First-Principles Study of the Structural, Mechanical, Electronic, and Thermodynamic Properties of AlCu2M (M = Ti, Cr, Zr, Sc, Hf, Mn, Pa, Lu, Pm) Ternary Intermetallic Compounds


Abstract
1. Introduction
2. Computational Methods
3. Results and Discussion
3.1. Structural Properties
3.2. Mechanical Properties
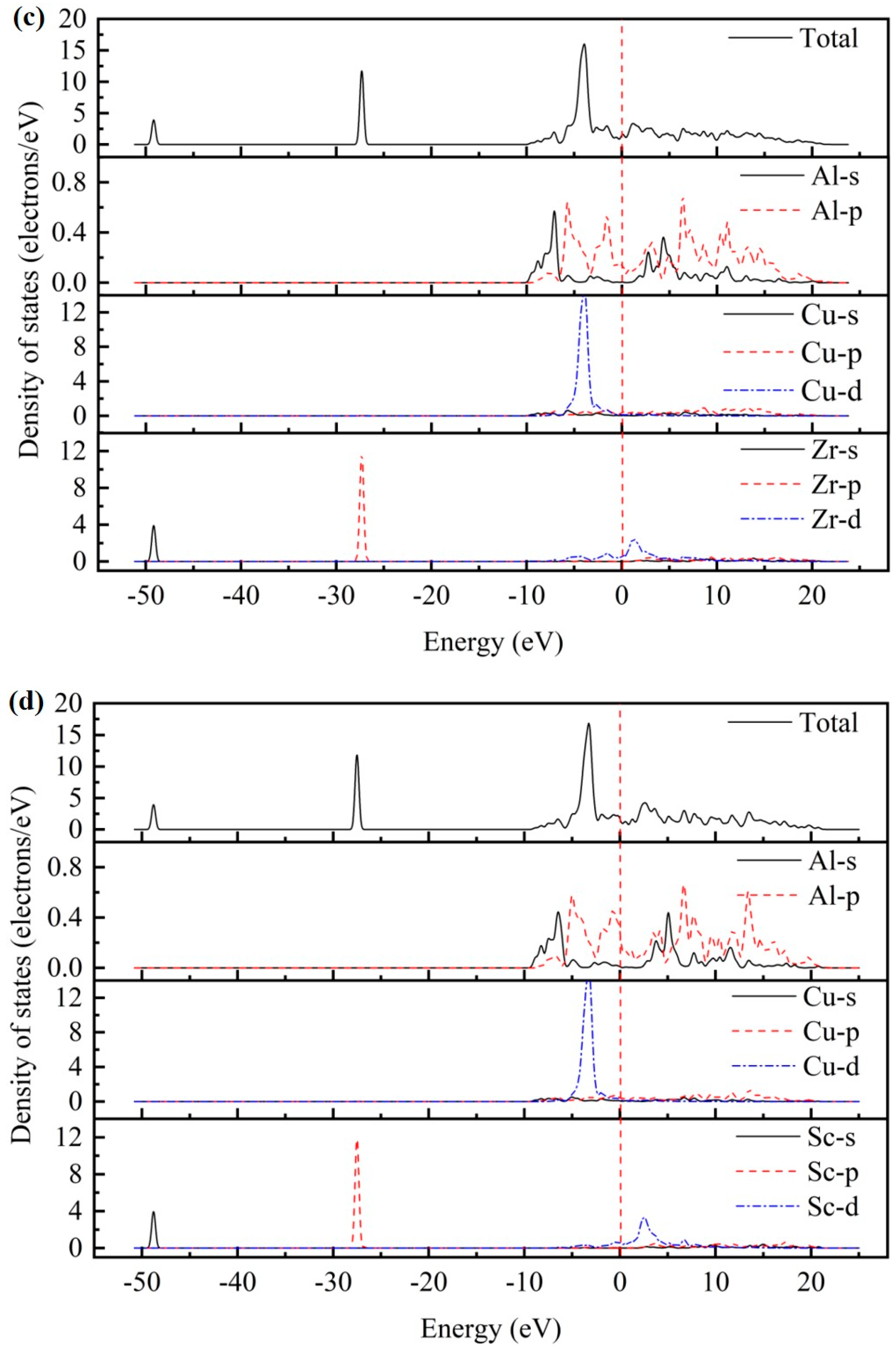
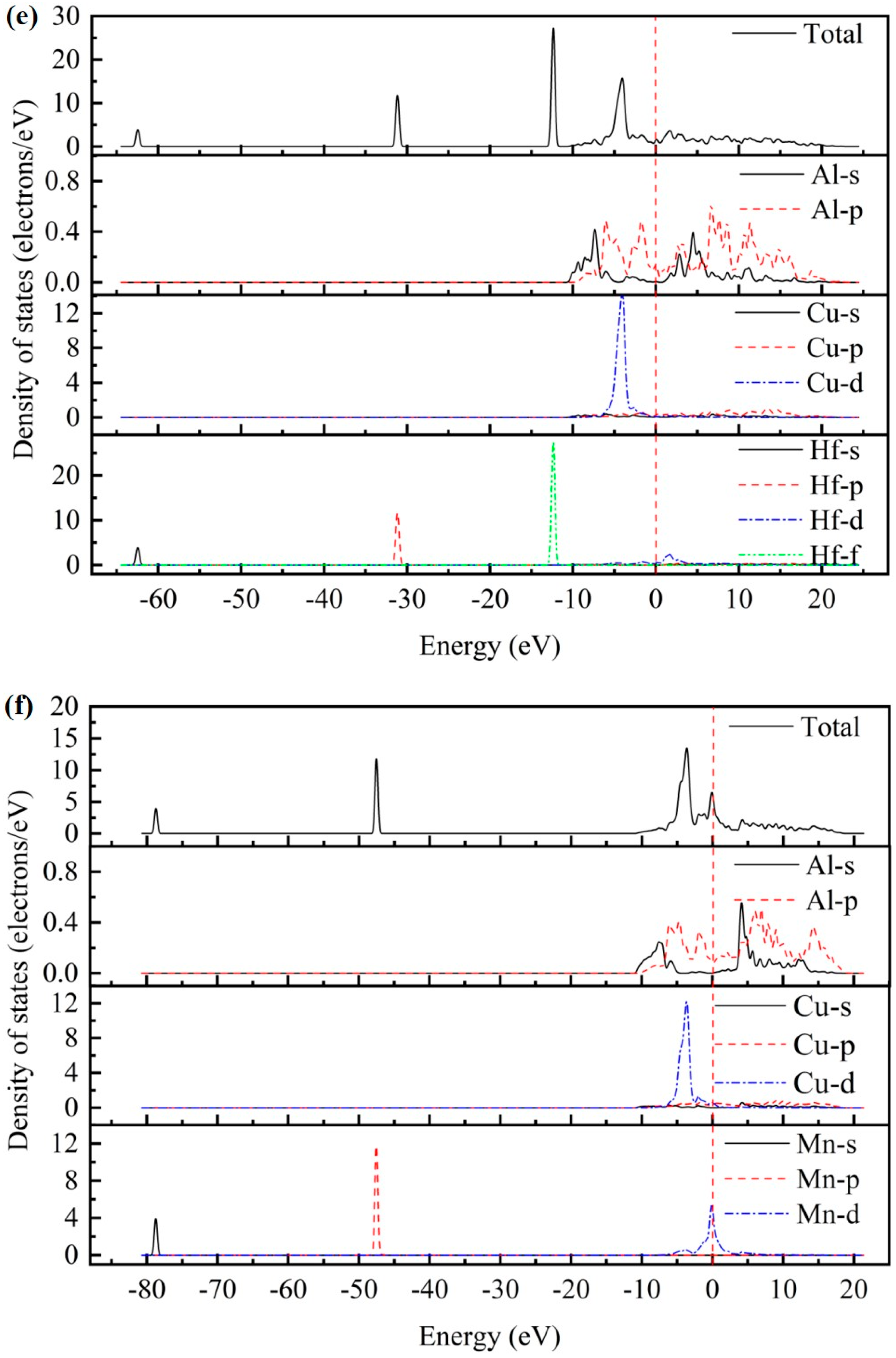
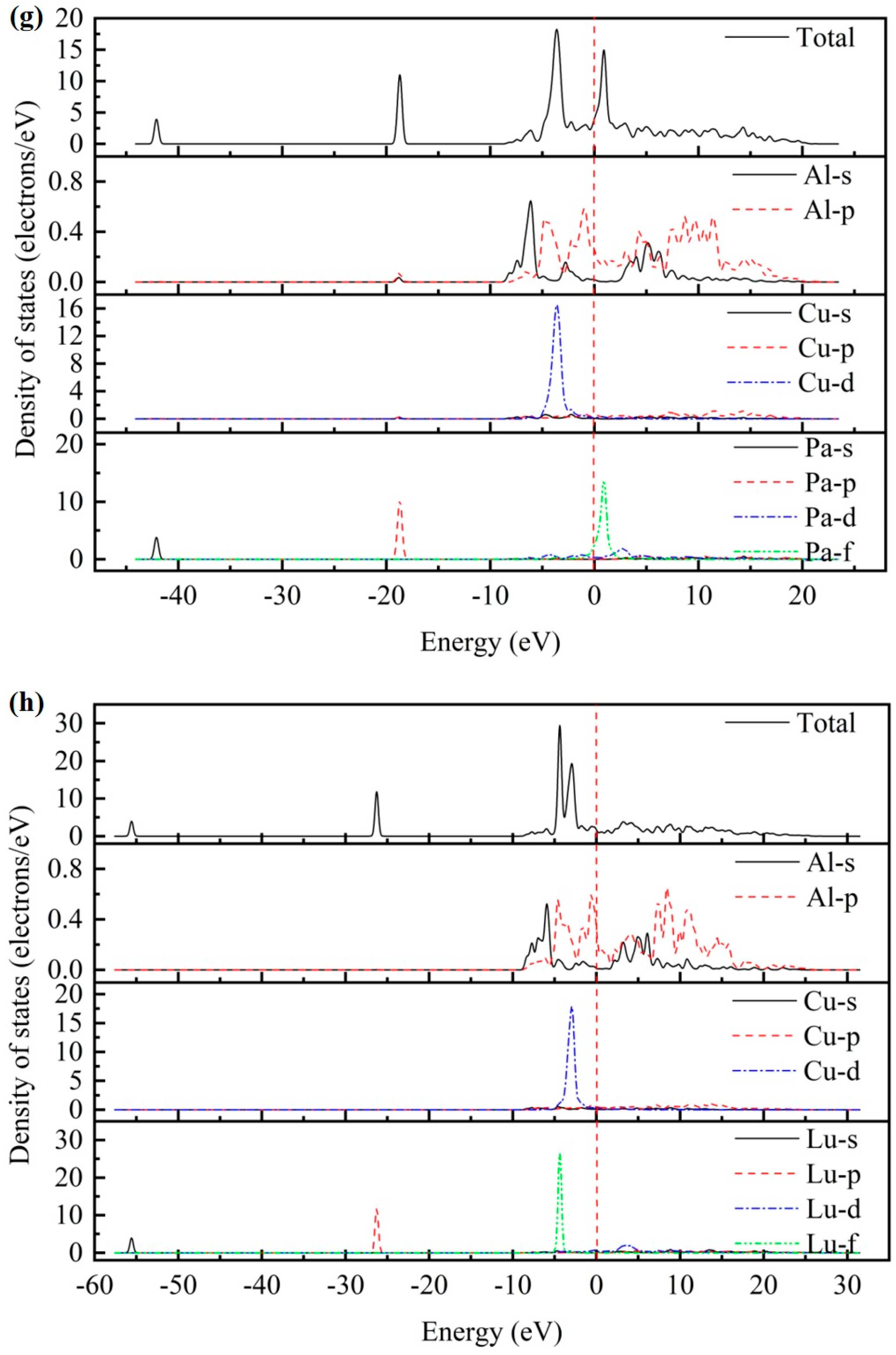
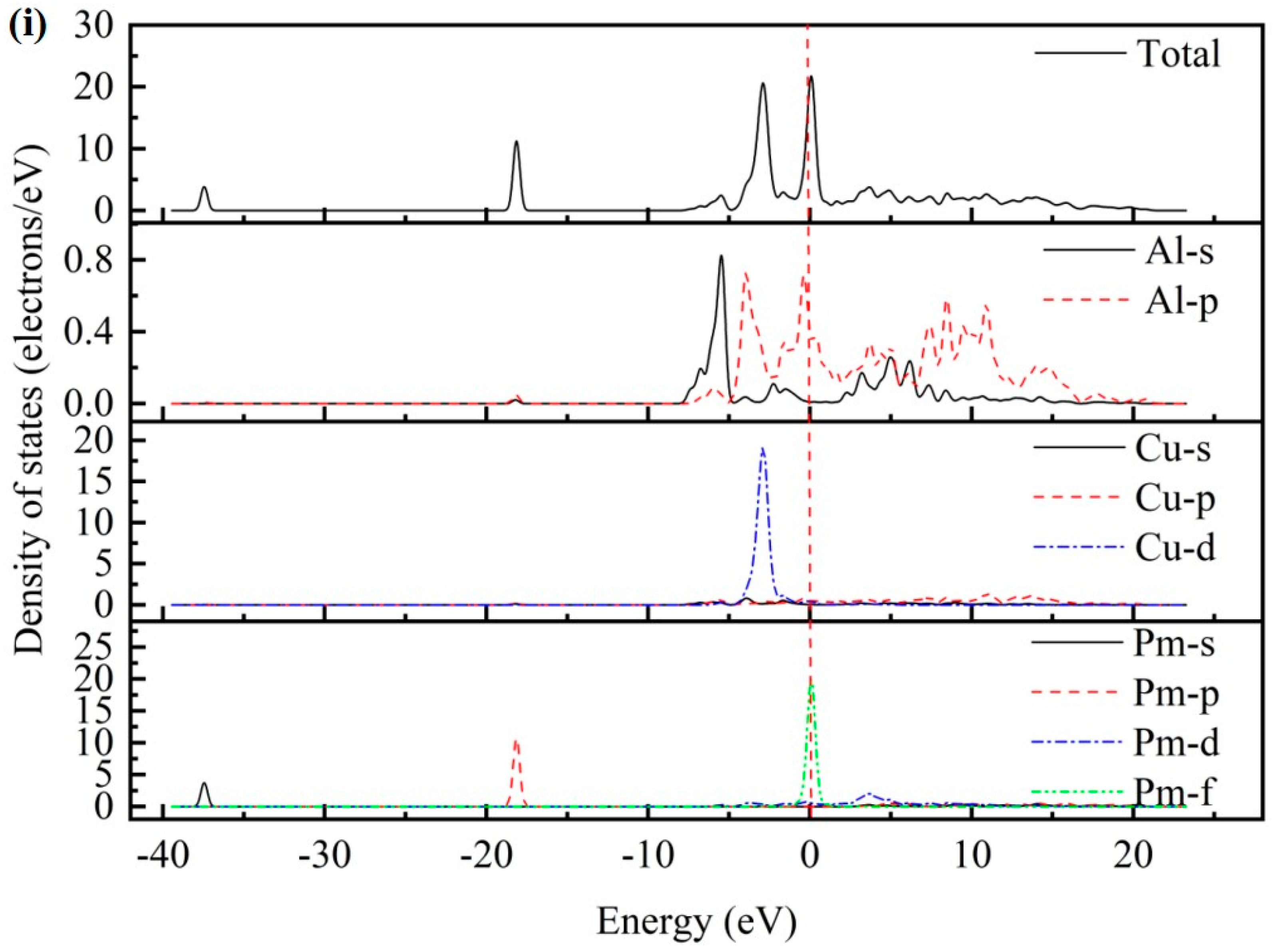
3.3. Electronic Properties
3.4. Thermodynamic Properties
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wen, M.C.; Hsu, Y.D.; Chen, M.C.; Yang, W.C.; Lee, S.L. Effects of heterogenization treatment on the hot-working temperature and mechanical properties of Al-Cu-Mg-Mn-(Zr) alloys. Materials 2023, 16, 4256. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.D.; Liu, W.S.; Xiao, D.H.; Huang, L.P. Influence of thermal exposure on the microstructure evolution and mechanical behaviors of an Al-Cu-Li alloy. Mater. Des. 2023, 227, 111767. [Google Scholar] [CrossRef]
- Belov, N.A.; Cherkasov, S.O.; Korotkova, N.O.; Motkov, M.M. The impact of thermomechanical treatment on structure, electrical resistance and hardness of Al-4%Cu-3%Mn alloy casted in an electromagnetic crystallizer. Phys. Met. Metallogr. 2024, 125, 203–210. [Google Scholar] [CrossRef]
- Bayram, U.; Marasli, N. Thermal conductivity and electrical resistivity dependences on growth rate in the directionally solidified Al-Cu-Ni eutectic alloy. J. Alloy Compd. 2018, 753, 695–702. [Google Scholar] [CrossRef]
- Deng, S.X.; Li, J.F.; Ning, H.; Lu, D.D.; Zeng, G.J.; Liu, Z.Z.; Xiang, H.; Que, J.R.; Liu, D.Y. Effect of Zr addition on the microstructure evolution and mechanical properties of extruded Al-Cu-Li-Mn alloys. Mater. Charact. 2023, 202, 113011. [Google Scholar] [CrossRef]
- Song, Z.X.; Li, Y.D.; Liu, W.J.; Yang, H.K.; Cao, Y.J.; Bi, G.L. Effect of La and Sc co-addition on the mechanical properties and thermal conductivity of as-cast Al-4.8%Cu alloys. Metals 2021, 11, 1866. [Google Scholar] [CrossRef]
- Ujjval, B.; Mahander, P.S.; Sukla, M.; Shyam, K.S.; Surendra, K.M.; Aloke, P.; Kamanio, C. The interplay of precipitation of ordered compounds and interfacial segregation in Al-Cu-Hf-Si alloys for high-temperature strength. Acta Mater. 2022, 240, 118355. [Google Scholar] [CrossRef]
- Jonathan, D.P.; Brian, K.M.; Lawrence, F.A.; Dongwon, S.; Patrick, S.; Matthew, F.C.; Amit, S. The synergistic role of Mn and Zr/Ti in producing θ’/L12 co-precipitates in Al-Cu alloys. Acta Mater. 2020, 194, 577–586. [Google Scholar] [CrossRef]
- Khisamov, R.K.; Khalikova, G.R.; Kistanov, A.A.; Korznikova, G.F.; Korznikova, E.A.; Nazarov, K.S.; Sergeev, S.N.; Shayakhmetov, R.U.; Timiryaev, R.R.; Yumaguzin, Y.M.; et al. Microstructure, microhardness and work function of in-situ Al-Cu composite processed by mechanical alloying by means of high-pressure torsion. Continuum Mech. Thermodyn. 2023, 35, 1433–1444. [Google Scholar] [CrossRef]
- Dong, L.M.; Han, Z.D.; Guan, Y.Z.; Li, W.; Zhang, X.Y. Mechanical properties and work function of L21 structure AlCu2X (X=Ti, Mn, Zr, or Hf) intermetallics. Mat. Sci. Eng. A 2012, 545, 13–19. [Google Scholar] [CrossRef]
- Pang, M.J.; Zhan, Y.Z.; Wang, H.Z.; Jiang, W.P.; Du, Y. Ab initio study of AlCu2M (M=Sc, Ti and Cr) ternary compounds under pressures. Comput. Mater. Sci. 2011, 50, 2930–2937. [Google Scholar] [CrossRef]
- Guo, Y.; Wang, Y.; Chen, H.T.; Xu, H.Y.; Hu, M.L.; Ji, Z.S. First-principles study on stability, electronic, mechanical and thermodynamic properties of Al-Cu-RE ternary compounds. Solid State Commun. 2019, 287, 63–67. [Google Scholar] [CrossRef]
- Huang, G.; Liu, L.; Zhang, L.; Jin, Z. Thermodynamic description of the Al-Cu-Yb ternary system supported by first-principles calculations. J. Min. Metall. B 2016, 52, 177–183. [Google Scholar] [CrossRef]
- Segall, M.D.; Lindan, P.J.D.; Probert, M.J.; Pickard, C.J.; Hasnip, P.J.; Clark, S.J.; Payne, M.C. First-principles simulation: Ideas, illustrations and the CASTEP code. J. Phys. Condens. Matter. 2002, 14, 2717. [Google Scholar] [CrossRef]
- Perdew, J.P. Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys. Rev. B 1986, 33, 8822–8824. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Pfrommer, B.G.; Côté, M.; Louie, S.G.; Cohen, M.L. Relaxation of crystals with the Quasi-Newton method. J. Comput. Phys. 1997, 131, 233–240. [Google Scholar] [CrossRef]
- Feynman, R.P. Forces in molecules. Phys. Rev. 1939, 56, 340–343. [Google Scholar] [CrossRef]
- Ruediger MZ, R.; Peter, C.S.; Alarich, W. Reaction of Hydrogen with the Heusler-Type Phases Cu2TiAl and Cu2ZrAl*. Z. Für Phys. Chem. 1989, 163, 103–108. [Google Scholar] [CrossRef]
- Buschow, K.H.J.; Engen, P.G.V. Magnetic and magneto-optical properties of heusler alloys based on aluminum and gallium. J. Magn. Magn. Mater. 1981, 25, 90–96. [Google Scholar] [CrossRef]
- Dwight, A.E.; Kimball, C.W. ScT2X and LnT2X compounds with the MnCu2al-type structure. J. Less-Common Metals 1987, 127, 179–182. [Google Scholar] [CrossRef]
- Takeuchi, A.; Yubuta, K.; Yokoyama, Y.; Makino, A.; Inoue, A. Noncrystalline atomic arrangements computationally created from crystalline compound by treating groups of atoms as hypothetical clusters. Intermetallics 2008, 16, 283–292. [Google Scholar] [CrossRef]
- Markiv, V.Y.; Voroshilov, Y.V.; Kripyakevich, P.I.; Cherkashin, E.E. New compounds of the MnCu2Al and MgZn2 types containing aluminum and gallium. Sov. Phys. Crystallogr. 1964, 9, 619–620. [Google Scholar]
- Jain, A.; Ong, S.P.; Hautier, G.; Chen, W.; Richards, W.D.; Dacek, S.; Cholia, S.; Gunter, D.; Skinner, D.; Ceder, G.; et al. Commentary: The Materials Project: A materials genome approach to accelerating materials innovation. APL Mater. 2013, 1, 011002. [Google Scholar] [CrossRef]
- Zubov, V.I.; Tretiakov, N.P.; Teixeira Rabelo, J.N.; Sanchez Ortiz, J.F. Calculations of the thermal expansion, cohesive energy and thermodynamic stability of a Van der Waals crystal-fullerene C60. Phys. Lett. A 1994, 194, 223–227. [Google Scholar] [CrossRef]
- Andrey, D.I.; Andrey, A.K.; Artem, A.I.; Elena, A.K. The nitriding effect on the stability and mechanical properties of the iron titan phase: First-principles investigation. Phys. Chem. Chem. Phys. 2023, 25, 24060. [Google Scholar] [CrossRef]
- Mouhat, F.; Coudert, F.X. Necessary and sufficient elastic stability conditions in various crystal systems. Phys. Rev. B 2014, 90, 224104. [Google Scholar] [CrossRef]
- Wang, A.J.; Shang, S.L.; Du, Y.; Kong, Y.; Zhang, L.J.; Chen, L. Structural and elastic properties of cubic and hexagonal TiN and AlN from first-principles calculations. Comput. Mater. Sci. 2010, 48, 705–709. [Google Scholar] [CrossRef]
- Guan, Y.Z.; Zhang, H.Y.; Li, W. First-principles study on alloying stability, electronic structure, and mechanical properties of Al-based intermetallics. Phys. B Condens. Matter 2011, 406, 1149–1153. [Google Scholar] [CrossRef]
- Voigt, W. Lehrbuch der Kristallphysik: Teubner-Leipzig; Macmillan: New York, NY, USA, 1928. [Google Scholar]
- Reuss, A. Berechnung del flieβgrenze von mischkristallen auf grund der plastizitätbedingung für einkristalle. J. Appl. Math. Mech. 1929, 9, 49–58. [Google Scholar] [CrossRef]
- Hill, R. The elastic behavior of crystalline aggregate. Proc. Phys. Soc. A 1952, 65, 349–354. [Google Scholar] [CrossRef]
- Guo, Y.; Wang, Y.; Chen, H.T.; Xu, H.Y.; Hu, M.L.; Ji, Z.S. Evaluation of stabilities and thermophysical properties of Si-Sr intermetallics based on first-principles analysis. Results Phys. 2020, 18, 103207. [Google Scholar] [CrossRef]
- Cao, L.; Zhang, X.D.; Wang, F. Structural, elastic, anisotropic, electronic, thermal properties and tensile strength of AlTM2Ti (TM = Ni, Fe, Cu, Co, Au) studied by first-principles calculations. Chem. Phys. Lett. 2023, 830, 140796. [Google Scholar] [CrossRef]
- Pan, Y.; Li, Y.; Zheng, Q. Influence of Ir concentration on the structure, elastic modulus and elastic anisotropy of Nb-Ir based compounds from first-principles calculations. J. Alloy Compd. 2019, 789, 860–866. [Google Scholar] [CrossRef]
- Pan, Y. RuAl2: Structure, electronic and elastic properties from first-principles. Mater. Res. Bull. 2017, 93, 56–62. [Google Scholar] [CrossRef]
- Pugh, S.F. XCII. Relations between the elastic moduli and the plastic properties of polycrystalline pure metals. Lond. Edinb. Dublin Philos. Mag. J. Sci. 1954, 45, 823–843. [Google Scholar] [CrossRef]
- Ravindran, P.; Fast, L.; Korzhavyi, P.A.; Johansson, B.; Wills, J.; Eriksson, O. Density functional theory for calculation of elastic properties of orthorhombic crystals: Applications to TiSi2. J. Appl. Phys. 1998, 84, 4891–4904. [Google Scholar] [CrossRef]
- Tian, Y.; Xu, B.; Zhao, Z. Microscopic theory of hardness and design of novel superhard crystals. Int. J. Refract. Met. Hard Mater. 2012, 33, 93–106. [Google Scholar] [CrossRef]
- Anderson, O.L. A simplified method for calculating the debye temperature from elastic constants. J. Phys. Chem. Solids 1963, 24, 909–917. [Google Scholar] [CrossRef]
- Schreiber, E.; Anderson, O.L.; Soga, N. Elastic Constants and Their Measurement; McGraw-Hill Companies: New York, NY, USA, 1974. [Google Scholar]
