

Thermo-Responsive Shape-Memory Dual-Cured Polymers Based on Vegetable Oils


Abstract
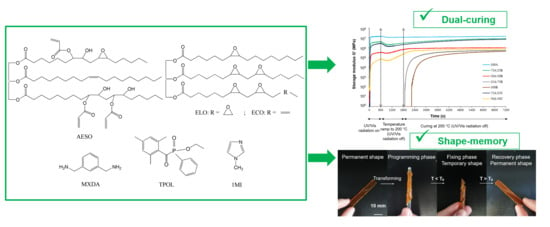
:
1. Introduction
- Tunable transition temperatures: by changing the composition of the polymer, it is frequently possible to modify the transition temperature at which an SMP undergoes shape-memory recovery;
- Mechanical flexibility: many SMPs can be employed in applications requiring materials to be bent, stretched, or conformed to various shapes because of their high mechanical flexibility;
- Lightweight characteristics: SMPs are frequently lightweight, which is useful in sectors like aerospace and automotive where weight reduction is essential;
- Lower energy consumption: the shape-memory effect can be used to execute mechanical activities, such as opening and closing valves, without the use of external energy sources;
- Biocompatibility: thermo-responsive SMPs can be produced from biocompatible and non-toxic starting materials, making them appropriate for biomedical and healthcare applications.
2. Materials and Methods
2.1. Materials
2.2. The Epoxidation of Camelia Oil
2.3. Preparation of Dual-Curable Resins and Cross-Linked Polymers
2.4. Kinetics of Dual Curing
2.5. Characterization Techniques
2.6. Shape-Memory Experiments
3. Results and Discussion
3.1. Monitoring Curing Kinetics by Rheometry
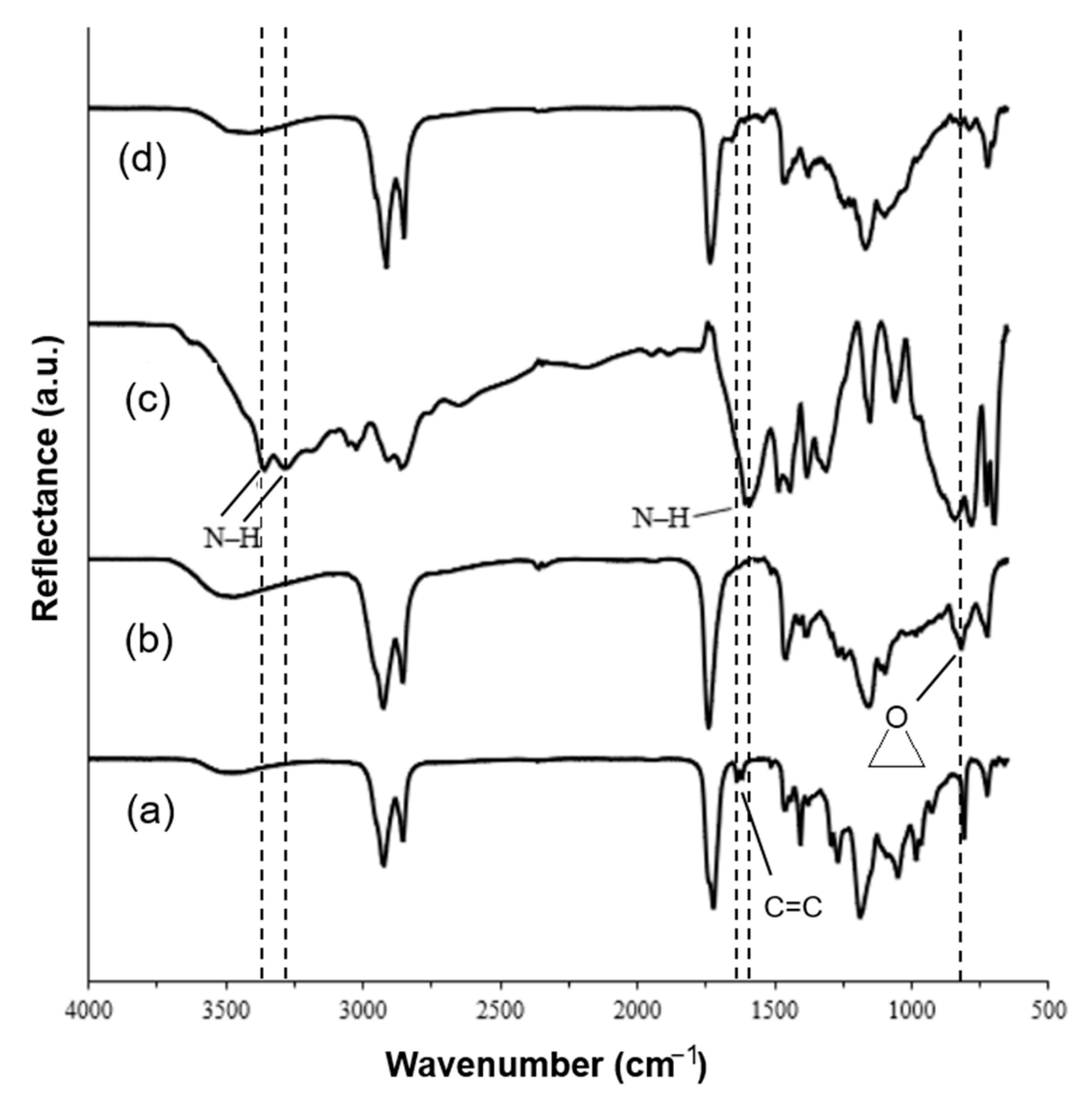
3.2. Characterization of Cross-Linked Polymer Structure
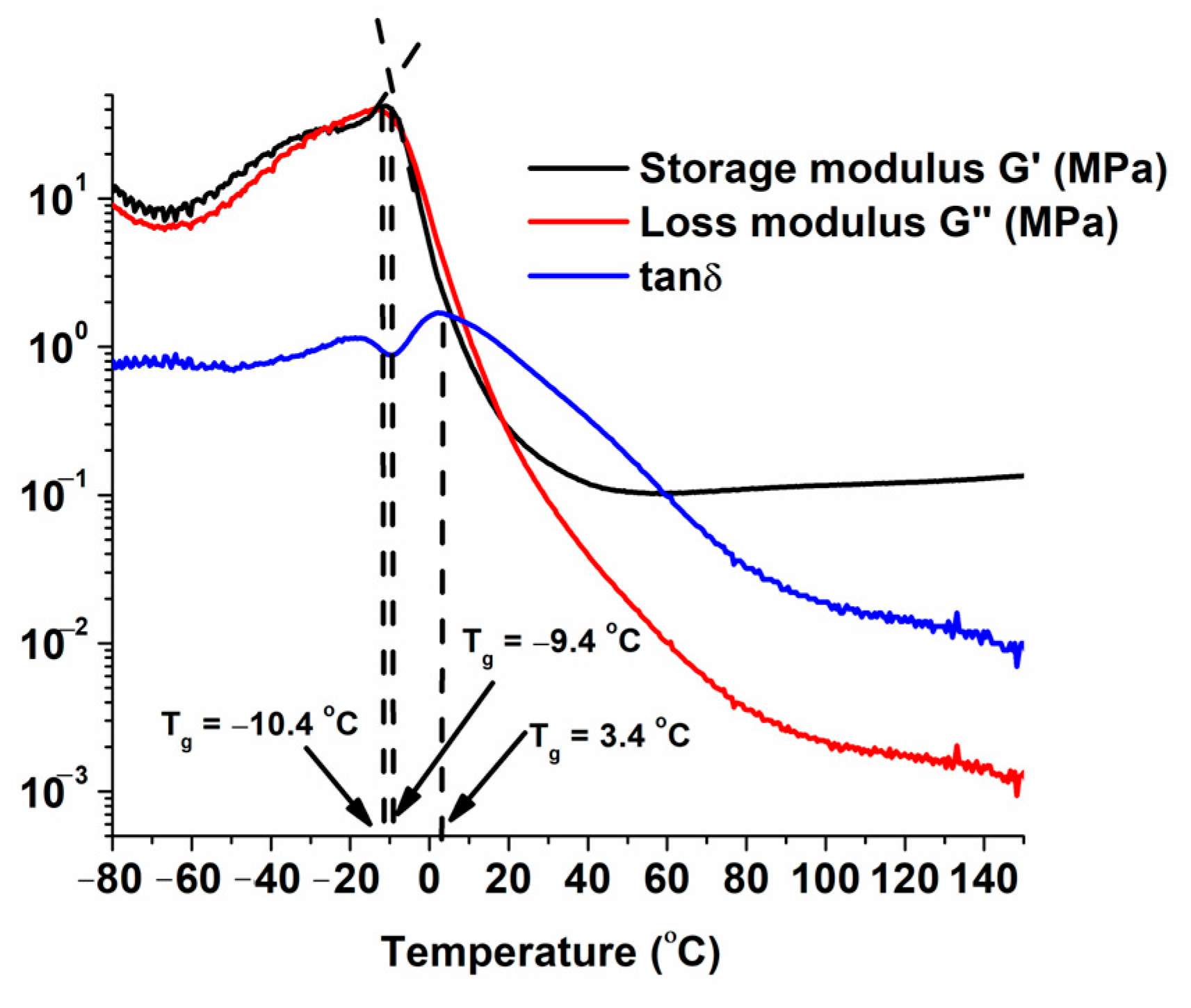
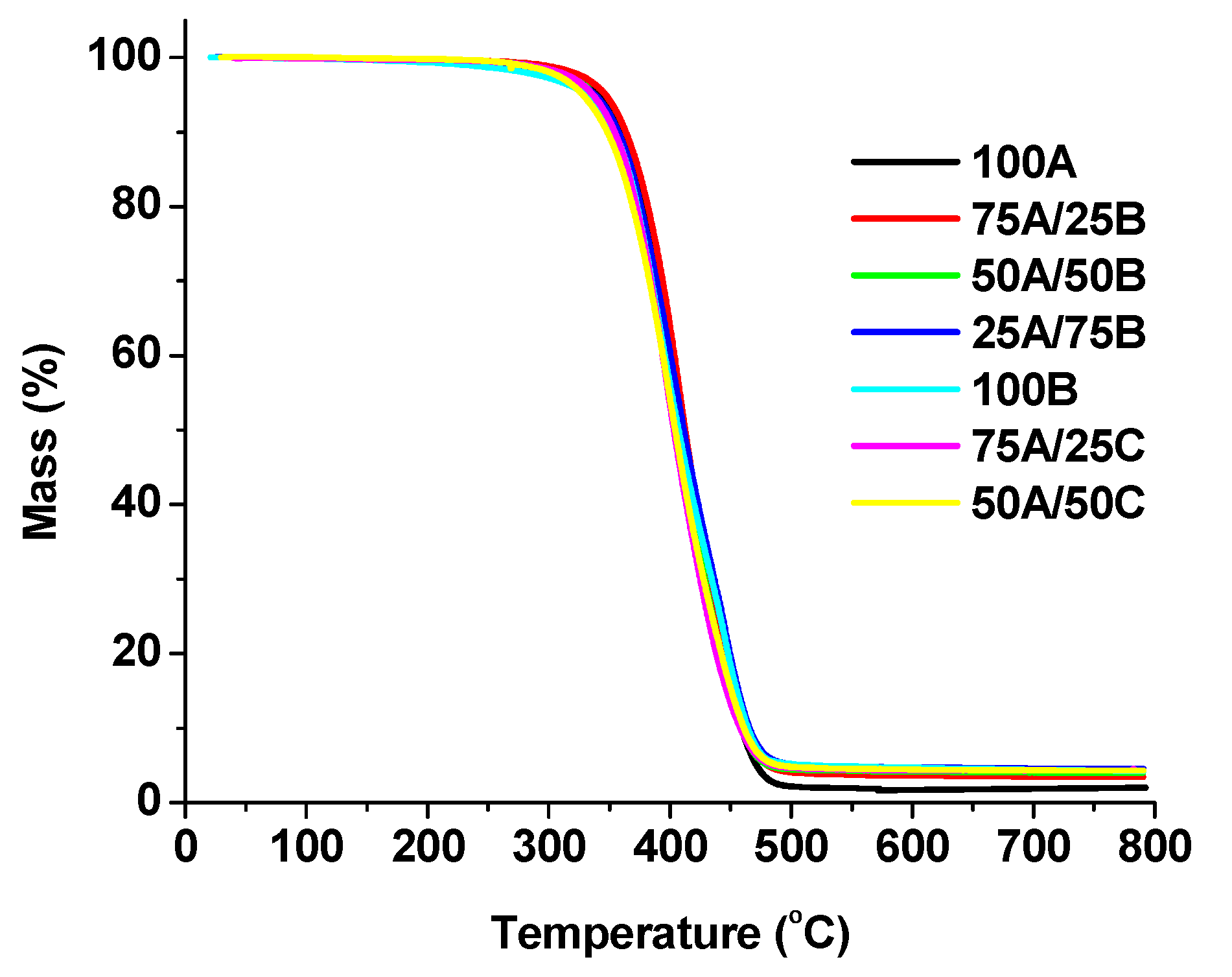
3.3. Thermal Characterization of the Materials
3.4. Mechanical Properties
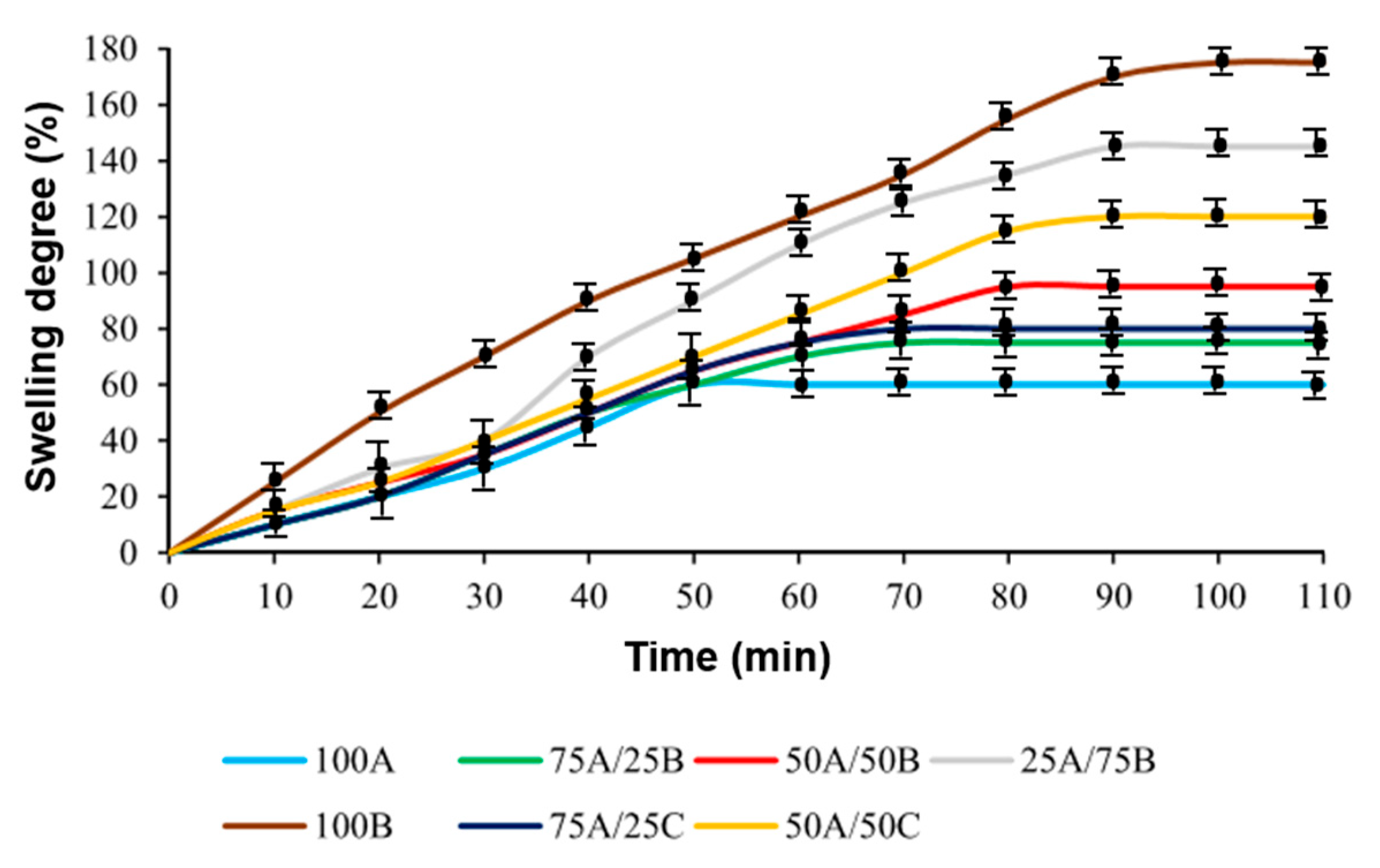
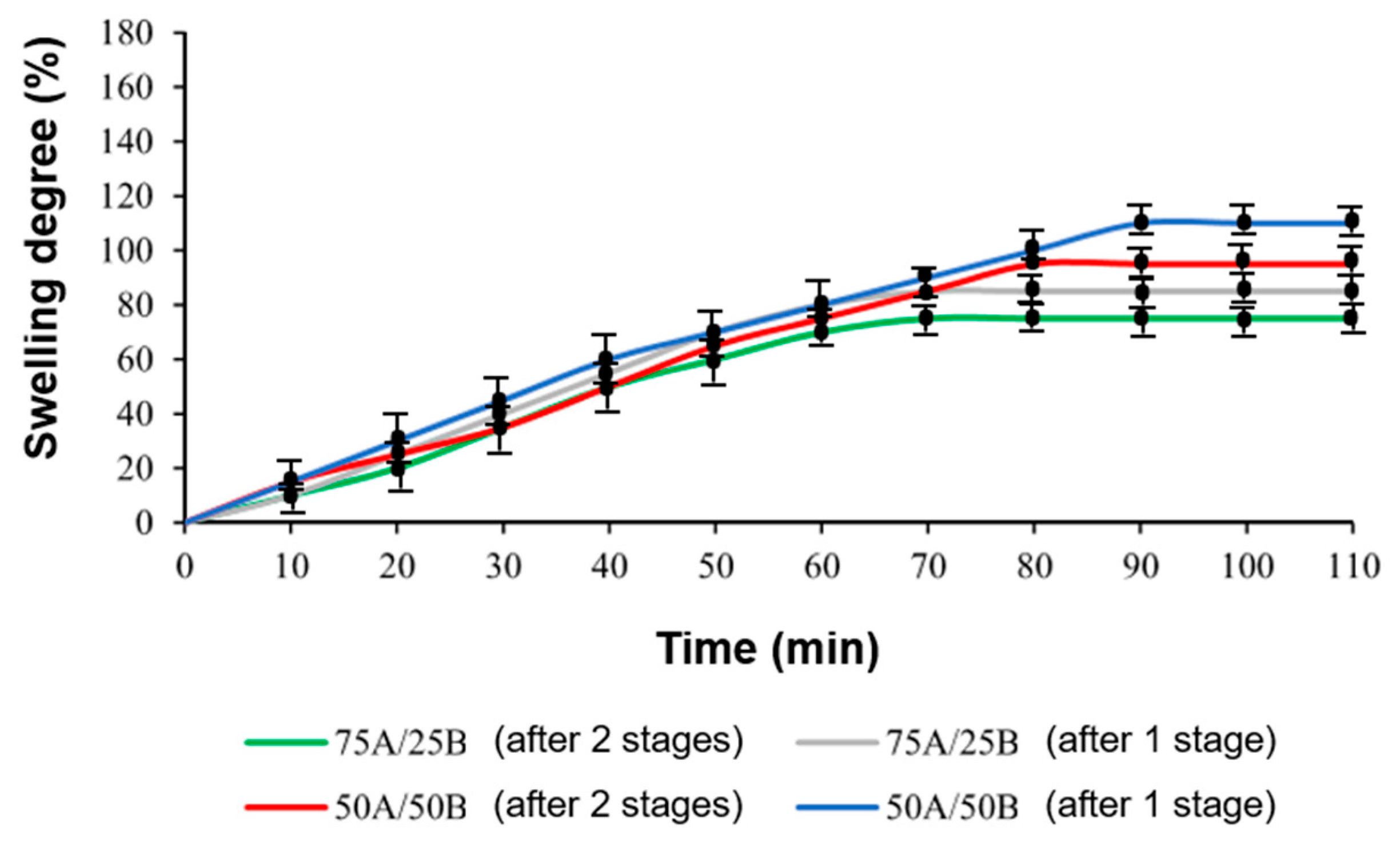
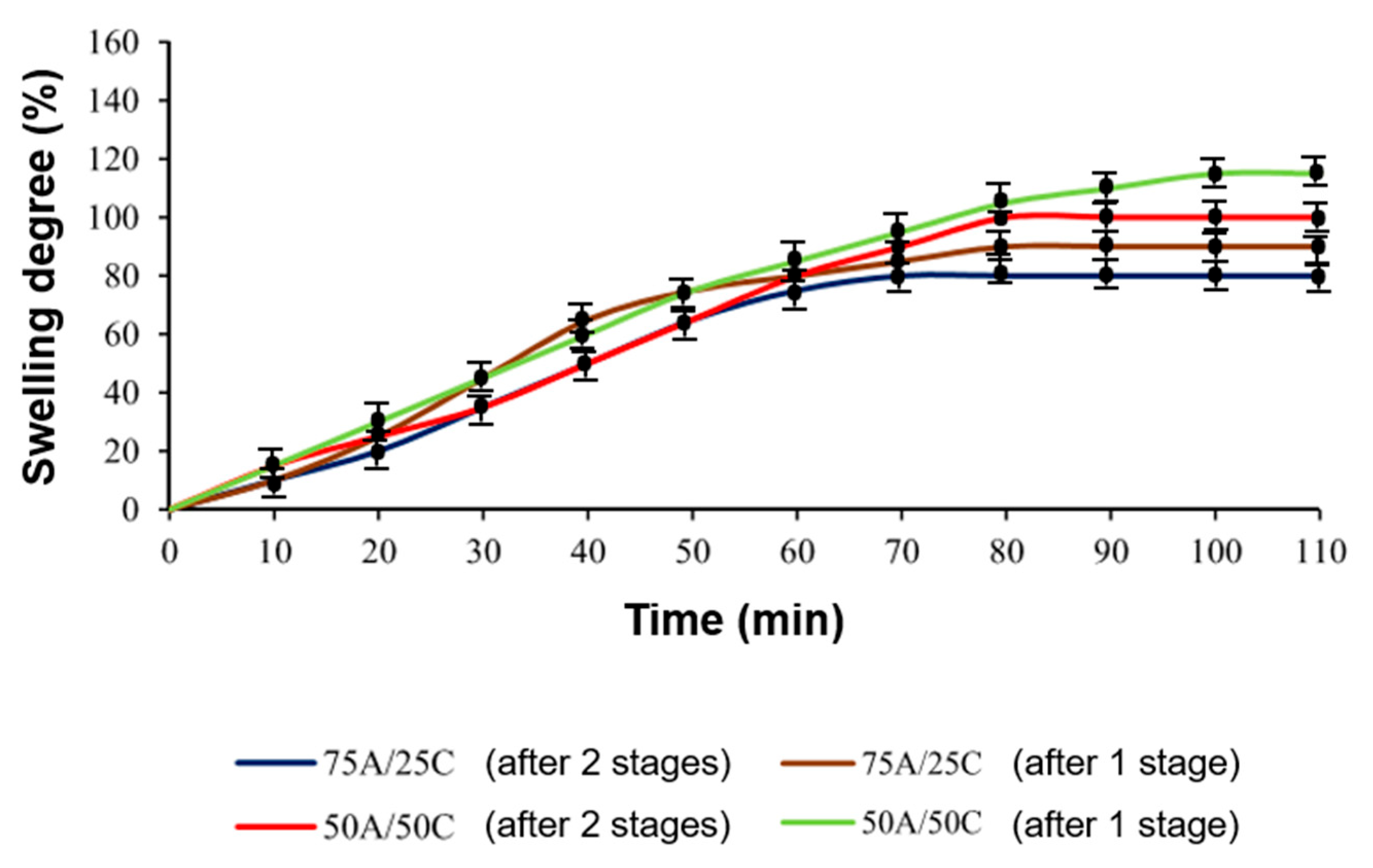
3.5. Shape-Memory Properties
4. Conclusions
- Epoxidized linseed oil can be replaced by epoxidized camelina oil in dual-curing systems, as polymers with epoxidized camelina oil fragments have rigidity insignificantly lower than those of polymers with epoxidized linseed oil fragments;
- By changing the amount of acrylated epoxidized soybean oil, epoxidized linseed oil, and epoxidized camelina oil in the resins, polymers with desired properties can be obtained;
- The second curing stage improved the rheological, mechanical, and thermal properties of the resulting polymers, which are suitable for a wide range of applications, such as electronics, biomedical devices, and robotics, because of thermo-responsive shape-memory properties.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Xia, Y.; He, Y.; Zhang, F.; Liu, Y.; Leng, J. A review of shape memory polymers and composites: Mechanisms, materials, and applications. Adv. Mater. 2021, 33, 2000713. [Google Scholar] [CrossRef] [PubMed]
- Idumah, C.I. Multifunctional properties optimization and stimuli-responsivity of shape memory polymeric nanoarchitectures and applications. Polym. Eng. Sci. 2023, 63, 1857–1873. [Google Scholar] [CrossRef]
- Yakacki, C.M.; Gall, K. Shape-Memory Polymers for Biomedical Applications. In Shape-Memory Polymers; Lendlein, A., Ed.; Springer: Berlin/Heidelberg, Germany, 2009; Volume 226, pp. 147–175. [Google Scholar] [CrossRef]
- Hu, J.; Zhu, Y.; Huang, H.; Lu, J. Recent advances in shape–memory polymers: Structure, mechanism, functionality, modeling and applications. Prog. Polym. Sci. 2012, 37, 1720–1763. [Google Scholar] [CrossRef]
- Zhao, Q.; Zou, W.; Luo, Y.; Xie, T. Shape memory polymer network with thermally distinct elasticity and plasticity. Sci. Adv. 2016, 2, e1501297. [Google Scholar] [CrossRef] [PubMed]
- Pilate, F.; Toncheva, A.; Dubois, P.; Raquez, J.M. Shape-memory polymers for multiple applications in the materials world. Eur. Polym. J. 2016, 80, 268–294. [Google Scholar] [CrossRef]
- Studer, K.; Decker, C.; Beck, E.; Schwalm, R. Thermal and photochemical curing of isocyanate and acrylate functionalized oligomers. Eur. Polym. J. 2005, 41, 157–167. [Google Scholar] [CrossRef]
- Ramis, X.; Fernández-Francos, X.; De la Flor, S.; Ferrando, F.; Serra, A. Click-based dual-curing thermosets and their applications. In Thermosets, 2nd ed.; Guo, Q., Ed.; Elsevier: Amsterdam, The Netherlands, 2018; pp. 511–541. [Google Scholar] [CrossRef]
- Mather, B.D.; Viswanathan, K.; Miller, K.M.; Long, T.E. Michael addition reactions in macromolecular design for emerging technologies. Prog. Polym. Sci. 2006, 31, 487–531. [Google Scholar] [CrossRef]
- Lahann, J. (Ed.) Click Chemistry for Biotechnology and Materials Science; John Wiley & Sons: Chichester, UK, 2009. [Google Scholar]
- Lowe, A.B.; Bowman, C.N. (Eds.) Thiol-x Chemistries in Polymer and Materials Science; Royal Society of Chemistry: Cambridge, UK, 2013. [Google Scholar]
- Carioscia, J.A.; Schneidewind, L.; O’Brien, C.; Ely, R.; Feeser, C.; Cramer, N.; Bowman, C.N. Thiol—Norbornene materials: Approaches to develop high Tg Thiol—Ene polymers. J. Polym. Sci. Part A Polym. Chem. 2007, 45, 5686–5696. [Google Scholar] [CrossRef]
- Belmonte, A.; Lama, G.C.; Gentile, G.; Cerruti, P.; Ambrogi, V.; Fernández-Francos, X.; De la Flor, S. Thermally-triggered free-standing shape-memory actuators. Eur. Polym. J. 2017, 97, 241–252. [Google Scholar] [CrossRef]
- Chen, Q.; Sukmanee, T.; Rong, L.; Yang, M.; Ren, J.; Ekgasit, S.; Advincula, R. A dual approach in direct ink writing of thermally cured shape memory rubber toughened epoxy. ACS Appl. Polym. Mater. 2020, 2, 5492–5500. [Google Scholar] [CrossRef]
- Pouladvand, A.R.; Mortezaei, M.; Fattahi, H.; Amraei, I.A. A novel custom-tailored epoxy prepreg formulation based on epoxy-amine dual-curable systems. Compos. Part A Appl. Sci. 2020, 132, 105852. [Google Scholar] [CrossRef]
- Konuray, A.O.; Fernández-Francos, X.; Serra, À.; Ramis, X. Sequential curing of amine-acrylate-methacrylate mixtures based on selective aza-Michael addition followed by radical photopolymerization. Eur. Polym. J. 2016, 84, 256–267. [Google Scholar] [CrossRef]
- Konuray, O.; Areny, N.; Morancho, J.M.; Fernandez-Francos, X.; Serra, A.; Ramis, X. Preparation and characterization of dual-curable off-stoichiometric amine-epoxy thermosets with latent reactivity. Polymer 2018, 146, 42–52. [Google Scholar] [CrossRef]
- Roig, A.; Ramis, X.; De la Flor, S.; Serra, A. Dual-cured thermosets from glycydil methacrylate obtained by epoxy-amine reaction and methacrylate homopolymerization. React. Funct. Polym. 2021, 159, 104822. [Google Scholar] [CrossRef]
- Morancho, J.M.; Ramis, X.; Fernández-Francos, X.; Salla, J.M.; Konuray, A.O.; Serra, À. Curing of off-stoichiometric amine–epoxy thermosets. J. Therm. Anal. Calorim. 2018, 133, 519–527. [Google Scholar] [CrossRef]
- Zhao, L.; Yu, R.; He, Y.; Zhang, M.; Tian, F.; Wang, L.; Zhao, Y.; Huang, W. 3D printed epoxy/acrylate hybrid polymers with excellent mechanical and shape memory properties via UV and thermal cationic dual-curing mechanism. Addit. Manuf. 2023, 79, 103904. [Google Scholar] [CrossRef]
- Yadav, A.; Singh, S.K.; Das, S.; Kumar, S.; Kumar, A. Shape recovery and mechanical properties investigation of carbon fiber dispersed bisphenol-A based epoxy composite. Smart Mater. Struct. 2023, 32, 095016. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, E.; Liu, J.; Qin, J.; Wu, M.; Yang, C.; Liang, L. Self-healing, reprocessable, degradable, thermadapt shape memory multifunctional polymers based on dynamic imine bonds and their application in nondestructively recyclable carbon fiber composites. Chem. Eng. J. 2023, 454, 139992. [Google Scholar] [CrossRef]
- Rochester, J.R.; Bolden, A.L.; Kwiatkowski, C.F. Prenatal exposure to bisphenol A and hyperactivity in children: A systematic review and meta-analysis. Environ. Int. 2018, 114, 343–356. [Google Scholar] [CrossRef]
- Onundi, Y.; Drake, B.A.; Malecky, R.T.; DeNardo, M.A.; Mills, M.R.; Kundu, S.; Ryabov, A.D.; Beach, E.S.; Horwitz, C.P.; Simonich, M.T.; et al. A multidisciplinary investigation of the technical and environmental performances of TAML/peroxide elimination of Bisphenol A compounds from water. Green Chem. 2017, 19, 4234–4262. [Google Scholar] [CrossRef]
- Yadav, S.K.; Schmalbach, K.M.; Kinaci, E.; Stanzione, J.F., III; Palmese, G.R. Recent advances in plant-based vinyl ester resins and reactive diluents. Eur. Polym. J. 2018, 98, 199–215. [Google Scholar] [CrossRef]
- Raczyk, M.; Popis, E.; Kruszewski, B.; Ratusz, K.; Rudzińska, M. Physicochemical quality and oxidative stability of linseed (Linum usitatissimum) and camelina (Camelina sativa) cold-pressed oils from retail outlets. Eur. J. Lipid. Sci. Technol. 2016, 118, 834–839. [Google Scholar] [CrossRef]
- Arshad, M.; Mohanty, A.K.; Van Acker, R.; Riddle, R.; Todd, J.; Khalil, H.; Misra, M. Valorization of camelina oil to biobased materials and biofuels for new industrial uses: A review. RSC Adv. 2022, 12, 27230–27245. [Google Scholar] [CrossRef] [PubMed]
