
Theoretical Investigation of a Novel Two-Dimensional Non-MXene Mo3C2 as a Prospective Anode Material for Li- and Na-Ion Batteries
Abstract
:1. Introduction
2. Computational Details
3. Results and Discussion
3.1. Structure and Stability
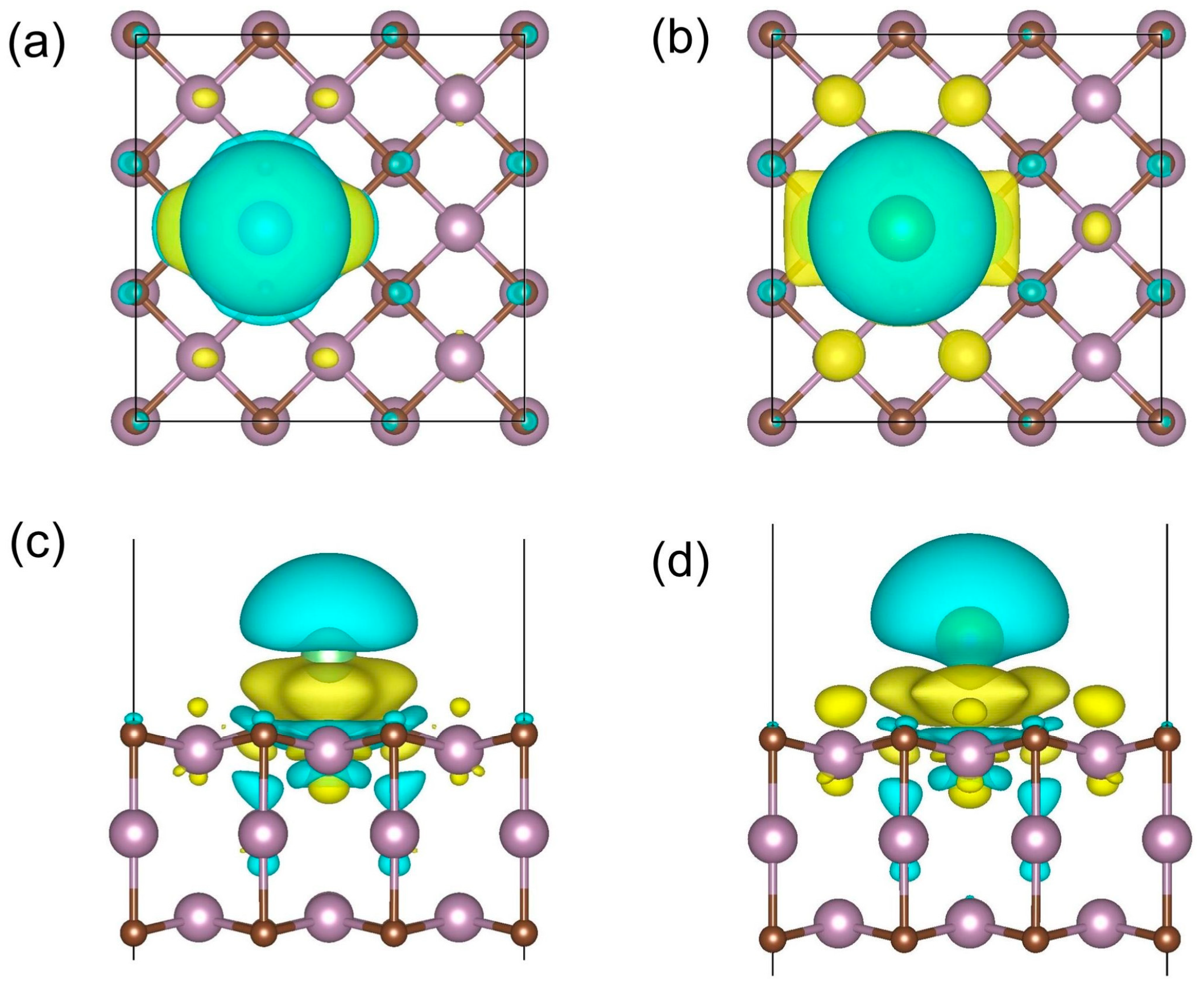
3.2. Lithiation and Sodiation Processes: Adsorption and Electronic Property
3.3. Li/Na Surface Diffusion