- Clarke, D.R. Materials selection guidelines for low thermal conductivity thermal barrier coatings. Surf. Coat. Technol. 2003, 163, 67–74. [Google Scholar] [CrossRef]
- Cahill, D.G.; Pohl, R.O. Lattice vibrations and heat transport in crystals and glasses. Ann. Rev. Phys. Chem. 1988, 39, 93–121. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | Method | Lattice Constants | V0 (Å3) | ΔH (eV/Atom) | Ref. |
|---|---|---|---|---|---|
| α (Å) | |||||
| AlCu2Ti | Cal. | 6.037 | 220.043 | −4.985 | Present |
| Cal. | 6.032 | 219.467 | −4.235 | [10] | |
| Exp. | 6.024 | 218.570 | - | [20] | |
| AlCu2Cr | Cal. | 5.869 | 202.160 | −5.283 | Present |
| Cal. | 5.891 | 204.429 | - | [11] | |
| Exp. | 5.809 | 196.020 | - | [21] | |
| AlCu2Zr | Cal. | 6.256 | 244.879 | −5.167 | Present |
| Cal. | 6.256 | 244.827 | −4.551 | [10] | |
| Exp. | 6.216 | 240.210 | - | [20] | |
| AlCu2Sc | Cal. | 6.223 | 241.031 | −4.546 | Present |
| Cal. | 6.243 | 243.327 | - | [11] | |
| Exp. | 6.199 | 238.210 | - | [22] | |
| AlCu2Hf | Cal. | 6.220 | 240.654 | −5.070 | Present |
| Cal. | 6.210 | 239.535 | −4.451 | [10] | |
| Exp. | 6.172 | 235.110 | - | [23] | |
| AlCu2Mn | Cal. | 5.850 | 200.283 | −5.139 | Present |
| Cal. | 5.817 | 196.844 | −3.419 | [10] | |
| Exp. | 5.968 | 212.560 | - | [24] | |
| AlCu2Pa | Cal. | 6.465 | 270.253 | −5.534 | Present |
| Cal. | 6.47 | 270.63 | - | [25] | |
| AlCu2Lu | Cal. | 6.373 | 258.859 | −4.720 | Present |
| Cal. | 6.29 | 249.33 | - | [25] | |
| AlCu2Pm | Cal. | 6.539 | 279.688 | −4.195 | Present |
| Cal. | 6.53 | 278.64 | - | [25] |
| Compounds | Elastic Constants Cij | Ref. | ||
|---|---|---|---|---|
| C11 | C12 | C44 | ||
| AlCu2Ti | 172.476 | 149.587 | 122.355 | Present |
| 163.314 | 120.827 | 96.918 | [10] | |
| 144.486 | 124.338 | 97.943 | [11] | |
| AlCu2Cr | 133.213 | 124.467 | 100.126 | Present |
| 157.5 | 115.3 | 62.7 | [30] | |
| AlCu2Zr | 167.383 | 133.152 | 70.846 | Present |
| 157.504 | 115.305 | 62.685 | [10] | |
| AlCu2Sc | 135.729 | 77.034 | 68.615 | Present |
| 155.252 | 78.727 | 75.987 | [10] | |
| AlCu2Hf | 166.777 | 152.559 | 72.617 | Present |
| 171.570 | 116.888 | 68.685 | [10] | |
| AlCu2Mn | 198.051 | 114.279 | 124.332 | Present |
| 225.069 | 121.565 | 107.411 | [10] | |
| AlCu2Pa | 157.043 | 75.478 | 22.908 | Present |
| AlCu2Lu | 126.094 | 64.315 | 42.077 | Present |
| AlCu2Pm | 117.970 | 57.364 | 3.993 | Present |
| Compounds | Elastic Modulus (GPa) | ν | B/G | HVT (GPa) | Ref. | ||
|---|---|---|---|---|---|---|---|
| B | G | E | |||||
| AlCu2Ti | 157.217 | 51.540 | 126.888 | 0.352 | 3.050 | 4.220 | Present |
| 134.989 | 54.481 | - | - | 2.478 | - | [10] | |
| 131.054 | 42.307 | 120.247 | 0.354 | 3.098 | - | [11] | |
| 129.22 | 37.38 | 102.278 | 0.368 | 3.457 | - | [35] | |
| AlCu2Cr | 127.383 | 36.042 | 90.967 | 0.397 | 4.501 | 2.771 | Present |
| 144.898 | 29.436 | 82.707 | 0.405 | 4.922 | - | [11] | |
| AlCu2Zr | 157.047 | 38.477 | 99.224 | 0.387 | 4.082 | 2.464 | Present |
| 129.371 | 41.237 | - | - | 3.137 | - | [10] | |
| AlCu2Sc | 142.302 | 57.078 | 135.106 | 0.324 | 2.493 | 5.705 | Present |
| 104.235 | 57.696 | 146.127 | 0.266 | 1.807 | - | [11] | |
| AlCu2Hf | 176.664 | 36.197 | 95.541 | 0.405 | 4.881 | 1.925 | Present |
| 135.115 | 47.971 | - | - | 2.817 | - | [10] | |
| AlCu2Mn | 142.203 | 80.458 | 175.265 | 0.262 | 1.767 | 10.757 | Present |
| 156.066 | 80.521 | - | - | 1.938 | - | [10] | |
| AlCu2Pa | 102.667 | 28.918 | 73.039 | 0.372 | 3.550 | 2.359 | Present |
| AlCu2Lu | 84.908 | 37.177 | 86.331 | 0.309 | 2.284 | 4.653 | Present |
| AlCu2Pm | 77.566 | 10.318 | 28.433 | 0.436 | 7.518 | 0.485 | Present |
| Compounds | ρ | V | ΘD | κmin | |||
|---|---|---|---|---|---|---|---|
| Vt | Vl | Vm | Clark | Cahill-Pohl | |||
| AlCu2Ti | 6.619 | 2790.550 | 5842.669 | 3768.708 | 481.329 | 0.968 | 0.227 |
