
Advanced Mass Spectrometric Techniques for the Comprehensive Study of Synthesized Silicon-Based Silyl Organic Compounds: Identifying Fragmentation Pathways and Characterization
 , , ,
, , ,  , , and
, , and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
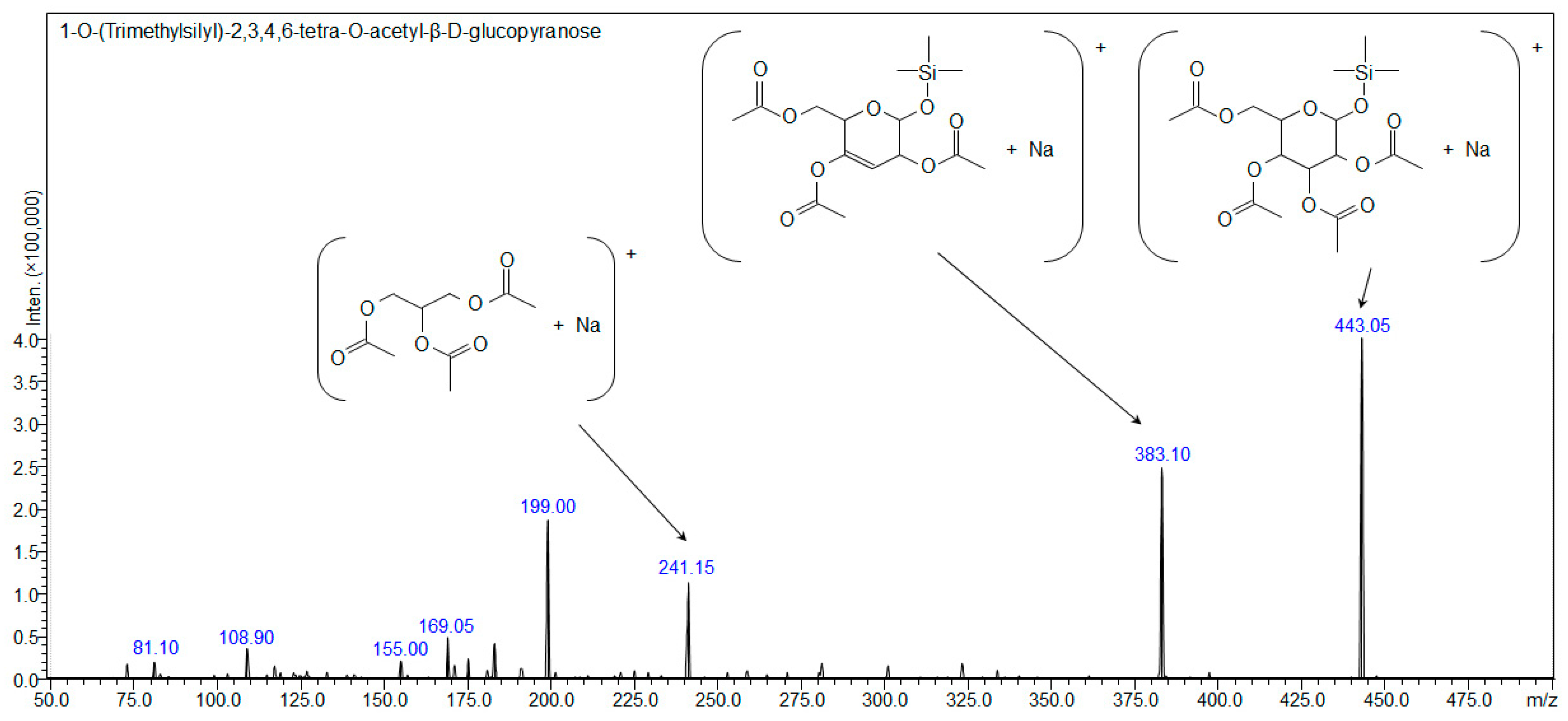
2.2. Synthesis of 1-O-(Trimethylsilyl)-2,3,4,6-tetra-O-acetyl-β-d-glucopyranose (Compound 1)
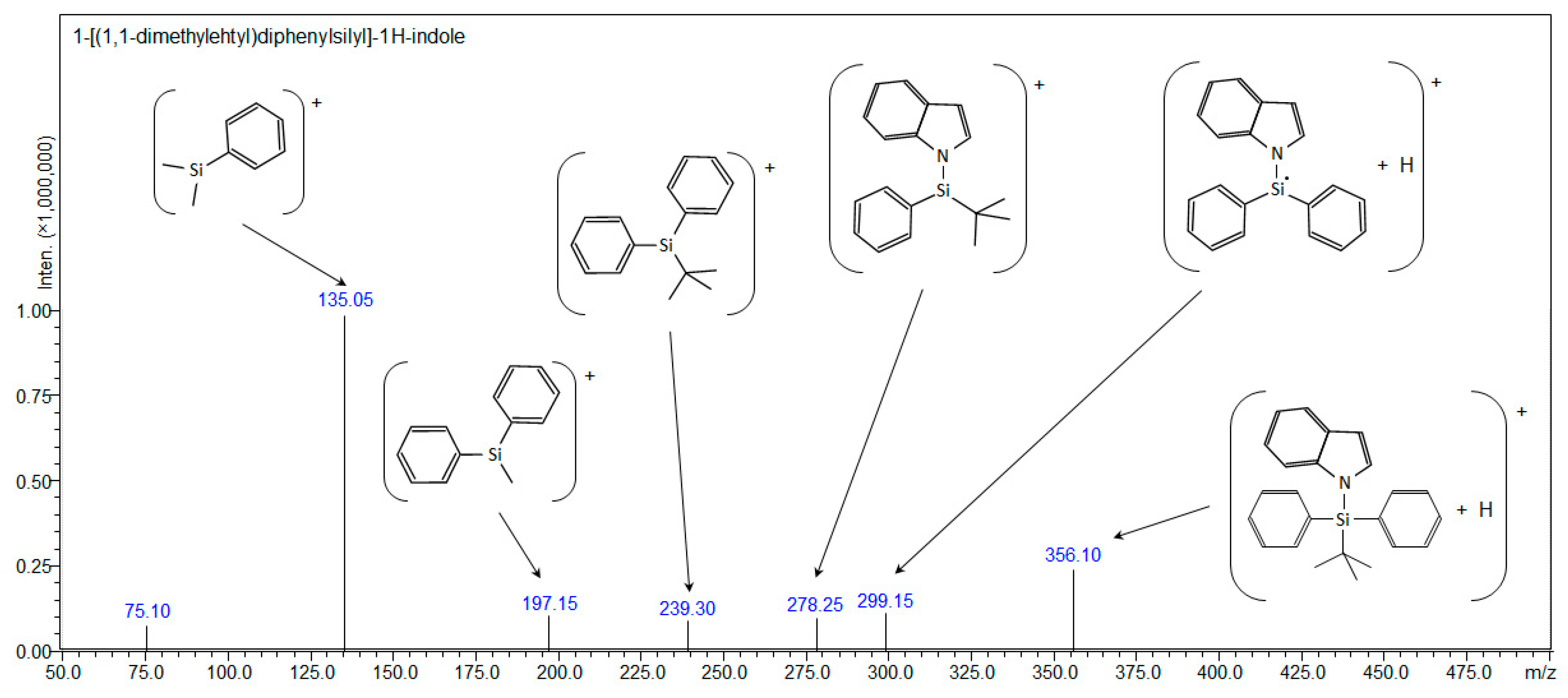
2.3. Synthesis of 1-[(1,1-Dimethylehtyl)diphenylsilyl]-1H-indole (Compound 2)
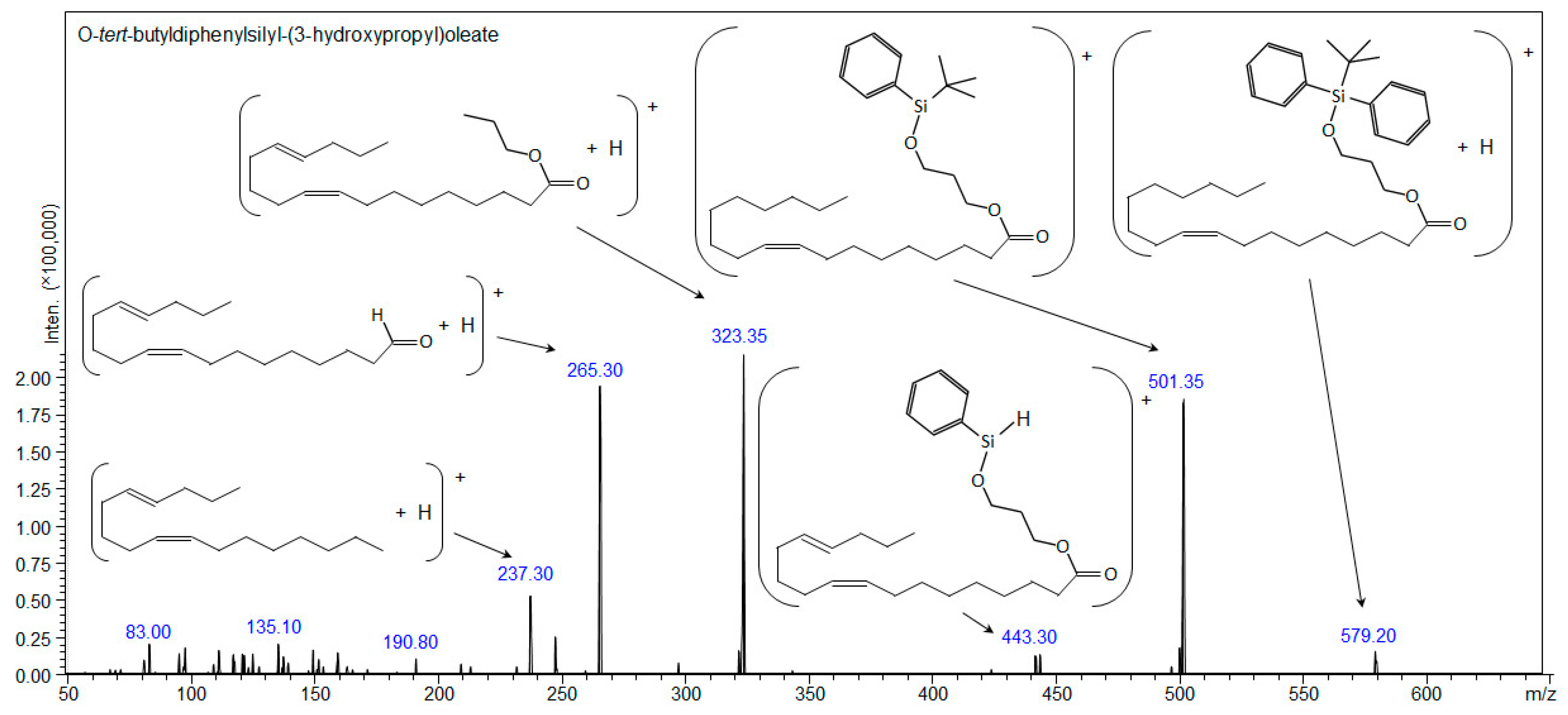
2.4. Synthesis of O-Tert-butyldiphenylsilyl-(3-hydroxypropyl)oleate (Compound 3)
2.5. Synthesis of 1-O-Tert-Butyldiphenylsilyl-myo-inositol (Compound 4)
2.6. NMR Analysis
2.7. Compound Analysis Using Mass Spectrometry Techniques
2.7.1. NALDI/SALDI MS Analysis on Silver Nanoparticles
2.7.2. FT-ICR-MS Analysis
2.7.3. QqQ ESI-MS/MS Analysis
3. Results and Discussion
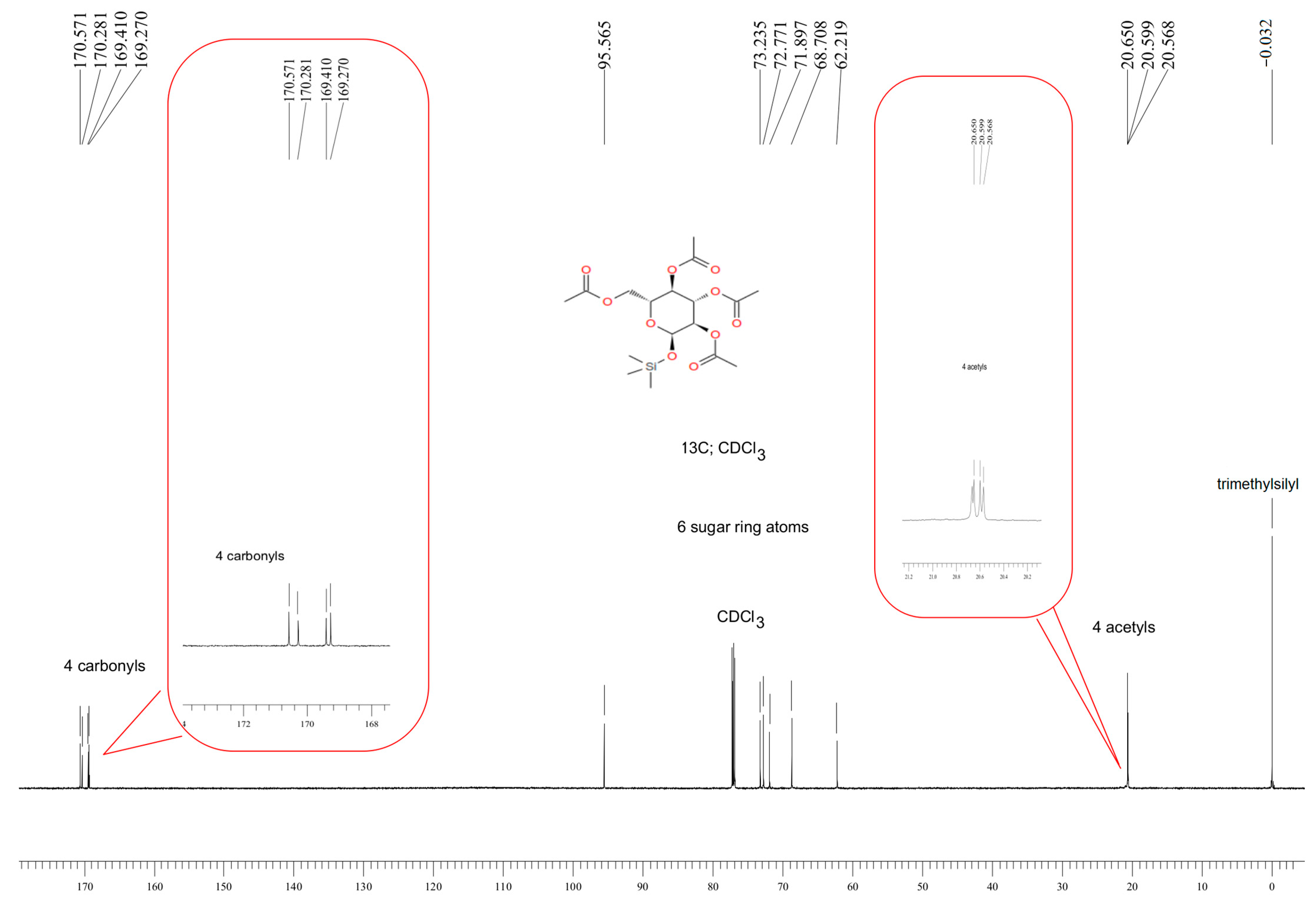
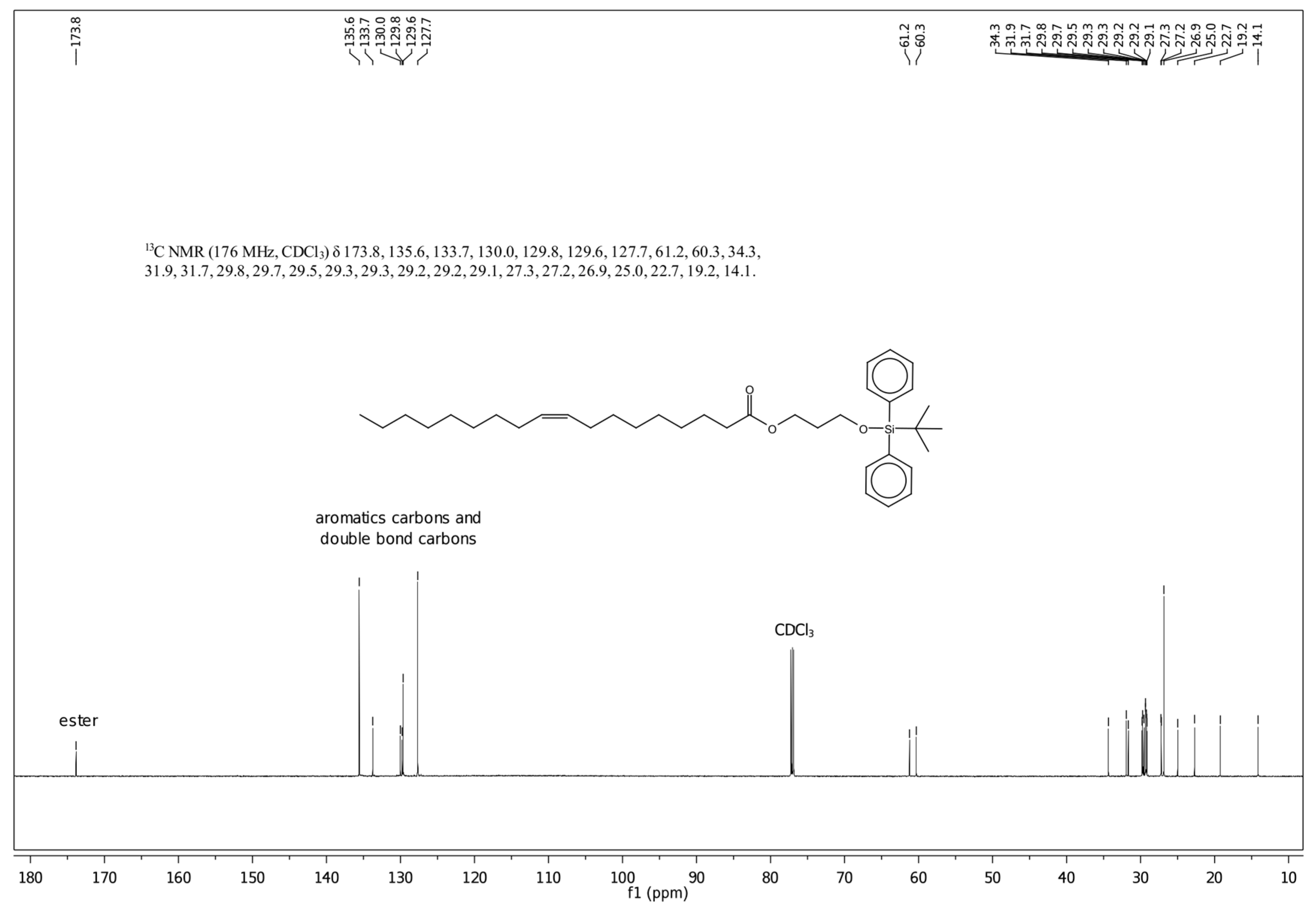
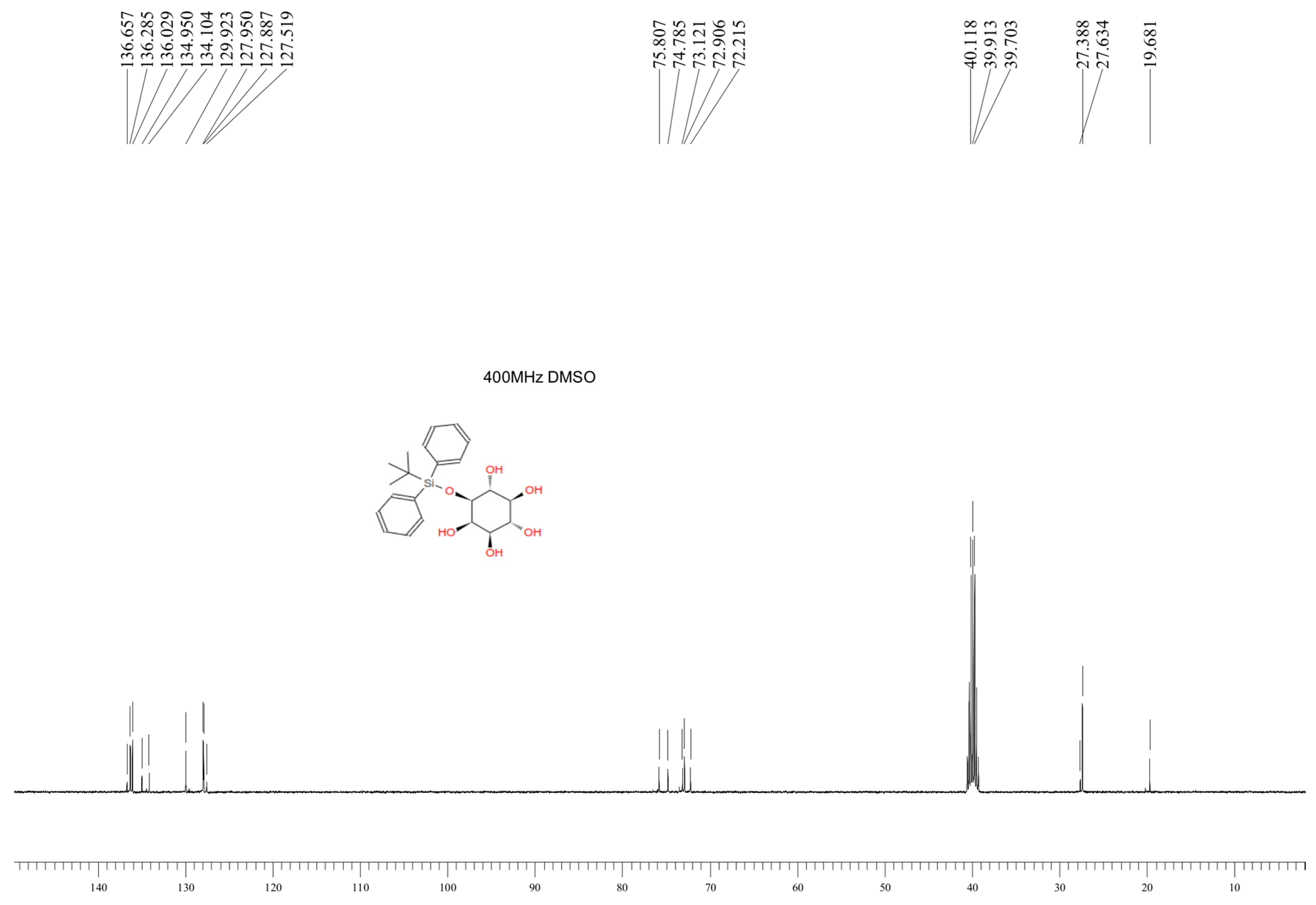
3.1. Characteristics of the Obtained Organic Compounds Using NMR Approach
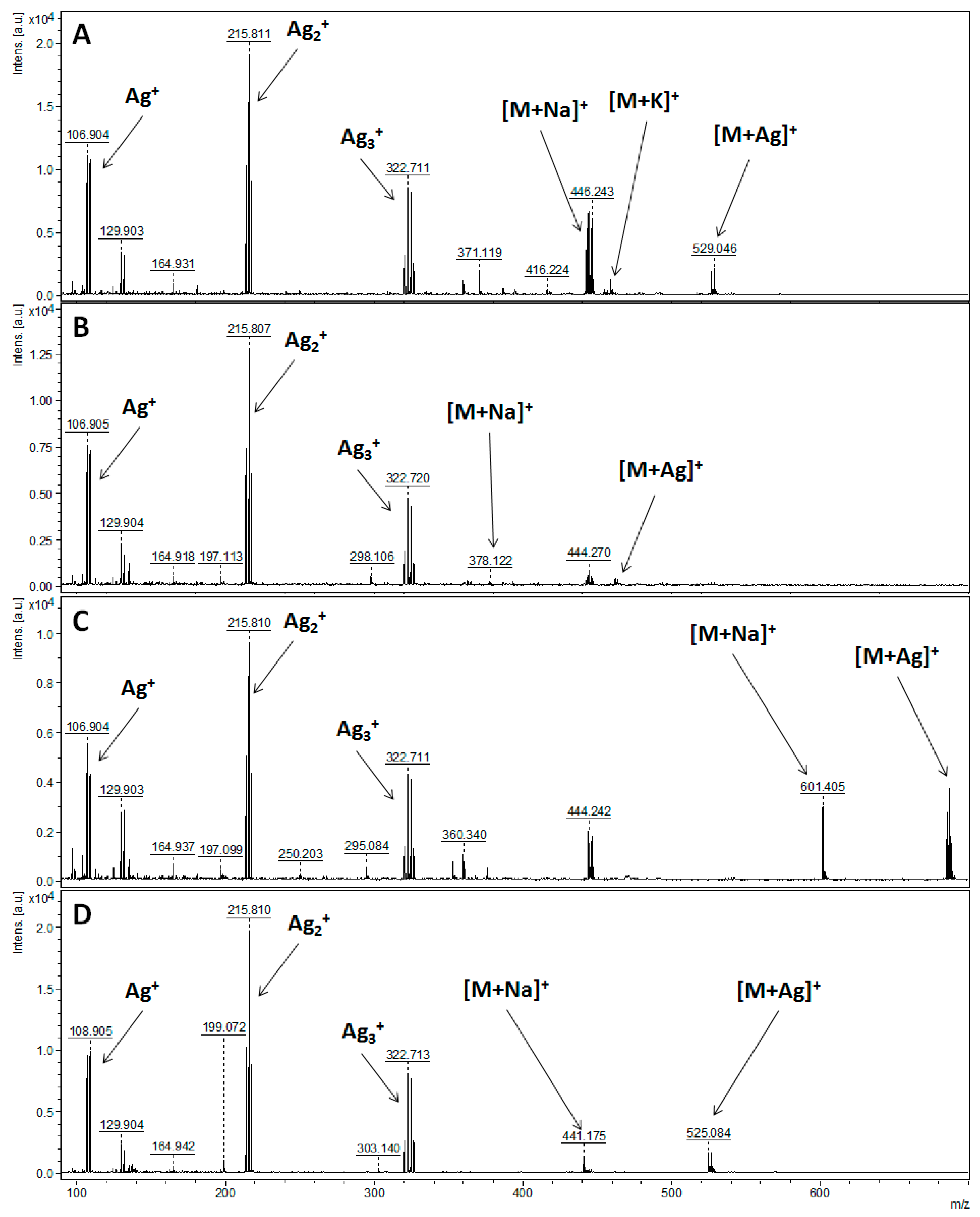
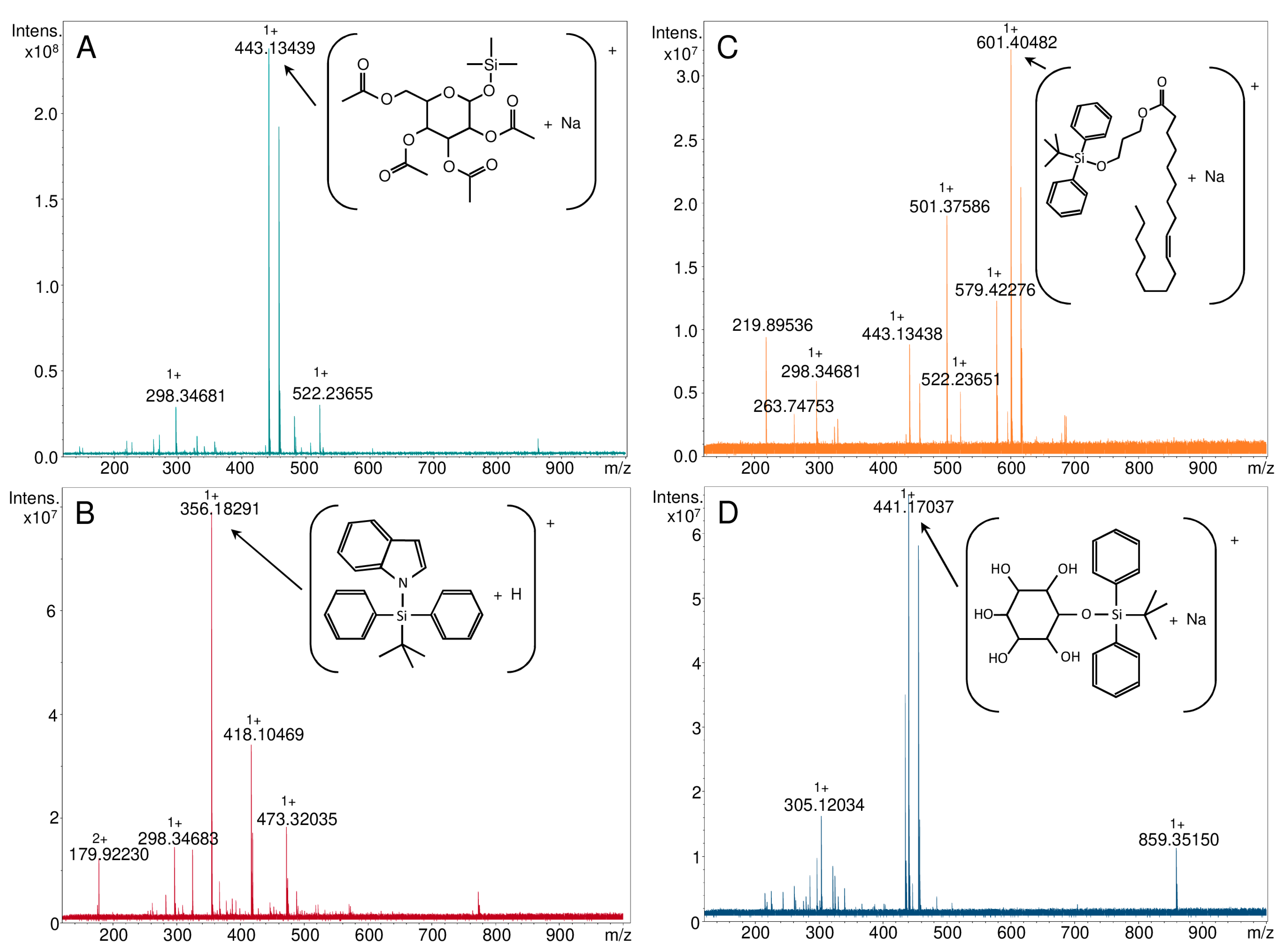
3.2. Characteristics of the Obtained Organic Compounds Using Mass Spectrometry Techniques
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bhuiyan, F.R.; Howlader, S.; Raihan, T.; Hasan, M. Plants Metabolites: Possibility of Natural Therapeutics against the COVID-19 Pandemic. Front. Med. 2020, 7, 444. [Google Scholar] [CrossRef]
- Hussein, R.A.; El-Anssary, A.A. Plants Secondary Metabolites: The Key Drivers of the Pharmacological Actions of Medicinal Plants. In Herbal Medicine; Builders, P.F., Ed.; IntechOpen: London, UK, 2018. [Google Scholar] [CrossRef]
- Singh, B.; Bhat, T.K.; Singh, B. Potential Therapeutic Applications of Some Antinutritional Plant Secondary Metabolites. J. Agric. Food Chem. 2003, 51, 5579–5597. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, F.; Palmeira-de-Oliveira, A.; das Neves, J.; Sarmento, B.; Amaral, M.H.; Oliveira, M.B. Medicago spp. Extracts as Promising Ingredients for Skin Care Products. Ind. Crops Prod. 2013, 49, 634–644. [Google Scholar] [CrossRef]
- Kasote, D.M.; Katyare, S.S.; Hegde, M.V.; Bae, H. Significance of Antioxidant Potential of Plants and Its Relevance to Therapeutic Applications. Int. J. Biol. Sci. 2015, 11, 982–991. [Google Scholar] [CrossRef]
- Yang, L.; Wen, K.-S.; Ruan, X.; Zhao, Y.-X.; Wei, F.; Wang, Q. Response of Plant Secondary Metabolites to Environmental Factors. Molecules 2018, 23, 762. [Google Scholar] [CrossRef]
- Luyckx, M.; Hausman, J.-F.; Lutts, S.; Guerriero, G. Silicon and Plants: Current Knowledge and Technological Perspectives. Front. Plant Sci. 2017, 8, 411. [Google Scholar] [CrossRef] [PubMed]
- Frew, A.; Weston, L.A.; Reynolds, O.L.; Gurr, G.M. The Role of Silicon in Plant Biology: A Paradigm Shift in Research Approach. Ann. Bot. 2018, 121, 1265–1273. [Google Scholar] [CrossRef]
- Ahanger, M.A.; Bhat, J.A.; Siddiqui, M.H.; Rinklebe, J.; Ahmad, P. Integration of Silicon and Secondary Metabolites in Plants: A Significant Association in Stress Tolerance. J. Exp. Bot. 2020, 71, 6758–6774. [Google Scholar] [CrossRef]
- Ma, J.F.; Miyake, Y.; Takahashi, E. Chapter 2 Silicon as a Beneficial Element for Crop Plants. In Studies in Plant Science; Datnoff, L.E., Snyder, G.H., Korndörfer, G.H., Eds.; Silicon in Agriculture; Elsevier: Amsterdam, The Netherlands, 2001; Volume 8, pp. 17–39. [Google Scholar]
- Currie, H.A.; Perry, C.C. Silica in Plants: Biological, Biochemical and Chemical Studies. Ann. Bot. 2007, 100, 1383–1389. [Google Scholar] [CrossRef]
- Coskun, D.; Deshmukh, R.; Sonah, H.; Menzies, J.G.; Reynolds, O.; Ma, J.F.; Kronzucker, H.J.; Bélanger, R.R. The Controversies of Silicon’s Role in Plant Biology. New Phytol. 2019, 221, 67–85. [Google Scholar] [CrossRef]
- Sahebi, M.; Hanafi, M.M.; Siti Nor Akmar, A.; Rafii, M.Y.; Azizi, P.; Tengoua, F.F.; Nurul Mayzaitul Azwa, J.; Shabanimofrad, M. Importance of Silicon and Mechanisms of Biosilica Formation in Plants. BioMed Res. Int. 2015, 2015, e396010. [Google Scholar] [CrossRef]
- Cabrera, Y.; Cabrera, A.; Larsen, F.H.; Felby, C. Solid-State 29Si NMR and FTIR Analyses of Lignin-Silica Coprecipitates. Holzforschung 2016, 70, 709–718. [Google Scholar] [CrossRef]
- Inanaga, S.; Okasaka, A.; Tanaka, S. Does Silicon Exist in Association with Organic Compounds in Rice Plant? Soil Sci. Plant Nutr. 1995, 41, 111–117. [Google Scholar] [CrossRef]
- Vedachalam, S.; Murugesh, N.; Chakraborty, P.; Karvembu, R.; Liu, X.-W. NHC Catalyzed Enantioselective Coates-Claisen Rearrangement: A Rapid Access to the Dihydropyran Core for Oleuropein Based Secoiridoids. New J. Chem. 2018, 42, 1832–1839. [Google Scholar] [CrossRef]
- Medina-Mercado, I.; Asomoza-Solís, E.O.; Martínez-González, E.; Ugalde-Saldívar, V.M.; Ledesma-Olvera, L.G.; Barquera-Lozada, J.E.; Gómez-Vidales, V.; Barroso-Flores, J.; Frontana-Uribe, B.A.; Porcel, S. Ascorbic Acid as an Aryl Radical Inducer in the Gold-Mediated Arylation of Indoles with Aryldiazonium Chlorides. Chem. Eur. J. 2020, 26, 634–642. [Google Scholar] [CrossRef]
- Watanabe, Y.; Uemura, T.; Yamauchi, S.; Tomita, K.; Saeki, T.; Ishida, R.; Hayashi, M. Regioselective Functionalization of Unprotected Myo-Inositol by Electrophilic Substitution. Tetrahedron 2013, 69, 4657–4664. [Google Scholar] [CrossRef]
- Nizioł, J.; Rode, W.; Zieliński, Z.; Ruman, T. Matrix-Free Laser Desorption–Ionization with Silver Nanoparticle-Enhanced Steel Targets. Int. J. Mass Spectrom. 2013, 335, 22–32. [Google Scholar] [CrossRef]
- Patiny, L.; Borel, A. ChemCalc: A Building Block for Tomorrow’s Chemical Infrastructure. J. Chem. Inf. Model. 2013, 53, 1223–1228. [Google Scholar] [CrossRef]
- Variaki, A.; Cummings, R.D.; Esko, J.D.; Freeze, H.; Hart, G.W.; Marth, J.D. Essentials of Glycobiology, 1st ed.; Cold Spring Harbor Laboratory Press: Plainview, NY, USA, 1999. [Google Scholar]
- Demchenko, A.V. Handbook of Chemical Glycosylation: Advances in Stereoselectivity and Therapeutic Relevance; Wiley-VCH: Hoboken, NJ, USA, 2008. [Google Scholar]
- Wang, Z. Carbohydrate-Based Drug Discovery; John Wiley & Sons: Hoboken, NJ, USA, 2003. [Google Scholar]
- Gribble, G.W. Indole Ring Synthesis: From Natural Products to Drug Discovery; John Wiley & Sons: Hoboken, NJ, USA, 2016. [Google Scholar]
- Holmberg, K. Surfactants and Polymers in Aqueous Solution; John Wiley & Sons: Hoboken, NJ, USA, 2002. [Google Scholar]
- Fahy, E.; Subramaniam, S.; Brown, H.A.; Glass, C.K.; Merrill, A.H., Jr.; Murphy, R.C.; Raetz, C.R.H.; Russell, D.W.; Seyama, Y.; Shaw, W.; et al. A comprehensive classification system for lipids. J. Lipid Res. 2005, 46, 839–861. [Google Scholar] [CrossRef]
- Michell, R.H. Inositol derivatives: Evolution and functions. Nat. Rev. Mol. Cell Biol. 2008, 9, 151–161. [Google Scholar] [CrossRef]
- Prysiazhnyi, V.; Dycka, F.; Kratochvil, J.; Sterba, J.; Stranak, V. Gas-Aggregated Ag Nanoparticles for Detection of Small Molecules Using LDI MS. Anal. Bioanal. Chem. 2020, 412, 1037–1047. [Google Scholar] [CrossRef]
- Stone, J.A. Gas-Phase Association Reactions of Trimethylsilylium ((CH3)3Si+) with Organic Bases. Mass Spectrom. Rev. 1997, 16, 25–49. [Google Scholar] [CrossRef]
- Harvey, D.J.; Vouros, P. Mass Spectrometric Fragmentation of Trimethylsilyl and Related Alkylsilyl Derivatives. Mass Spectrom. Rev. 2020, 39, 105–211. [Google Scholar] [CrossRef]
- Allevi, P.; Anastasia, M.; Ciuffreda, P.; Bigatti, E.; Macdonald, P. Stereoselective glucosidation of podophyllum lignans. A new simple synthesis of etoposide. J. Org. Chem. 1993, 58, 4175–4178. [Google Scholar] [CrossRef]
- Cheng, Z.D.; Cui, Y.L.; Mao, J.W. 2-Acetylamino-1,3,4,6-tetra-O-(trimethylsilyl)-2-deoxy-a-D-glucopyranose. Acta Cryst. 2013, 69, o917. [Google Scholar] [CrossRef]
- Anderson, R.J.; Gainsford, G.J. 1D-1-O-tert-Butyldiphenylsilyl-2,3,6-Otris(methoxymethylene)-myo-inositol 4,5-bis(dibenzylphosphate). Acta Cryst. 2012, 68, o900. [Google Scholar] [CrossRef]
- Dauzonne, D.; O’Neil, I.A.; Renaud, A. Preparation and reactions of 4-(trimethylsilyl)indole. J. Org. Chem. 1984, 49, 4409–4415. [Google Scholar] [CrossRef]
- Behr, A.; Naendrup, F.; Obst, D. The synthesis of silicon oleochemicals by hydrosilylation of unsaturated fatty acid derivatives. Eur. J. Lipid Sci. Technol. 2002, 104, 161–166. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parent Ion | Theoretical m/z | NALDI-MS | ESI-FT-ICR MS | LC-QqQ-ESI-MS/MS |