- Balanuca, B.; Stan, R.; Hanganu, A.; Lungu, A.; Iovu, H. Design of new camelina oil-based hydrophilic monomers for novel polymeric materials. J. Am. Oil. Chem. Soc. 2015, 92, 881–891. [Google Scholar] [CrossRef]
- Kasetaite, S.; Ostrauskaite, J.; Grazuleviciene, V.; Svediene, J.; Bridziuviene, D. Camelina oil-and linseed oil-based polymers with bisphosphonate crosslinks. J. Appl. Polym. Sci. 2014, 131, 1–8. [Google Scholar] [CrossRef]
- Kim, N.; Li, Y.; Sun, X.S. Epoxidation of Camelina sativa oil and peel adhesion properties. Ind. Crops Prod. 2015, 64, 1–8. [Google Scholar] [CrossRef]
- Sung, J.; Sun, X.S. Cardanol modified fatty acids from camelina oils for flexible bio-based acrylates coatings. Prog. Org. Coat. 2018, 123, 242–253. [Google Scholar] [CrossRef]
- Di Mauro, C.; Malburet, S.; Genua, A.; Graillot, A.; Mija, A. Sustainable series of new epoxidized vegetable oil-based thermosets with chemical recycling properties. Biomacromolecules 2020, 21, 3923–3935. [Google Scholar] [CrossRef]
- Di Mauro, C.; Malburet, S.; Graillot, A.; Mija, A. Recyclable, repairable, and reshapable (3R) thermoset materials with shape memory properties from bio-based epoxidized vegetable oils. ACS Appl. Bio Mater. 2020, 3, 8094–8104. [Google Scholar] [CrossRef]
- Malburet, S.; Di Mauro, C.; Noè, C.; Mija, A.; Sangermano, M.; Graillot, A. Sustainable access to fully biobased epoxidized vegetable oil thermoset materials prepared by thermal or UV-cationic processes. RSC Adv. 2020, 10, 41954–41966. [Google Scholar] [CrossRef]
