

Highly Efficient Photoinitiation Systems Based on Dibenzo[a,c]phenazine Sensitivity to Visible Light for Dentistry

Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Synthesis
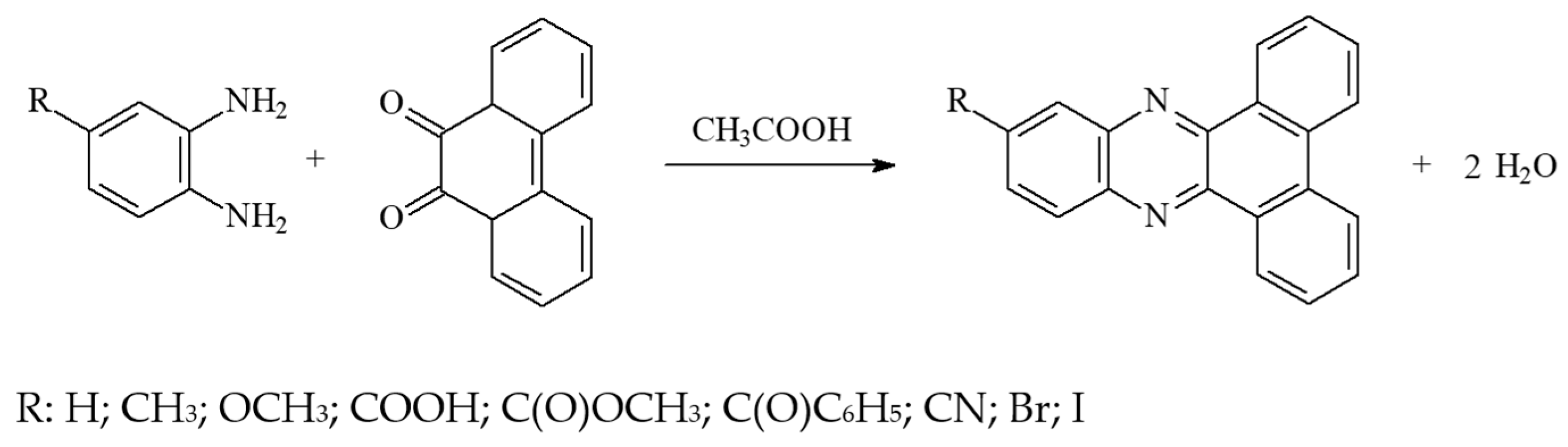
2.2.1. Procedure for Obtaining Dibenzo[a,c]phenazine (DBPh1) and Its Derivatives (DBPh2–DBPh9)
DBPh1: Dibenzo[a,c]phenazine
DBPh2: 11-methyldibenzo[a,c]phenazine
DBPh3: 11-methoxydibenzo[a,c]phenazine
DBPh4: Dibenzo[a,c]phenazine-2-carboxylic Acid
DBPh5: Dibenzo[a,c]phenazine-2-carboxylic Acid Methyl Ester
DBPh6: 11-benzoyldibenzo[a,c]phenazine
DBPh7: 11-carbonitriledibenzo[a,c]phenazine
DBPh8: 11-bromodibenzo[a,c]phenazine
DBPh9: 11-iododibenzo[a,c]phenazine
2.3. Methods
3. Results and Discussion
3.1. Selection and Modification of the Compounds Tested
3.2. Photophysical Data
3.3. Photopolymerization
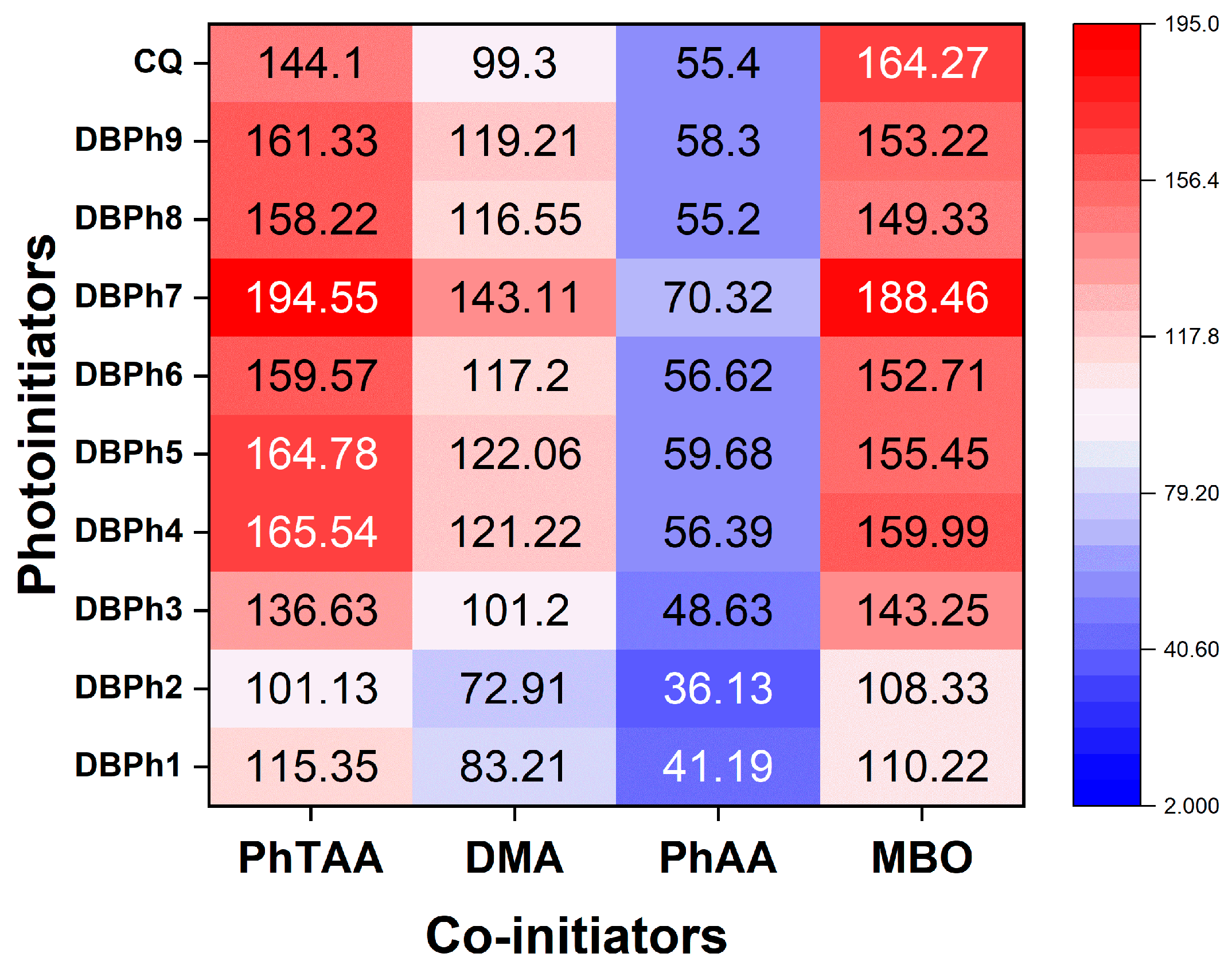
3.3.1. Role of the Photoinitiator
3.3.2. Role of the Co-Initiator
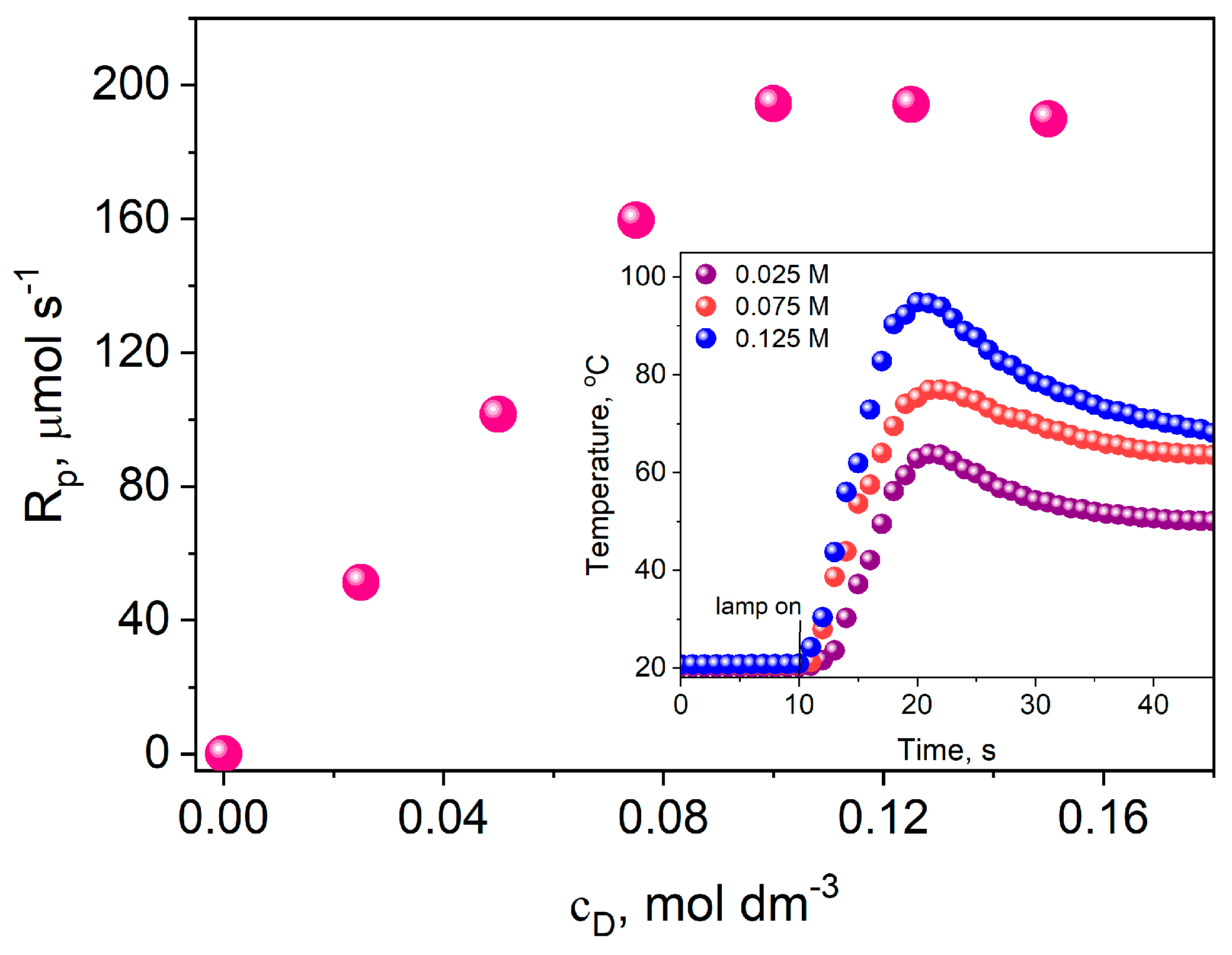
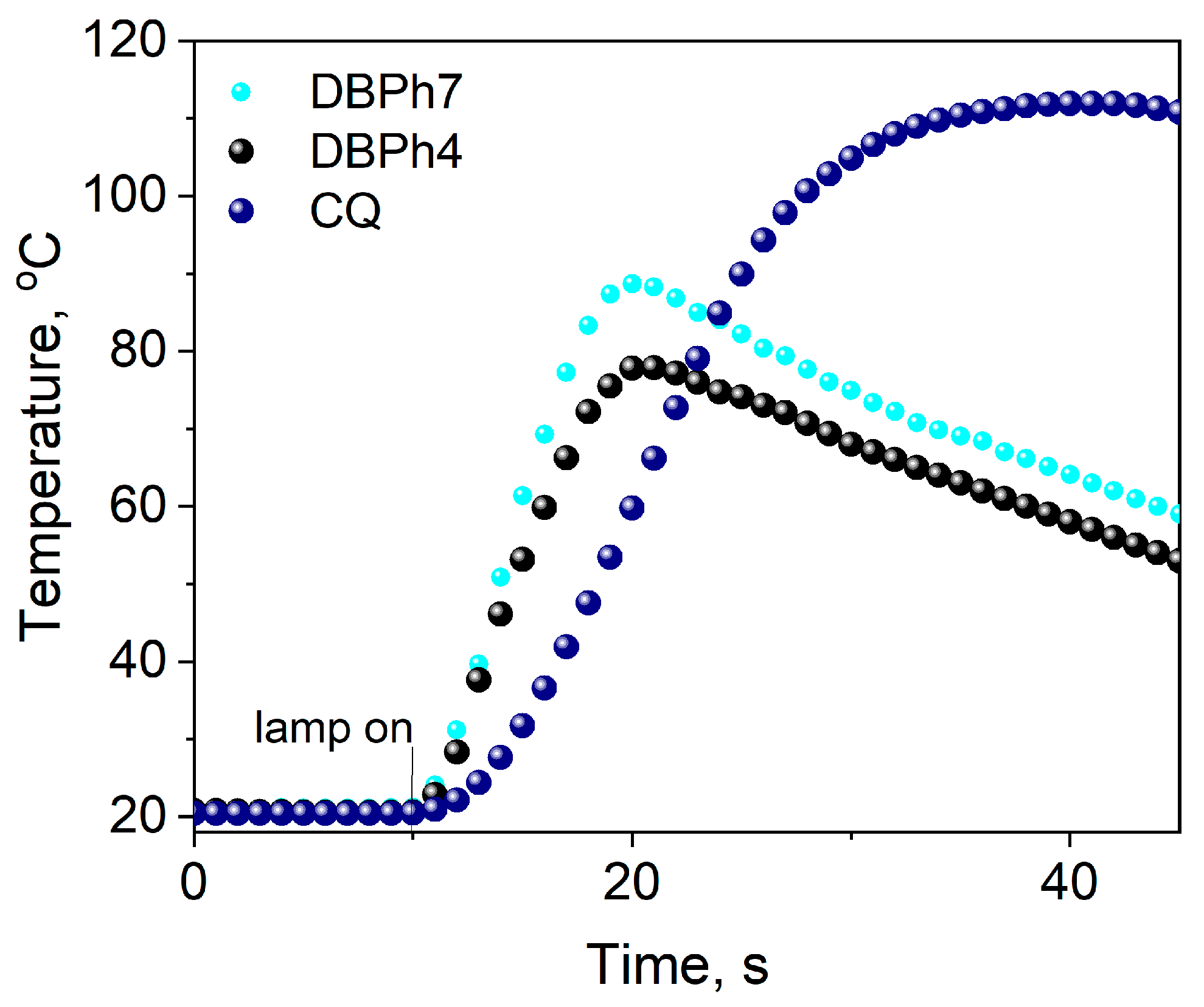
3.3.3. Influence of the Light Intensity of the Dental Lamp
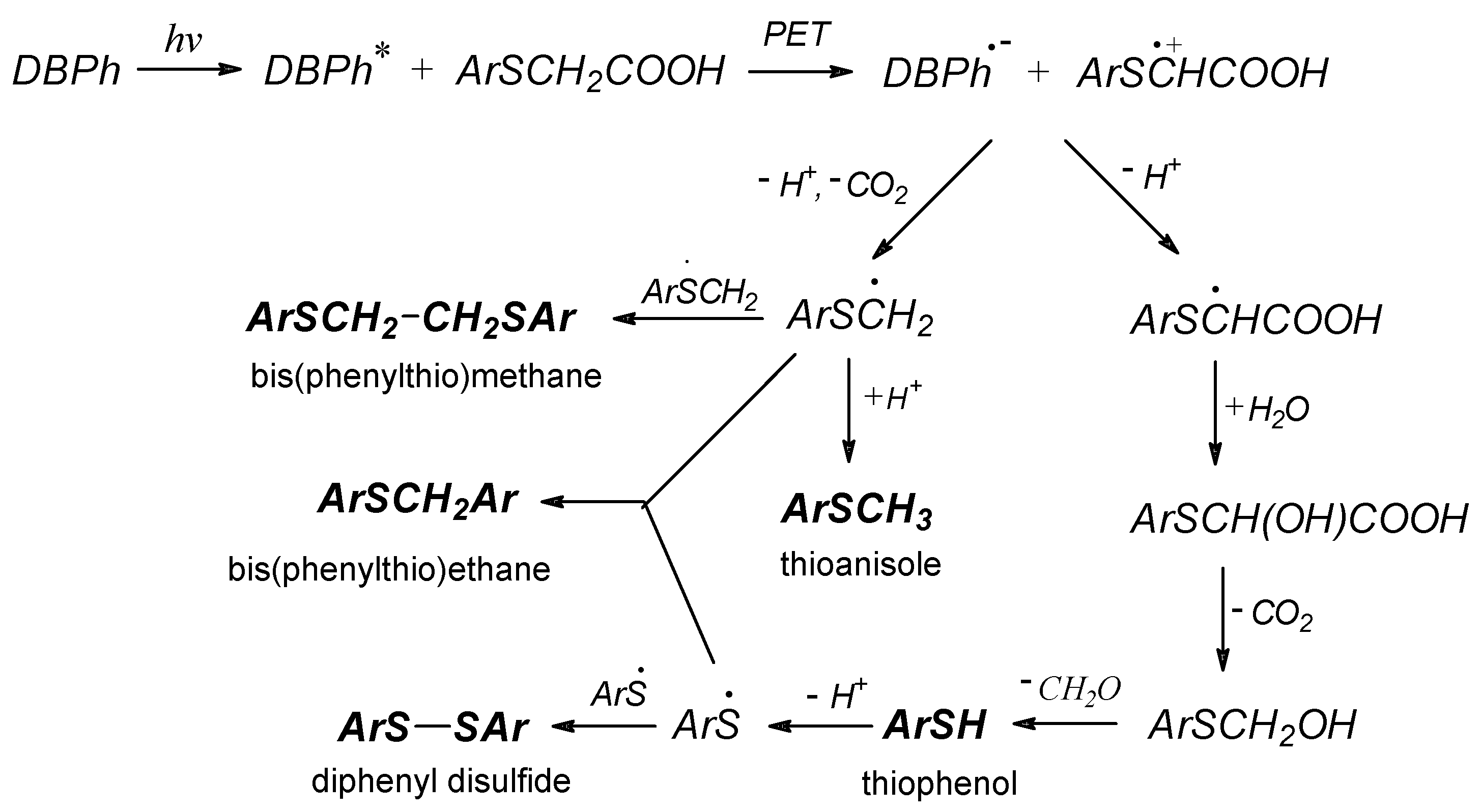
3.3.4. Photoinitiation via the Hydrogen-Atom Transfer Mechanism
3.3.5. Comparative Experiments with a Commercial Photoinitiator
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Allen, N.S.; Rånby, B.G.; Rabek, J.F.; Högskolan, K.T. New Trends in the Photochemistry of Polymers; Springer: Dordrecht, The Netherlands, 1985; ISBN 978-0-85334-365-3. [Google Scholar]
- Yagci, Y.; Jockusch, S.; Turro, N.J. Photoinitiated Polymerization: Advances, Challenges, and Opportunities. Macromolecules 2010, 43, 6245–6260. [Google Scholar] [CrossRef]
- Dumur, F. Recent Advances on Phenothiazine-Based Oxime Esters as Visible Light Photoinitiators of Polymerization. Eur. Polym. J. 2024, 202, 112597. [Google Scholar] [CrossRef]
- Oliveira, P.F.M.; Haruta, N.; Chamayou, A.; Guidetti, B.; Baltas, M.; Tanaka, K.; Sato, T.; Baron, M. Comprehensive Experimental Investigation of Mechanically Induced 1,4-Diazines Synthesis in Solid State. Tetrahedron 2017, 73, 2305–2310. [Google Scholar] [CrossRef]
- Tawagi, E.; Ganesh, T.; Cheng, H.-L.M.; Santerre, J.P. Synthesis of Degradable-Polar-Hydrophobic-Ionic Co-Polymeric Microspheres by Membrane Emulsion Photopolymerization: In Vitro and in Vivo Studies. Acta Biomater. 2019, 89, 279–288. [Google Scholar] [CrossRef] [PubMed]
- Messaddeq, S.H.; Bonnet, A.-S.; Santagnelli, S.H.; Salek, G.; Colmenares, Y.N.; Messaddeq, Y. Photopolymerized Hybrids Containing TiO2 Nanoparticles for Gradient-Index Lens. Mater. Chem. Phys. 2019, 236, 121793. [Google Scholar] [CrossRef]
- Wei, L.; Shang, S.; Zheng, Y.; Liu, J.; Zhu, P. Iridescent Structural Colors Printing on Cellulose Fabrics with Robust Structural Coloration. Dye. Pigment. 2024, 221, 111824. [Google Scholar] [CrossRef]
- Dumur, F. The Future of Visible Light Photoinitiators of Polymerization for Photocrosslinking Applications. Eur. Polym. J. 2023, 187, 111883. [Google Scholar] [CrossRef]
- Mavila, S.; Sinha, J.; Hu, Y.; Podgórski, M.; Shah, P.K.; Bowman, C.N. High Refractive Index Photopolymers by Thiol–Yne “Click” Polymerization. ACS Appl. Mater. Interfaces 2021, 13, 15647–15658. [Google Scholar] [CrossRef]
- Imran, O.Q.; Kim, N.K.; Bodkin, L.N.; Dwulet, G.E.; Feng, X.; Kawabata, K.; Elimelech, M.; Gin, D.L.; Osuji, C.O. Nanoscale Thickness Control of Nanoporous Films Derived from Directionally Photopolymerized Mesophases. Adv. Mater. Interfaces 2021, 8, 2001977. [Google Scholar] [CrossRef]
- Chen, H.; Noirbent, G.; Zhang, Y.; Sun, K.; Liu, S.; Brunel, D.; Gigmes, D.; Graff, B.; Morlet-Savary, F.; Xiao, P.; et al. Photopolymerization and 3D/4D Applications Using Newly Developed Dyes: Search around the Natural Chalcone Scaffold in Photoinitiating Systems. Dye. Pigment. 2021, 188, 109213. [Google Scholar] [CrossRef]
- Vivero-Lopez, M.; Xu, X.; Muras, A.; Otero, A.; Concheiro, A.; Gaisford, S.; Basit, A.W.; Alvarez-Lorenzo, C.; Goyanes, A. Anti-Biofilm Multi Drug-Loaded 3D Printed Hearing Aids. Mater. Sci. Eng. C 2021, 119, 111606. [Google Scholar] [CrossRef] [PubMed]
- Jandt, K.D.; Mills, R.W.; Blackwell, G.B.; Ashworth, S.H. Depth of Cure and Compressive Strength of Dental Composites Cured with Blue Light Emitting Diodes (LEDs). Dent. Mater. 2000, 16, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Esposito Corcione, C.; Striani, R.; Frigione, M. UV-Cured Siloxane-Modified Methacrylic System Containing Hydroxyapatite as Potential Protective Coating for Carbonate Stones. Prog. Org. Coat. 2013, 76, 1236–1242. [Google Scholar] [CrossRef]
- Striani, R.; Esposito Corcione, C.; Dell’Anna Muia, G.; Frigione, M. Durability of a Sunlight-Curable Organic–Inorganic Hybrid Protective Coating for Porous Stones in Natural and Artificial Weathering Conditions. Prog. Org. Coat. 2016, 101, 1–14. [Google Scholar] [CrossRef]
- Sun, K.; Xu, Y.; Dumur, F.; Morlet-Savary, F.; Chen, H.; Dietlin, C.; Graff, B.; Lalevée, J.; Xiao, P. In Silico Rational Design by Molecular Modeling of New Ketones as Photoinitiators in Three-Component Photoinitiating Systems: Application in 3D Printing. Polym. Chem. 2020, 11, 2230–2242. [Google Scholar] [CrossRef]
- Kowalska, A.; Sokolowski, J.; Bociong, K. The Photoinitiators Used in Resin Based Dental Composite—A Review and Future Perspectives. Polymers 2021, 13, 470. [Google Scholar] [CrossRef] [PubMed]
- Lalevee, J.; Fouassier, J.P. Dyes and Chromophores in Polymer Science; ISTE Ltd. and John Wiley & Sons Inc.: Hoboken, NJ, USA, 2015. [Google Scholar]
- Ma, Q.; Zhang, Y.; Launay, V.; Le Dot, M.; Liu, S.; Lalevée, J. How to Overcome the Light Penetration Issue in Photopolymerization? An Example for the Preparation of High Content Iron-Containing Opaque Composites and Application in 3D Printing. Eur. Polym. J. 2022, 165, 111011. [Google Scholar] [CrossRef]
- Dickens, S.H.; Stansbury, J.W.; Choi, K.M.; Floyd, C.J.E. Photopolymerization Kinetics of Methacrylate Dental Resins. Macromolecules 2003, 36, 6043–6053. [Google Scholar] [CrossRef]
- Noè, C.; Hakkarainen, M.; Sangermano, M. Cationic UV-Curing of Epoxidized Biobased Resins. Polymers 2021, 13, 89. [Google Scholar] [CrossRef]
- Dikova, T.; Maximov, J.; Todorov, V.; Georgiev, G.; Panov, V. Optimization of Photopolymerization Process of Dental Composites. Processes 2021, 9, 779. [Google Scholar] [CrossRef]
- Lalevée, J.; Allonas, X.; Jradi, S.; Fouassier, J.-P. Role of the Medium on the Reactivity of Cleavable Photoinitiators in Photopolymerization Reactions. Macromolecules 2006, 39, 1872–1879. [Google Scholar] [CrossRef]
- Andrzejewska, E. Free Radical Photopolymerization of Multifunctional Monomers. In Three-Dimensional Microfabrication Using Two-Photon Polymerization: Fundamentals, Technology, and Applications; William Andrew Publishing: Norwich, NY, USA, 2016; pp. 62–81. ISBN 978-0-323-35321-2. [Google Scholar]
- Pączkowski, J.; Neckers, D.C. Photoinduced Electron Transfer Initiating Systems for Free-Radical Polymerization. In Electron Transfer in Chemistry; Wiley: Hoboken, NJ, USA, 2008; Volume 5. [Google Scholar]
- Zhang, J.; Zivic, N.; Dumur, F.; Xiao, P.; Graff, B.; Gigmes, D.; Fouassier, J.P.; Lalevée, J. A Benzophenone-Naphthalimide Derivative as Versatile Photoinitiator of Polymerization under near UV and Visible Lights. J. Polym. Sci. Part A Polym. Chem. 2015, 53, 445–451. [Google Scholar] [CrossRef]
- Lalevée, J.; Telitel, S.; Tehfe, M.A.; Fouassier, J.P.; Curran, D.P.; Lacôte, E. N-Heterocyclic Carbene Boranes Accelerate Type I Radical Photopolymerizations and Overcome Oxygen Inhibition. Angew. Chem. Int. Ed. 2012, 51, 5958–5961. [Google Scholar] [CrossRef] [PubMed]
- Paczkowski, J.; Kucybala, Z. Generalization of the Kinetic Scheme for a Dye-Photosensitized Free-Radical Polymerization Initiating System via an Intermolecular Electron-Transfer Process. Application of Marcus Theory. Macromolecules 1995, 28, 269–273. [Google Scholar] [CrossRef]
- Wrzyszczynski, A.; Bartoszewicz, J.; Hug, G.L.; Marciniak, B.; Paczkowski, J. Photochemical Studies of a Photodissociative Initiator Based on a Benzophenone Derivative Possessing a Thioether Moiety. J. Photochem. Photobiol. A Chem. 2003, 155, 253–259. [Google Scholar] [CrossRef]
- Abu-Abdoun, I.I.; Thijs, L.; Neckers, D.C. Nonketonic Perester Photoinitiators. Macromolecules 1984, 17, 282–288. [Google Scholar] [CrossRef]
- Sakota, N.; Kishiue, K.; Shimada, S.; Minoura, Y. Photosensitized Copolymerization of Optically Active N-l-Menthylmaleimide with Styrene and Methyl Methacrylate. J. Polym. Sci. Polym. Chem. Ed. 1974, 12, 1787–1797. [Google Scholar] [CrossRef]
- Müller, S.M.; Schlögl, S.; Wiesner, T.; Haas, M.; Griesser, T. Recent Advances in Type I Photoinitiators for Visible Light Induced Photopolymerization. ChemPhotoChem 2022, 6, e202200091. [Google Scholar] [CrossRef]
- Pyszka, I.; Jędrzejewska, B. Photoinitiation Abilities of Indeno- and Indoloquinoxaline Derivatives and Mechanical Properties of Dental Fillings Based on Multifunctional Acrylic Monomers and Glass Ionomer. Polymer 2023, 266, 125625. [Google Scholar] [CrossRef]
- Pyszka, I.; Jędrzejewska, B. Acenaphthoquinoxaline Derivatives as Dental Photoinitiators of Acrylates Polymerization. Materials 2021, 14, 4881. [Google Scholar] [CrossRef]
- Kucybała, Z.; Wrzyszczyński, A. Photolysis of N-[(4-Benzoyl)Benzenesulfonyl]Benzenesulfonamide. J. Photochem. Photobiol. A Chem. 2002, 153, 109–112. [Google Scholar] [CrossRef]
- Fouassier, J.P.; Allonas, X.; Burget, D. Photopolymerization Reactions under Visible Lights: Principle, Mechanisms and Examples of Applications. Prog. Org. Coat. 2003, 47, 16–36. [Google Scholar] [CrossRef]
- Pyszka, I.; Jędrzejewska, B. Design of Dyes Based on the Quinoline or Quinoxaline Skeleton towards Visible Light Photoinitiators. Int. J. Mol. Sci. 2024, 25, 4289. [Google Scholar] [CrossRef]
- Ghorbani, F.; Harry, S.A.; Capilato, J.N.; Pitts, C.R.; Joram, J.; Peters, G.N.; Tovar, J.D.; Smajlagic, I.; Siegler, M.A.; Dudding, T.; et al. Carbonyl-Directed Aliphatic Fluorination: A Special Type of Hydrogen Atom Transfer Beats Out Norrish II. J. Am. Chem. Soc. 2020, 142, 14710–14724. [Google Scholar] [CrossRef]
- Zhu, J.L.; Schull, C.R.; Tam, A.T.; Rentería-Gómez, Á.; Gogoi, A.R.; Gutierrez, O.; Scheidt, K.A. Photoinduced Acylations Via Azolium-Promoted Intermolecular Hydrogen Atom Transfer. J. Am. Chem. Soc. 2023, 145, 1535–1541. [Google Scholar] [CrossRef]
- Kabatc, J.; Kucybała, Z.; Pietrzak, M.; Ścigalski, F.; Pączkowski, J. Free Radical Polymerization Initiated via Photoinduced Intermolecular Electron Transfer Process: Kinetic Study 3. Polymer 1999, 40, 735–745. [Google Scholar] [CrossRef]
- Jiang, X.; Luo, J.; Yin, J. A Novel Amphipathic Polymeric Thioxanthone Photoinitiator. Polymer 2009, 50, 37–41. [Google Scholar] [CrossRef]
- Tang, J.; Xu, X.; Shen, X.; Kuang, C.; Chen, H.; Shi, M.; Huang, N. Ketocoumarin-Based Photoinitiators for High-Sensitivity Two-Photon Lithography. ACS Appl. Polym. Mater. 2023, 5, 2956–2963. [Google Scholar] [CrossRef]
- Pyszka, I.; Kucybała, Z.; Pączkowski, J. Reinvestigation of the Mechanism of the Free Radical Polymerization Photoinitiation Process by Camphorquinone–Coinitiator Systems: New Results. Macromol. Chem. Phys. 2004, 205, 2371–2375. [Google Scholar] [CrossRef]
- Cook, W.D. Photopolymerization Kinetics of Dimethacrylates Using the Camphorquinone/Amine Initiator System. Polymer 1992, 33, 600–609. [Google Scholar] [CrossRef]
- Jakubiak, J.; Allonas, X.; Fouassier, J.P.; Sionkowska, A.; Andrzejewska, E.; Linden, L.Å.; Rabek, J.F. Camphorquinone–Amines Photoinitating Systems for the Initiation of Free Radical Polymerization. Polymer 2003, 44, 5219–5226. [Google Scholar] [CrossRef]
- Jędrzejewska, B.; Wejnerowska, G. Highly Effective Sensitizers Based on Merocyanine Dyes for Visible Light Initiated Radical Polymerization. Polymers 2020, 12, 1242. [Google Scholar] [CrossRef] [PubMed]
- Pyszka, I.; Kucybała, Z.; Jędrzejewska, B. Effective Singlet Oxygen Sensitizers Based on the Phenazine Skeleton as Efficient Light Absorbers in Dye Photoinitiating Systems for Radical Polymerization of Acrylates. Materials 2021, 14, 3085. [Google Scholar] [CrossRef] [PubMed]
- Jędrzejewska, B.; Ośmiałowski, B. Difluoroboranyl Derivatives as Efficient Panchromatic Photoinitiators in Radical Polymerization Reactions. Polym. Bull. 2018, 75, 3267–3281. [Google Scholar] [CrossRef]
- Strzelczyk, R.; Podsiadły, R. Naphthoylenebenzimidazolone Dyes as One-Component Photoinitiators. Color. Technol. 2017, 133, 178–183. [Google Scholar] [CrossRef]
- Kabatc, J. The Influence of a Radical Structure on the Kinetics of Photopolymerization. J. Polym. Sci. Part A Polym. Chem. 2017, 55, 1575–1589. [Google Scholar] [CrossRef]
- Balcerak, A.; Kabatc, J. The Photooxidative Sensitization of Bis(p-Substituted Diphenyl)Iodonium Salts in the Radical Polymerization of Acrylates. RSC Adv. 2019, 9, 28490–28499. [Google Scholar] [CrossRef] [PubMed]
- Mousawi, A.A.; Dietlin, C.; Graff, B.; Morlet-Savary, F.; Toufaily, J.; Hamieh, T.; Fouassier, J.P.; Chachaj-Brekiesz, A.; Ortyl, J.; Lalevée, J. Meta-Terphenyl Derivative/Iodonium Salt/9H-Carbazole-9-Ethanol Photoinitiating Systems for Free Radical Promoted Cationic Polymerization upon Visible Lights. Macromol. Chem. Phys. 2016, 217, 1955–1965. [Google Scholar] [CrossRef]
- Allen, N.S. Photochemistry and Photophysics of Polymer Materials; Wiley: Hoboken, NJ, USA, 2010. [Google Scholar]
- Ferracane, J.L. Current Trends in Dental Composites. Crit. Rev. Oral Biol. Med. 1995, 6, 302–318. [Google Scholar] [CrossRef]
- Ferracane, J.L. Resin Composite—State of the Art. Dent. Mater. 2011, 27, 29–38. [Google Scholar] [CrossRef]
- Carlier, L.; Baron, M.; Chamayou, A.; Couarraze, G. Use of Co-Grinding as a Solvent-Free Solid State Method to Synthesize Dibenzophenazines. Tetrahedron Lett. 2011, 52, 4686–4689. [Google Scholar] [CrossRef]
- Niknam, K.; Zolfigol, M.A.; Tavakoli, Z.; Heydari, Z. Metal Hydrogen Sulfates M(HSO4)n: As Efficient Catalysts for the Synthesis of Quinoxalines in EtOH at Room Temperature. J. Chin. Chem. Soc. 2008, 55, 1373–1378. [Google Scholar] [CrossRef]
- Go, A.; Lee, G.; Kim, J.; Bae, S.; Lee, B.M.; Kim, B.H. One-Pot Synthesis of Quinoxalines from Reductive Coupling of 2-Nitroanilines and 1,2-Diketones Using Indium. Tetrahedron 2015, 71, 1215–1226. [Google Scholar] [CrossRef]
- Kawauchi, D.; Noda, K.; Komatsu, Y.; Yoshida, K.; Ueda, H.; Tokuyama, H. Aerobic Dehydrogenation of N-Heterocycles with Grubbs Catalyst: Its Application to Assisted-Tandem Catalysis to Construct N-Containing Fused Heteroarenes. Chem. A Eur. J. 2020, 26, 15793–15798. [Google Scholar] [CrossRef] [PubMed]
- Tamarkin, D.; Yoram, C.; Rabinovitz, M. Ring Closure of Heterocyclic Systems by Potassium-Graphite Intercalat-C8K. Synthesis 2002, 1987, 196–197. [Google Scholar] [CrossRef]
- Patil, M.U.; Shinde, S.K.; Patil, S.P.; Patil, S.S. [BBSA-DBN][HSO4]: A Novel –SO3H Functionalized Bronsted Acidic Ionic Liquid for Easy Access of Quinoxalines. Res. Chem. Intermed. 2020, 46, 4923–4938. [Google Scholar] [CrossRef]
- Karami, B.; Khodabakhshi, S. A novel and simple synthesis of some new and known dibenzo phenazine and quinoxaline derivatives using lead dichloride. J. Chil. Chem. Soc. 2013, 58, 1655–1658. [Google Scholar] [CrossRef]
- Wilson, J.; Hunt, F. Iminodiacetic Acid Derivatives of Benzimidazole. Synthesis of N-(Benzimidazol-2-Ylmethyl)Iminodiacetic Acids. Aust. J. Chem. 1983, 36, 2317–2325. [Google Scholar] [CrossRef]
- Pyszka, I.; Skowroński, Ł.; Jędrzejewska, B. Study on New Dental Materials Containing Quinoxaline-Based Photoinitiators in Terms of Exothermicity of the Photopolymerization Process. Int. J. Mol. Sci. 2023, 24, 2752. [Google Scholar] [CrossRef]
- Chen, J.; Chen, Y.; Wu, Y.; Wang, X.; Yu, Z.; Xiao, L.; Liu, Y.; Tian, H.; Yao, J.; Fu, H. Modulated Emission from Dark Triplet Excitons in Aza-Acene Compounds: Fluorescence versus Phosphorescence. New J. Chem. 2017, 41, 1864–1871. [Google Scholar] [CrossRef]
- Jabali, B.; Abu Ali, H. New Zinc(II) Complexes of the Non-Steroidal Anti-Inflammatory Drug (Indomethacin) and Various Nitrogen Donor Ligands. Synthesis, Characterization and Biological Activity. Polyhedron 2016, 117, 249–258. [Google Scholar] [CrossRef]
- Jaman, Z.; Karim, M.R.; Siddiquee, T.A.; Mirza, A.H.; Ali, M.A. Synthesis of 5-Substituted 2, 9-Dimethyl-1,10-Phenanthroline Dialdehydes and Their Schiff Bases with Sulfur-Containing Amines. Int. J. Org. Chem. 2013, 3, 214–219. [Google Scholar] [CrossRef]
- Ajani, O.O.; Nlebemuo, M.T.; Adekoya, J.A.; Ogunniran, K.O.; Siyanbola, T.O.; Ajanaku, C.O. Chemistry and Pharmacological Diversity of Quinoxaline Motifs as Anticancer Agents. Acta Pharm. 2019, 69, 177–196. [Google Scholar] [CrossRef] [PubMed]
- Nie, J.; Andrzejewska, E.; Rabek, J.F.; Lindén, L.Å.; Fouassier, J.P.; Paczkowski, J.; Scigalski, F.; Wrzyszczynski, A. Effect of Peroxides and Hydroperoxides on the Camphorquinone-Initiated Photopolymerization. Macromol. Chem. Phys. 1999, 200, 1692–1701. [Google Scholar] [CrossRef]
- Nie, J.; Lindén, L.Å.; Rabek, J.F.; Ekstrand, J. Photocuring Kinetic Studies of New Dental Restorative Resins Based on Poly(Ethylene Glycol) Diacrylate and Tris[2-(Acryloyloxy)-Ethyl]Isocyanurate. Die Angew. Makromol. Chem. 1998, 257, 47–52. [Google Scholar] [CrossRef]
- Corrigan, N.; Yeow, J.; Judzewitsch, P.; Xu, J.; Boyer, C. Seeing the Light: Advancing Materials Chemistry through Photopolymerization. Angew. Chem. Int. Ed. 2019, 58, 5170–5189. [Google Scholar] [CrossRef]
- Kucybała, Z.; Pyszka, I.; Pączkowski, J. Development of New Dyeing Photoinitiators for Free Radical Polymerization Based on the 1H-Pyrazolo[3,4-b]Quinoxaline Skeleton. Part 2. J. Chem. Soc. Perkin Trans. 2 2000, 1559–1567. [Google Scholar] [CrossRef]
- Kucybała, Z.; Przyjazna, B.; Linden, L.-Å.; Paczkowski, J. Development of New Dyeing Photoinitiators for Free Radical Polymerization Based on 1H-Pyrazolo[3,4-b]Quinoline Skeleton. IV. Polym. Bull. 2000, 45, 327–334. [Google Scholar] [CrossRef]
- Pyszka, I.; Kucybała, Z. Quinolineimidazopyridinium Derivatives as Visible-Light Photoinitiators of Free Radical Polymerization. Polymer 2007, 48, 959–965. [Google Scholar] [CrossRef]
- Kabatc, J.; Gruszewska, M.; Jędrzejewska, B.; Pączkowski, J. Novel 6-Bromo-3-Ethyl-2-Styrylbenzothiazolium n-Butyl-Triphenylborates as Photoinitiators of Trimethylolopropane Triacrylate (TMPTA) Polymerization. Polym. Bull. 2007, 58, 691–701. [Google Scholar] [CrossRef]
- Podsiadły, R.; Sokołowska, J.; Kolińska, J.; Grzelakowska, A. Synthesis and Photochemical Reaction of Benzo[a]Quinoxalino[2,3-c]Phenazine Dyes. Color. Technol. 2017, 133, 498–505. [Google Scholar] [CrossRef]
- Podsiadły, R.; Szymczak, A.M.; Podemska, K. The Synthesis of Novel, Visible-Wavelength, Oxidizable Polymerization Sensitizers Based on the 8-Halogeno-5,12-Dihydroquinoxalino[2,3-b]Quinoxaline Skeleton. Dye. Pigment. 2009, 82, 365–371. [Google Scholar] [CrossRef]
- OSTER, G. Dye-Sensitized Photopolymerization. Nature 1954, 173, 300–301. [Google Scholar] [CrossRef]
- Linden, S.M.; Neckers, D.C. Fundamental Properties of Rose Bengal. 25. Bleaching Studies of Rose Bengal Onium Salts. J. Am. Chem. Soc. 1988, 110, 1257–1260. [Google Scholar] [CrossRef]
- Polykarpov, A.Y.; Hassoon, S.; Neckers, D.C. Tetramethylammonium Tetraorganylborates as Coinitiators with 5,7-Diiodo-3-Butoxy-6-Fluorone in Visible Light Polymerization of Acrylates. Macromolecules 1996, 29, 8274–8276. [Google Scholar] [CrossRef]
- Hansch, C.; Leo, A.; Taft, R.W. A Survey of Hammett Substituent Constants and Resonance and Field Parameters. Chem. Rev. 1991, 91, 165–195. [Google Scholar] [CrossRef]
- Pyszka, I.; Kucybała, Z. The Effect of Co-Initiator Structure on Photoinitiating Efficiency of Photoredox Couples Composed of Quinoline[2,3-b]-1H-Imidazo[1,2-a]Pyridiniumbromide and Phenoxyacetic Acid or N,N-Dimethylaniline Derivatives. Polym. Bull. 2008, 61, 553–562. [Google Scholar] [CrossRef]
- Borjigin, T.; Schmitt, M.; Giacoletto, N.; Rico, A.; Bidotti, H.; Nechab, M.; Zhang, Y.; Graff, B.; Morlet-Savary, F.; Xiao, P.; et al. The Blue-LED-Sensitive Naphthoquinone-Imidazolyl Derivatives as Type II Photoinitiators of Free Radical Photopolymerization. Adv. Mater. Interfaces 2023, 10, 2202352. [Google Scholar] [CrossRef]
- Brimage, D.R.G.; Davidson, R.S.; Steiner, P.R. Use of Heterocyclic Compounds as Photosensitisers for the Decarboxylation of Carboxylic Acids. J. Chem. Soc. Perkin Trans. 1 1973, 526–529. [Google Scholar] [CrossRef]
- Bartholomew, R.F.; Davidson, R.S. The Photosensitised Oxidation of Amines. Part I. The Use of Benzophenone as a Sensitiser. J. Chem. Soc. C 1971, 2342–2346. [Google Scholar] [CrossRef]
- Davidson, R.S.; Harrison, K.; Steiner, P.R. The Photosensitised Decarboxylation of Carboxylic Acids by Aromatic Ketones. J. Chem. Soc. C 1971, 3480–3482. [Google Scholar] [CrossRef]
- Davidson, R.S.; Steiner, P.R. The Photosensitized Decarboxylation of Carboxylic Acids by Benzophenone and Quinones. J. Chem. Soc. C 1971, 1682–1689. [Google Scholar] [CrossRef]
- Gruber, H.F. Photoinitiators for Free Radical Polymerization. Prog. Polym. Sci. 1992, 17, 953–1044. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dye | Structure | λmax | ε | Dye | Structure | λmax | ε |
|---|---|---|---|---|---|---|---|
| DBPh1 | 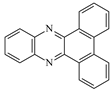 | 375 395 | 9700 11,400 | DBPh6 |  | 385 406 | 14,900 20,500 |
| DBPh2 |  | 378 398 | 11,500 18,300 | DBPh7 |  | 393 413 | 19,400 20,600 |
| DBPh3 |  | 384 405 | 15,600 21,100 | DBPh8 |  | 382 403 | 20,500 25,600 |
| DBPh4 | 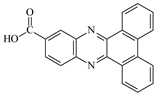 | 377 399 | 23,500 27,000 | DBPh9 | 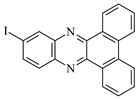 | 385 406 | 20,400 26,900 |
| DBPh5 | 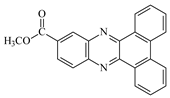 | 383 404 | 19,200 28,300 | CQ |  | 472 | 40 |
| Compound | Structure | σ, ppm |
|---|---|---|
| thioanisole | C6H5SCH3 | 2.47 |
| thiophenol | C6H5SH | 3.95 |
| Diphenyl disulfide | C6H5S-SC6H5 | - |
| bis(phenylthio)methane | C6H5S-CH2-SC6H5 | 4.47 |
| bis(phenylthio)ethane | C6H5S-CH2-CH2-SC6H5 | 3.12 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pyszka, I.; Jędrzejewska, B. Highly Efficient Photoinitiation Systems Based on Dibenzo[a,c]phenazine Sensitivity to Visible Light for Dentistry. Materials 2024, 17, 2597. https://doi.org/10.3390/ma17112597
Pyszka I, Jędrzejewska B. Highly Efficient Photoinitiation Systems Based on Dibenzo[a,c]phenazine Sensitivity to Visible Light for Dentistry. Materials. 2024; 17(11):2597. https://doi.org/10.3390/ma17112597
Chicago/Turabian StylePyszka, Ilona, and Beata Jędrzejewska. 2024. "Highly Efficient Photoinitiation Systems Based on Dibenzo[a,c]phenazine Sensitivity to Visible Light for Dentistry" Materials 17, no. 11: 2597. https://doi.org/10.3390/ma17112597