|---|---|---|---|---|
| Experimental m/z | ||||
| Compound 1: 1-O-(Trimethylsilyl)-2,3,4,6-tetra-O-acetyl-β-d-glucopyranose | ||||
| [C17H28O10Si+H]+ | 421.15245 | - | - | - |
| [C17H28O10Si+Na]+ | 443.13439 | 443.139 | 443.13439 | 443.10 |
| [C17H28O10Si+K]+ | 459.10833 | 459.115 | - | - |
| [C17H28O10Si+107Ag]+ | 527.04972 | 527.049 | - | - |
| [C17H28O10Si+109Ag]+ | 529.04938 | 529.046 | - | - |
| Compound 2: 1-[(1,1-dimethylehtyl)diphenylsilyl]-1H-indole | ||||
| [C24H25NSi+H]+ | 356.18290 | - | 356.18291 | 356.10 |
| [C24H25NSi+Na]+ | 378.16485 | 378.122 | - | - |
| [C24H25NSi+107Ag]+ | 462.08017 | 462.085 | - | - |
| [C24H25NSi+109Ag]+ | 464.07983 | 464.087 | - | - |
| Compound 3: O-tert-butyldiphenylsilyl-(3-hydroxypropyl)oleate | ||||
| [C37H58O3Si+H]+ | 579.42280 | - | 579.42276 | 579.20 |
| [C37H58O3Si+Na]+ | 601.40474 | 601.405 | 601.40482 | - |
| [C37H58O3Si+107Ag]+ | 685.32007 | 685.320 | - | - |
| [C37H58O3Si+109Ag]+ | 687.31973 | 687.325 | - | - |
| Compound 4: 1-O-tert-Butyldiphenylsilyl-myo-inositol | ||||
| [C22H30O6Si+H]+ | 419.18844 | - | - | - |
| [C22H30O6Si+Na]+ | 441.17039 | 441.175 | 441.17037 | 441.15 |
| [C22H30O6Si+107Ag]+ | 525.08571 | 525.084 | - | - |
| [C22H30O6Si+109Ag]+ | 527.08537 | 527.084 | - | - |