- Taung Mai, L.L.; Aung, M.M.; Muhamad Saidi, S.A.; H’ng, P.S.; Rayung, M.; Jaafar, A.M. Non edible oil-based epoxy resins from Jatropha oil and their shape memory behaviors. Polymers 2021, 13, 2177. [Google Scholar] [CrossRef] [PubMed]
- Grauzeliene, S.; Navaruckiene, A.; Skliutas, E.; Malinauskas, M.; Serra, A.; Ostrauskaite, J. Vegetable oil-based thiol-ene/thiol-epoxy resins for laser direct writing 3D micro-/nano-lithography. Polymers 2021, 13, 872. [Google Scholar] [CrossRef] [PubMed]
- Sereikaite, V.; Navaruckiene, A.; Jaras, J.; Skliutas, E.; Ladika, D.; Gray, D.; Malinauskas, M.; Talacka, V.; Ostrauskaite, J. Functionalized soybean oil-and vanillin-based dual cure photopolymerizable system for light-based 3D structuring. Polymers 2022, 14, 5361. [Google Scholar] [CrossRef] [PubMed]
- Di Mauro, C.; Genua, A.; Mija, A. Kinetical Study, Thermo-Mechanical Characteristics and Recyclability of Epoxidized Camelina Oil Cured with Antagonist Structure (Aliphatic/Aromatic) or Functionality (Acid/Amine) Hardeners. Polymers 2021, 13, 2503. [Google Scholar] [CrossRef] [PubMed]
- Green, W.A. Industrial Photoinitiators: A Technical Guide; CRC Press: Boca Raton, FL, USA, 2010. [Google Scholar]
- Guzman, D.; Ramis, X.; Fernandez-Francos, X.; De la Flor, S.; Serra, A. New bio-based materials obtained by thiol-ene/thiol-epoxy dual curing click procedures from eugenol derivates. Eur. Polym. J. 2017, 93, 530–544. [Google Scholar] [CrossRef]
- Remeikyte, A.; Ostrauskaite, J.; Grazuleviciene, V. Synthesis and properties of photocross-linked polymers of epoxidized linseed oil with different reactive diluents. J. Appl. Polym. Sci. 2013, 129, 1290–1298. [Google Scholar] [CrossRef]
- ISO 527-3:2018; Plastics—Determination of Tensile Properties—Part 3: Test Conditions for Films and Sheets. ISO: Geneva, Switzerland, 2018.
- Gu, S.; Jana, S. Effects of polybenzoxazine on shape memory properties of polyurethanes with amorphous and crystalline soft segments. Polymers 2014, 6, 1008–1025. [Google Scholar] [CrossRef]
- Meyers, M.A.; Chawla, K.K. Mechanical Behavior of Materials, 2nd ed.; Cambridge University Press: Cambridge, UK, 2008. [Google Scholar]
- Kašėtaitė, S.; Ostrauskaitė, J.; Gražulevičienė, V. DMTA Analysis of glycerol diglycidyl ether based photocross-linked polymers. In Proceedings of the 3rd World Congress on Mechanical, Chemical, and Material Engineering (MCM’17), Rome, Italy, 8–10 June 2017. [Google Scholar]
- Rana, A.; Evitts, R.W. Synthesis and characterization of acrylated epoxidized flaxseed oil for biopolymeric applications. Int. Polym. Process. 2015, 30, 331–336. [Google Scholar] [CrossRef]
- Grauzeliene, S.; Kazlauskaite, B.; Skliutas, E.; Malinauskas, M.; Ostrauskaite, J. Photocuring and digital light processing 3D printing of vitrimer composed of 2-hydroxy-2-phenoxypropyl acrylate and acrylated epoxidized soybean oil. Express Polym. Lett. 2023, 17, 54–68. [Google Scholar] [CrossRef]
- Grauzeliene, S.; Schuller, A.S.; Delaite, C.; Ostrauskaite, J. Biobased vitrimer synthesized from 2-hydroxy-3-phenoxypropyl acrylate, tetrahydrofurfuryl methacrylate and acrylated epoxidized soybean oil for digital light processing 3D printing. Eur. Polym. J. 2023, 198, 112424. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Component | Composition | Polymerization |
|---|---|---|
| A | AESO + 3 mol.% TPOL | Radical photopolymerization |
| B | 2 mol ELO + 3 mol MXDA + 1 wt.% 1MI | Cationic thermal polymerization |
| C | 4 mol ECO + 5 mol MXDA + 1 wt.% 1MI | Cationic thermal polymerization |
| Resin | Amount of Component A (wt.%) | Amount of Component B (wt.%) | Amount of Component C (wt.%) |
|---|---|---|---|
| 100A | 100 | 0 | 0 |
| 75A/25B | 75 | 25 | 0 |
| 50A/50B | 50 | 50 | 0 |
| 25A/75B | 25 | 75 | 0 |
| 100B | 0 | 100 | 0 |
| 75A/25C | 75 | 0 | 25 |
| 50A/50C | 50 | 0 | 50 |
| Resin | 100A | 75A/25B | 50A/50B | 25A/75B | 100B | 75A/25C | 50A/50C | |
|---|---|---|---|---|---|---|---|---|
| Characteristic | ||||||||
| Gel point tgel (s) | 2 | 6 | 14 | 2030 | 1728 | 6 | 14 | |
| Maximum storage modulusG′(MPa) | 16.21 | 8.55 | 1.01 | 0.56 | 0.42 | 7.19 | 0.81 | |
| Polymer | Tg a (°C) | Tdec. −10% b (°C) | Residue c (%) | Photo of the Sample |
|---|---|---|---|---|
| 100A (after 2nd curing stage) | −13.9 | 362 | 1.9 |  |
| 75A/25B (after 2nd curing stage) | −9.0 | 364 | 3.4 |  |
| 75A/25B (after 1st curing stage) | −17.3 | 359 | 3.5 |  |
| 50A/50B (after 2nd curing stage) | −24.7 | 356 | 3.7 |  |
| 50A/50B (after 1st curing stage) | −31.2 | 354 | 4.0 |  |
| 25A/75B (after 2nd curing stage) | −10.2 | 357 | 4.5 |  |
| 100B (after 2nd curing stage) | −10.4 | 354 | 4.2 |  |
| 75A/25C (after 2nd curing stage) | −8.1 | 355 | 4.3 |  |
| 75A/25C (after 1st curing stage) | −32.9 | 350 | 4.7 |  |
| 50A/50C (after 2nd curing stage) | −20.9 | 348 | 4.3 |  |
| 50A/50C (after 1st curing stage) | −44.9 | 347 | 4.4 |  |
| Polymer | Young’s Modulus (MPa) | Tensile Strength (MPa) | Elongation at Break (%) |
|---|---|---|---|
| 100A (after 2nd curing stage) | 87.84 ± 5.97 | 4.18 ± 0.61 | 9.64 ± 0.62 |
| 75A/25B (after 2nd curing stage) | 10.78 ± 1.35 | 0.94 ± 0.12 | 10.13 ± 1.23 |
| 75A/25B (after 1st curing stage) | 9.72 ± 0.79 | 0.86 ± 0.07 | 11.71 ± 0.90 |
| 50A/50B (after 2nd curing stage) | 1.13 ± 0.15 | 0.01 ± 0.23∙10−2 | 12.19 ± 0.55 |
| 25A/75B (after 2nd curing stage) | 0.49 ± 0.08 | 0.01 ± 0.17∙10−2 | 31.14 ± 2.71 |
| 75A/25C (after 2nd curing stage) | 10.15 ± 0.67 | 0.90 ± 0.13 | 9.82 ± 1.37 |
| 75A/25C (after 1st curing stage) | 9.48 ± 0.83 | 0.62 ± 0.16 | 9.95 ± 1.21 |
| 50A/50C (after 2nd curing stage) | 1.01 ± 0.11 | 0.01 ± 0.19∙10−2 | 10.67 ± 0.67 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Petrauskas, R.; Grauzeliene, S.; Ostrauskaite, J. Thermo-Responsive Shape-Memory Dual-Cured Polymers Based on Vegetable Oils. Materials 2024, 17, 24. https://doi.org/10.3390/ma17010024
Petrauskas R, Grauzeliene S, Ostrauskaite J. Thermo-Responsive Shape-Memory Dual-Cured Polymers Based on Vegetable Oils. Materials. 2024; 17(1):24. https://doi.org/10.3390/ma17010024
Chicago/Turabian StylePetrauskas, Rokas, Sigita Grauzeliene, and Jolita Ostrauskaite. 2024. "Thermo-Responsive Shape-Memory Dual-Cured Polymers Based on Vegetable Oils" Materials 17, no. 1: 24. https://doi.org/10.3390/ma17010024
APA StylePetrauskas, R., Grauzeliene, S., & Ostrauskaite, J. (2024). Thermo-Responsive Shape-Memory Dual-Cured Polymers Based on Vegetable Oils. Materials, 17(1), 24. https://doi.org/10.3390/ma17010024