| AlCu2Cr | 6.678 | 2323.171 | 5125.528 | 3165.179 | 402.728 | 0.810 | 0.189 |
| AlCu2Zr | 7.175 | 2315.789 | 5388.834 | 3179.895 | 391.013 | 0.762 | 0.175 |
| AlCu2Sc | 6.012 | 3081.312 | 6027.450 | 4106.714 | 510.415 | 0.992 | 0.238 |
| AlCu2Hf | 9.866 | 1915.424 | 4774.732 | 2652.930 | 327.756 | 0.644 | 0.146 |
| AlCu2Mn | 6.932 | 3406.952 | 5999.286 | 4428.178 | 567.786 | 1.120 | 0.279 |
| AlCu2Pa | 9.464 | 1748.006 | 3862.900 | 2382.164 | 276.413 | 0.507 | 0.117 |
| AlCu2Lu | 8.443 | 2098.392 | 3990.930 | 2779.609 | 327.185 | 0.600 | 0.145 |
| AlCu2Pm | 7.150 | 1201.278 | 3573.853 | 1690.776 | 193.957 | 0.356 | 0.079 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, Y.; Jiang, B.; Zhang, X.; Li, S. First-Principles Study of the Structural, Mechanical, Electronic, and Thermodynamic Properties of AlCu2M (M = Ti, Cr, Zr, Sc, Hf, Mn, Pa, Lu, Pm) Ternary Intermetallic Compounds. Materials 2024, 17, 3441. https://doi.org/10.3390/ma17143441
Guo Y, Jiang B, Zhang X, Li S. First-Principles Study of the Structural, Mechanical, Electronic, and Thermodynamic Properties of AlCu2M (M = Ti, Cr, Zr, Sc, Hf, Mn, Pa, Lu, Pm) Ternary Intermetallic Compounds. Materials. 2024; 17(14):3441. https://doi.org/10.3390/ma17143441
Chicago/Turabian StyleGuo, Yu, Bo Jiang, Xun Zhang, and Shikang Li. 2024. "First-Principles Study of the Structural, Mechanical, Electronic, and Thermodynamic Properties of AlCu2M (M = Ti, Cr, Zr, Sc, Hf, Mn, Pa, Lu, Pm) Ternary Intermetallic Compounds" Materials 17, no. 14: 3441. https://doi.org/10.3390/ma17143441
APA StyleGuo, Y., Jiang, B., Zhang, X., & Li, S. (2024). First-Principles Study of the Structural, Mechanical, Electronic, and Thermodynamic Properties of AlCu2M (M = Ti, Cr, Zr, Sc, Hf, Mn, Pa, Lu, Pm) Ternary Intermetallic Compounds. Materials, 17(14), 3441. https://doi.org/10.3390/ma17143441