| [C22H30O6Si-H]- | 417.17389 | - | - | 417.20 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rogowska, A.; Szultka-Młyńska, M.; Kanawati, B.; Pomastowski, P.; Arendowski, A.; Gołębiowski, A.; Schmitt-Kopplin, P.; Fordymacka, M.; Sukiennik, J.; Krzywik, J.; et al. Advanced Mass Spectrometric Techniques for the Comprehensive Study of Synthesized Silicon-Based Silyl Organic Compounds: Identifying Fragmentation Pathways and Characterization. Materials 2023, 16, 3563. https://doi.org/10.3390/ma16093563
Rogowska A, Szultka-Młyńska M, Kanawati B, Pomastowski P, Arendowski A, Gołębiowski A, Schmitt-Kopplin P, Fordymacka M, Sukiennik J, Krzywik J, et al. Advanced Mass Spectrometric Techniques for the Comprehensive Study of Synthesized Silicon-Based Silyl Organic Compounds: Identifying Fragmentation Pathways and Characterization. Materials. 2023; 16(9):3563. https://doi.org/10.3390/ma16093563
Chicago/Turabian StyleRogowska, Agnieszka, Małgorzata Szultka-Młyńska, Basem Kanawati, Paweł Pomastowski, Adrian Arendowski, Adrian Gołębiowski, Phillipe Schmitt-Kopplin, Marta Fordymacka, Jarosław Sukiennik, Julia Krzywik, and et al. 2023. "Advanced Mass Spectrometric Techniques for the Comprehensive Study of Synthesized Silicon-Based Silyl Organic Compounds: Identifying Fragmentation Pathways and Characterization" Materials 16, no. 9: 3563. https://doi.org/10.3390/ma16093563