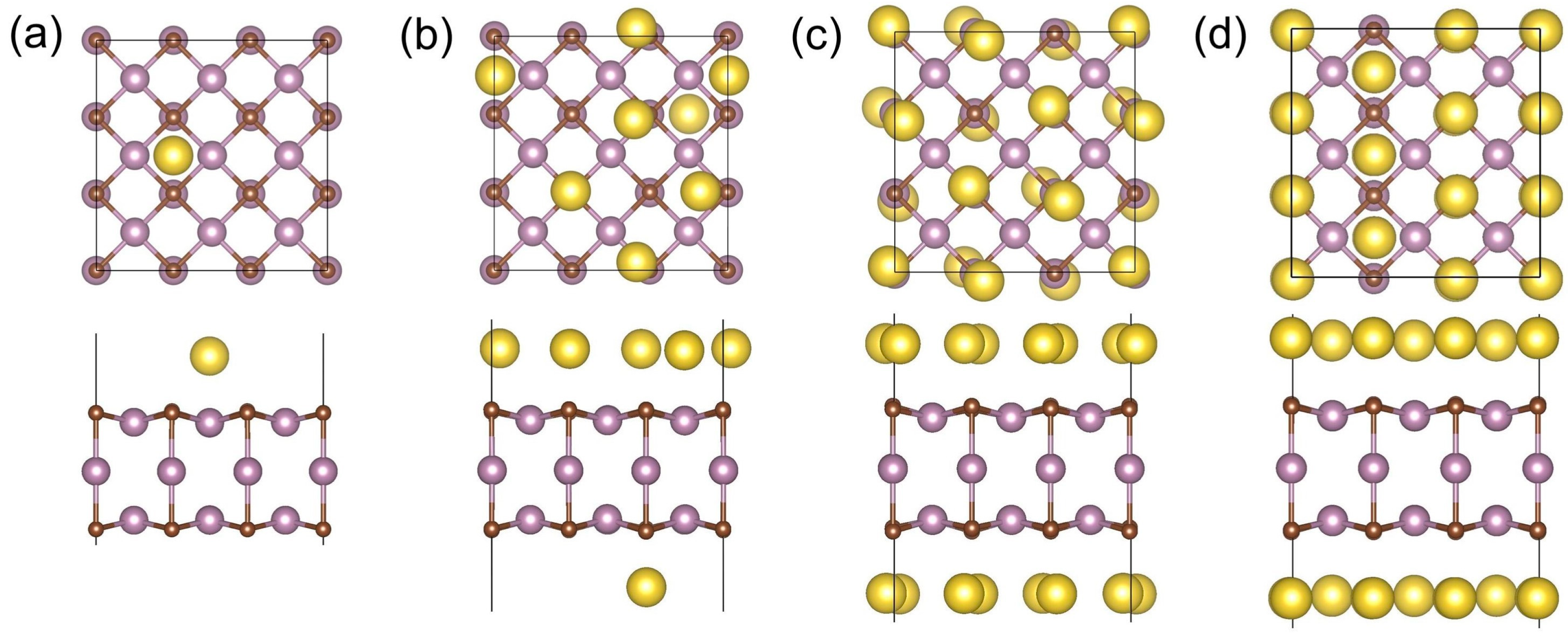
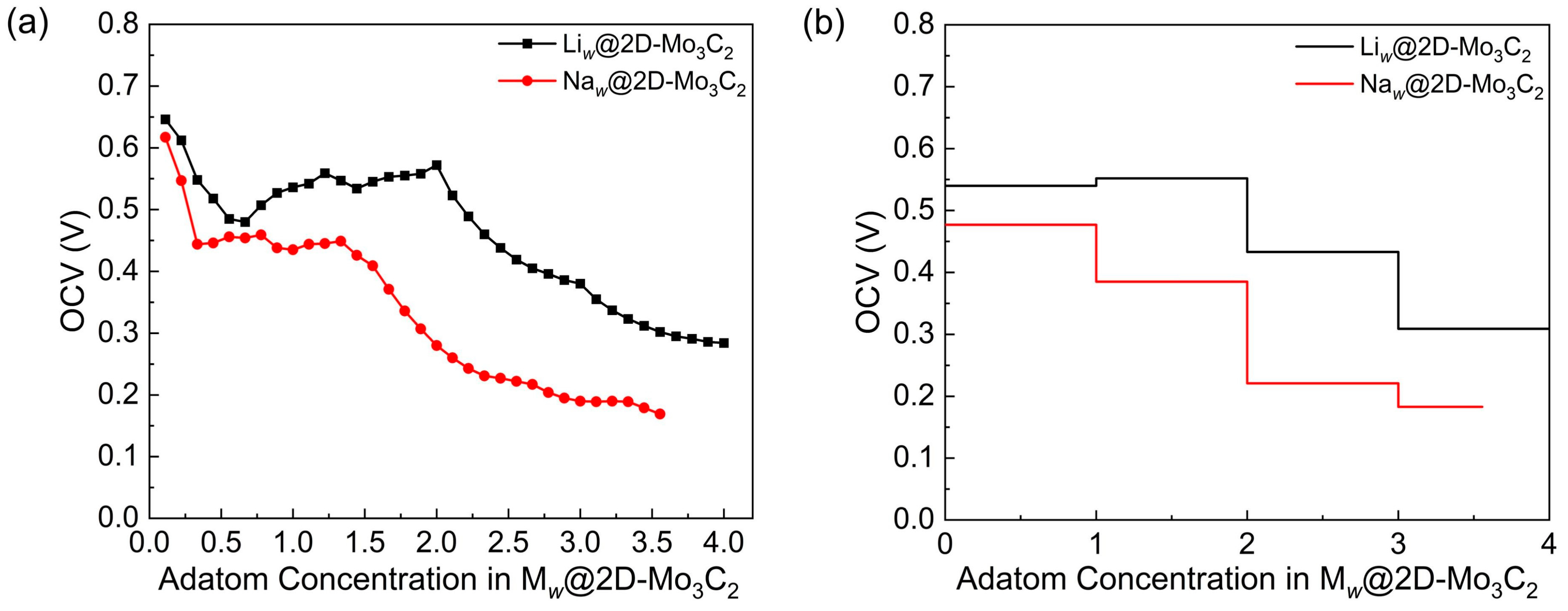
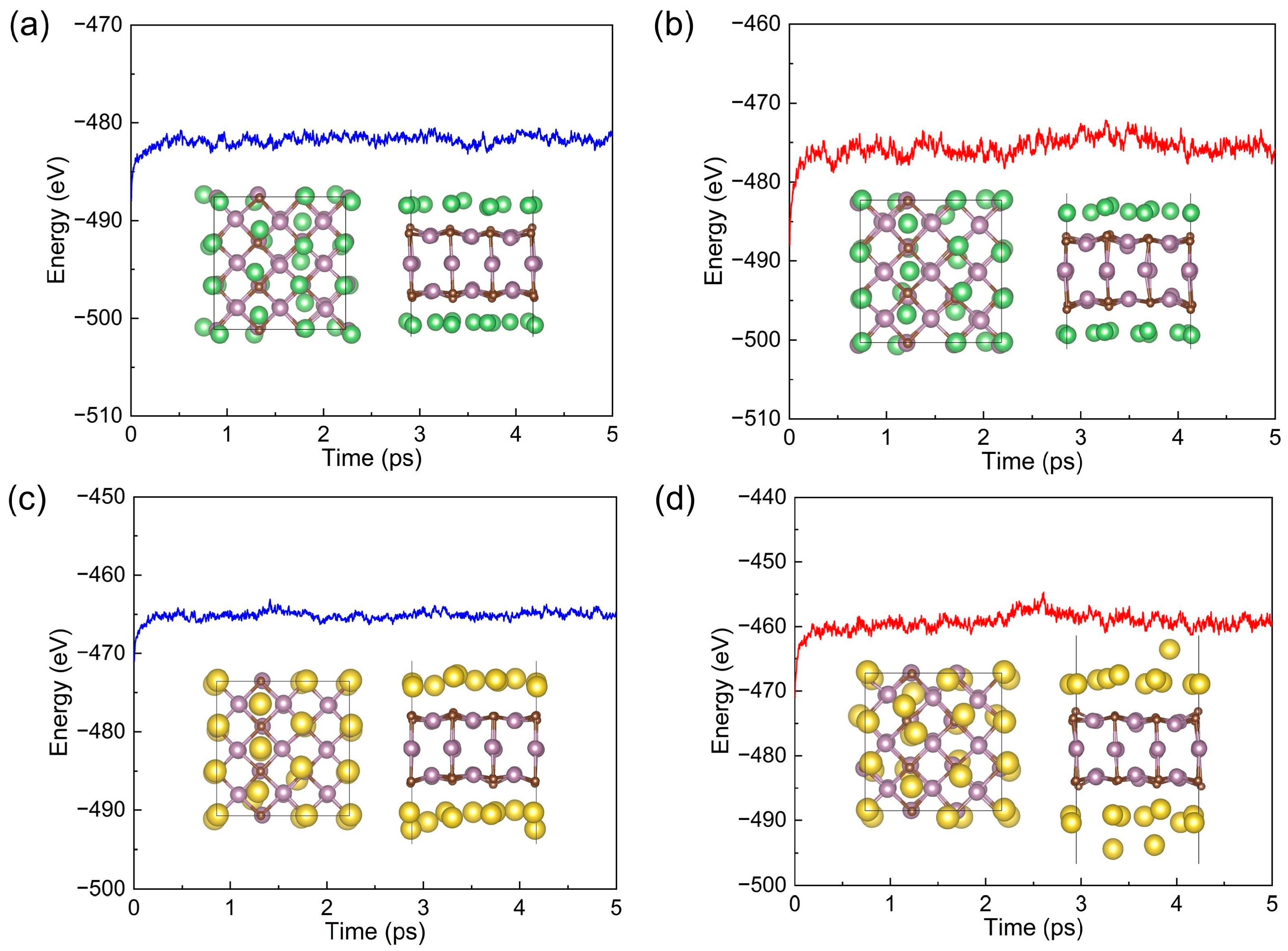
3.4. Theoretical Storage Capacity, Open-Circuit Voltage, and Thermal Stability
4. Summary
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wang, C.; Guan, S.; Zhang, H.; Shen, R.; Yuan, H.; Li, B. Perspectives on two-dimensional ultra-thin materials in energy catalysis and storage. APL Mater. 2023, 11, 050902. [Google Scholar] [CrossRef]
- Baig, N. Two-dimensional nanomaterials: A critical review of recent progress, properties, applications, and future directions. Compos. Part A-Appl. S. 2023, 165, 107362. [Google Scholar] [CrossRef]
- Hu, Z.; Liu, Q.; Chou, S.L.; Dou, S.X. Two-dimensional material-based heterostructures for rechargeable batteries. Cell Rep. Phys. Sci. 2021, 2, 100286. [Google Scholar] [CrossRef]
- Tarascon, J.M. Na-ion versus Li-ion batteries: Complementarity rather than competitiveness. Joule 2020, 4, 1616–1620. [Google Scholar] [CrossRef]
- Abraham, K.M. How comparable are sodium-ion batteries to lithium-ion counterparts? ACS Energy Lett. 2020, 5, 3544–3547. [Google Scholar] [CrossRef]
- Zhao, D.; Jiang, S.; