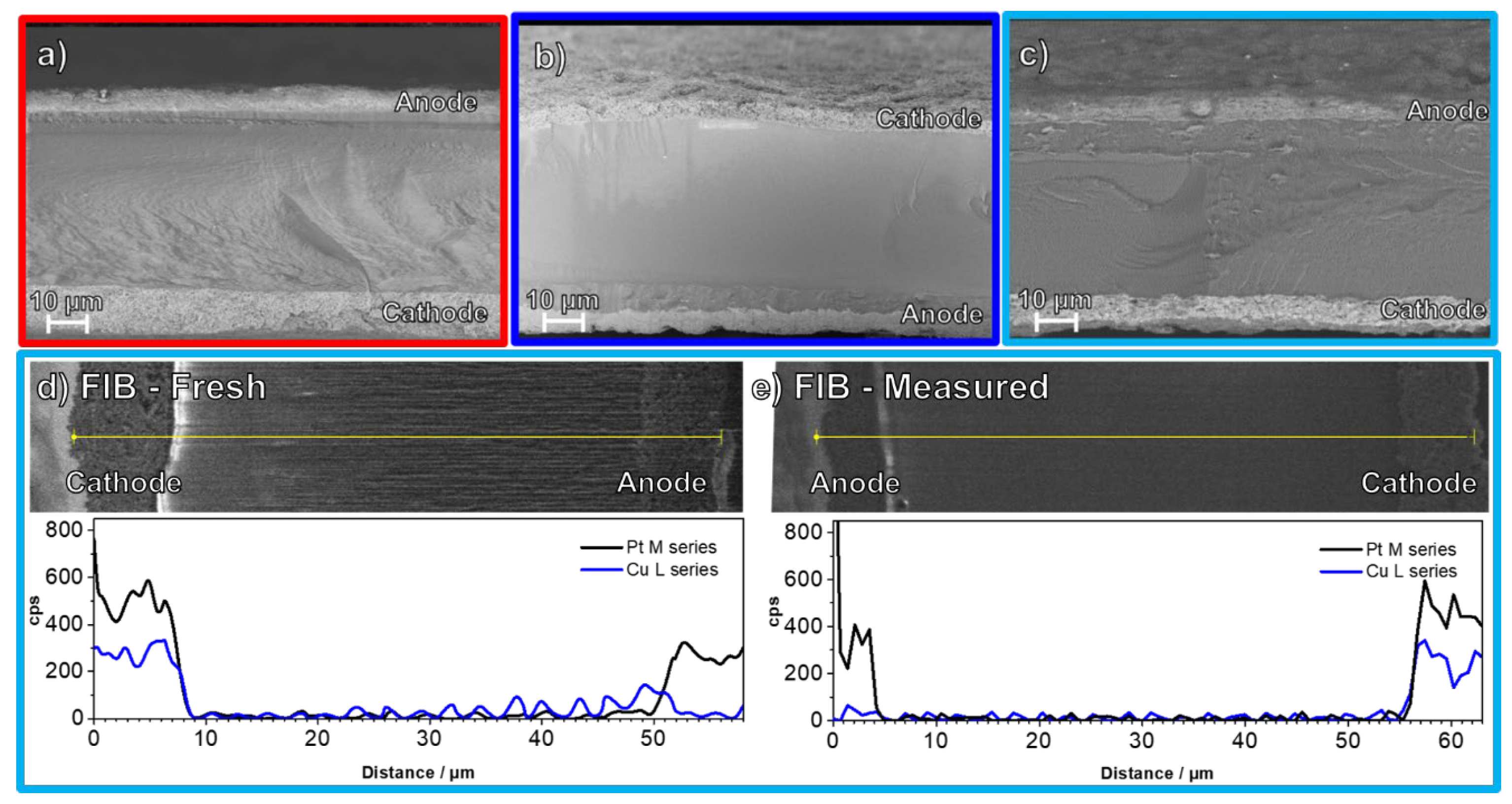
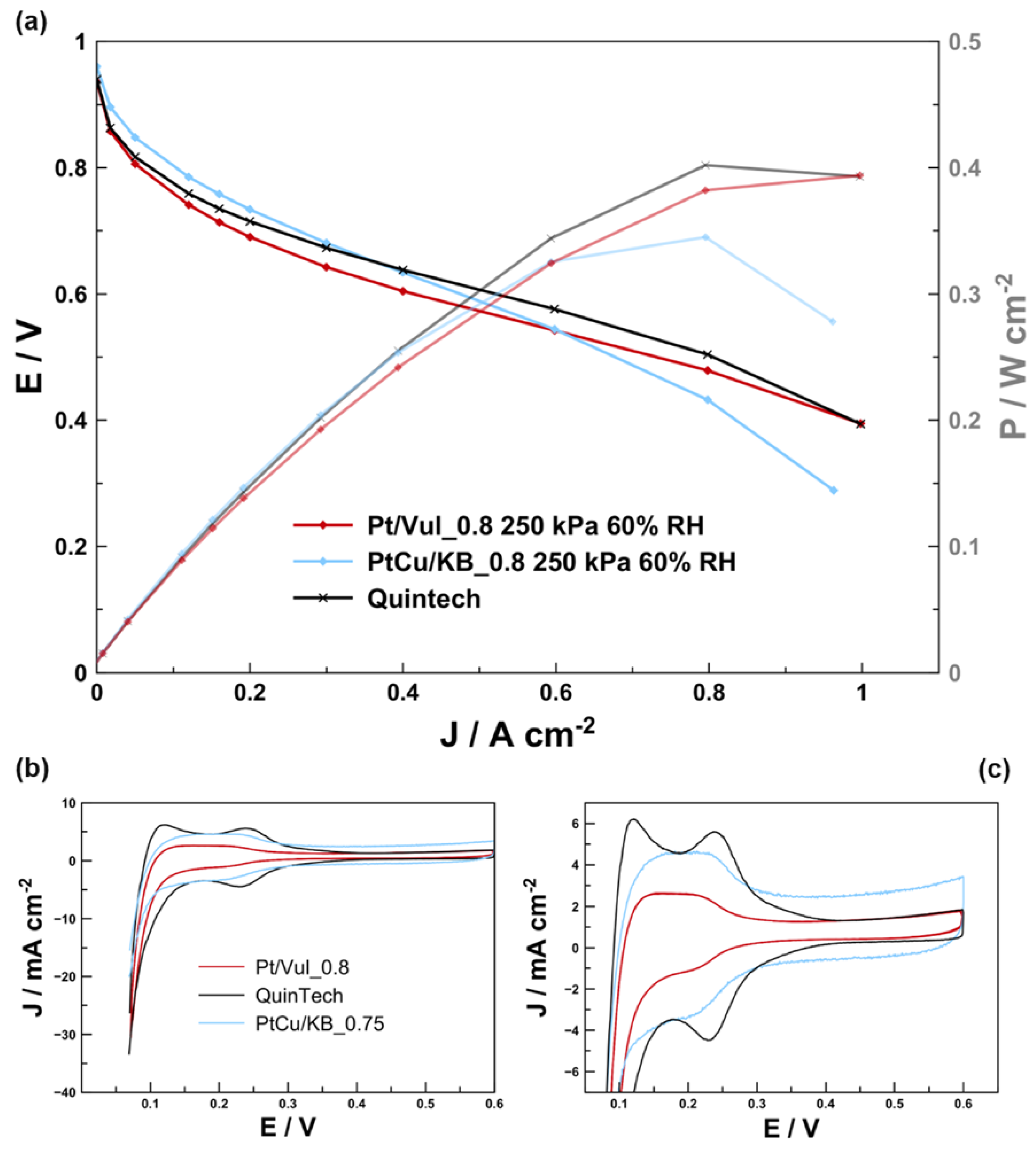

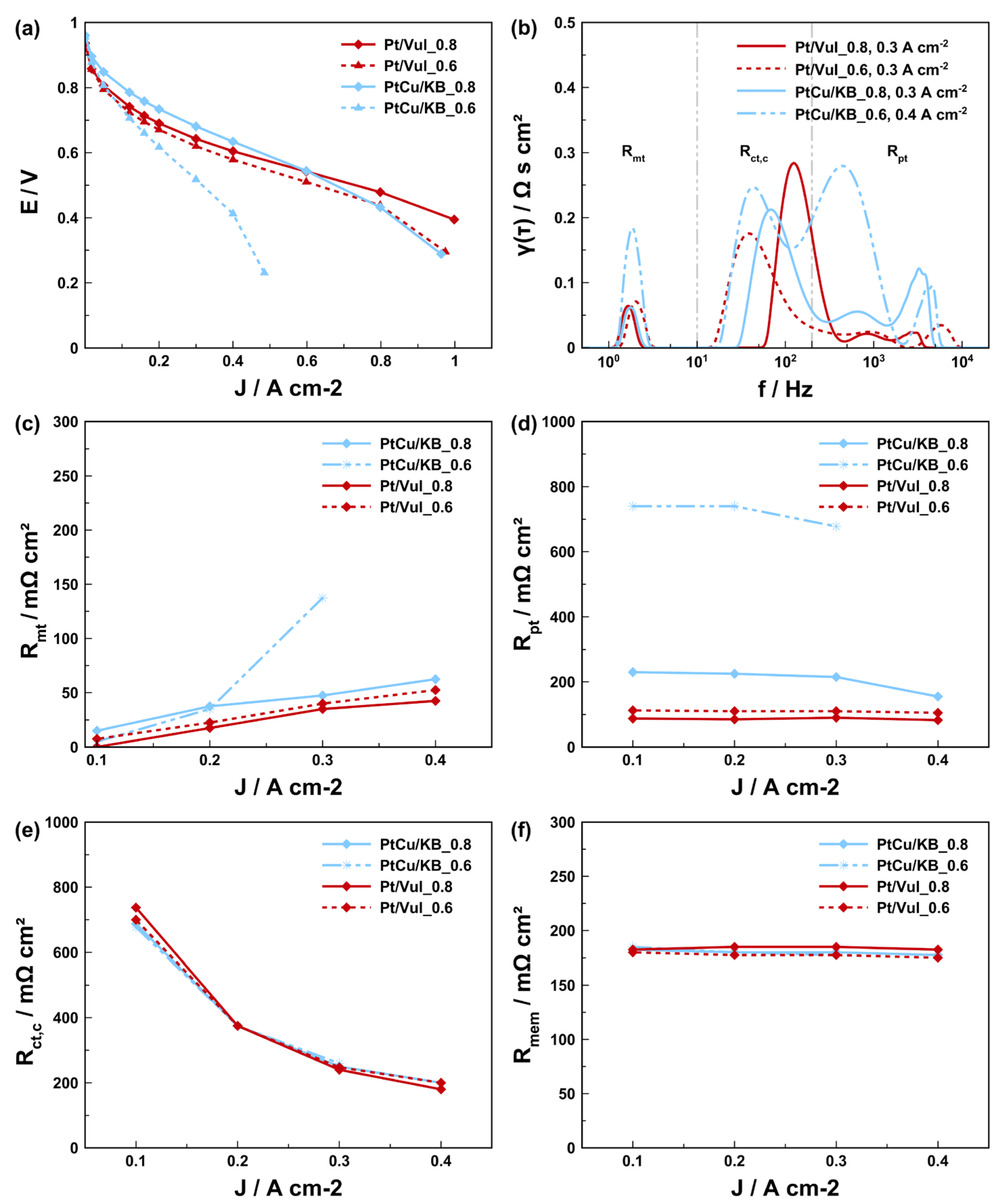

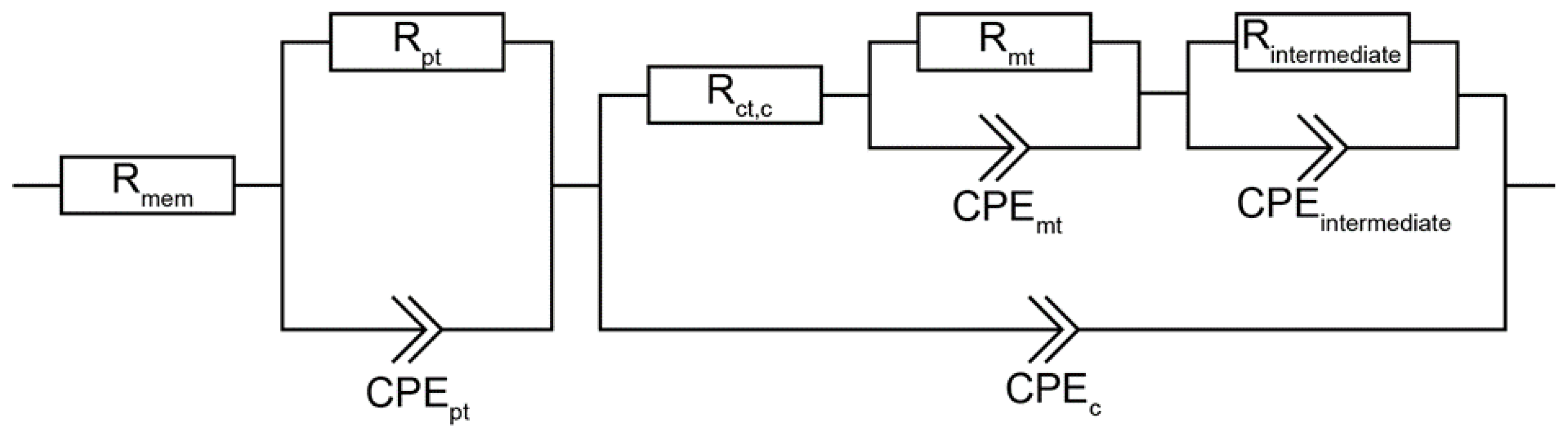{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CCM Code | Cathode Catalyst | Pt-Loading Loading/mg cm2 | Cathode I/C | Cathode Catalyst Layer Thickness/µm | Anode Catalyst | Pt-Loading Loading/mg cm2 |
|---|---|---|---|---|---|---|
| Pt/Vul_0.8 | Pt/Vul 40% (HiSPec 4000) | 0.125 | 0.8 | 4.4 ± 0.3 | Pt/Vul 20% (HiSPec 3000) | 0.05 |
| Pt/Vul_0.6 | Pt/Vul 20% (HiSPec 3000) | 0.125 | 0.6 | 11.7 ± 0.3 | Pt/Vul 20% (HiSPec 3000) | 0.05 |
| PtCu/KB_0.8 | PtCu3/KB 43% | 0.125 | 0.8 | 8.4 ± 0.2 | Pt/Vul 20% (HiSPec 3000) | 0.05 |
| PtCu/KB_0.8 * | PtCu3/KB 43% | 0.125 | 0.8 | 8.4 ± 0.2 | Pt/Vul 20% (HiSPec 3000) | 0.05 |
| PtCu/KB_0.6 | PtCu3/KB 43% | 0.125 | 0.6 | n.a. | Pt/Vul 20% (HiSPec 3000) | 0.05 |
| QuinTech | Pt/C | 0.6 | n.a. | n.a. | Pt/C | 0.3 |
| CCM Code | Fuel/Oxidant | θ/°C | p/kPa | RH/% | V/mL min−1 | CCM Code |
|---|---|---|---|---|---|---|
| Pt/Vul_0.8 | H2/air | 80 | 250 | 60 | 600 | Pt/Vul_0.8 |
| Pt/Vul_0.6 | H2/air | 80 | 250 | 60 | 600 | Pt/Vul_0.6 |
| Pt/Vul_0.6 | H2/air | 80 | 100 | 60 | 600 | Pt/Vul_0.6 |
| Pt/Vul_0.6 | H2/air | 80 | 100 | 100 | 600 | Pt/Vul_0.6 |
| Pt/Vul_0.6 | H2/air | 80 | 100 | 60 | 800 | Pt/Vul_0.6 |
| PtCu/KB_0.8 | H2/air | 80 | 250 | 60 | 600 | PtCu/KB_0.8 |
| CCM Code | ECSA/m² gPt−1 |
|---|---|
| Pt/Vul_0.8 | 32.8 |
| Quintech | 31.0 |
| PtCu/KB_0.8 | 57.9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grandi, M.; Gatalo, M.; Kamšek, A.R.; Kapun, G.; Mayer, K.; Ruiz-Zepeda, F.; Šala, M.; Marius, B.; Bele, M.; Hodnik, N.; et al. Mechanistic Study of Fast Performance Decay of PtCu Alloy-based Catalyst Layers for Polymer Electrolyte Fuel Cells through Electrochemical Impedance Spectroscopy. Materials 2023, 16, 3544. https://doi.org/10.3390/ma16093544
Grandi M, Gatalo M, Kamšek AR, Kapun G, Mayer K, Ruiz-Zepeda F, Šala M, Marius B, Bele M, Hodnik N, et al. Mechanistic Study of Fast Performance Decay of PtCu Alloy-based Catalyst Layers for Polymer Electrolyte Fuel Cells through Electrochemical Impedance Spectroscopy. Materials. 2023; 16(9):3544. https://doi.org/10.3390/ma16093544
Chicago/Turabian StyleGrandi, Maximilian, Matija Gatalo, Ana Rebeka Kamšek, Gregor Kapun, Kurt Mayer, Francisco Ruiz-Zepeda, Martin Šala, Bernhard Marius, Marjan Bele, Nejc Hodnik, and et al. 2023. "Mechanistic Study of Fast Performance Decay of PtCu Alloy-based Catalyst Layers for Polymer Electrolyte Fuel Cells through Electrochemical Impedance Spectroscopy" Materials 16, no. 9: 3544. https://doi.org/10.3390/ma16093544
APA StyleGrandi, M., Gatalo, M., Kamšek, A. R., Kapun, G., Mayer, K., Ruiz-Zepeda, F., Šala, M., Marius, B., Bele, M., Hodnik, N., Bodner, M., Gaberšček, M., & Hacker, V. (2023). Mechanistic Study of Fast Performance Decay of PtCu Alloy-based Catalyst Layers for Polymer Electrolyte Fuel Cells through Electrochemical Impedance Spectroscopy. Materials, 16(9), 3544. https://doi.org/10.3390/ma16093544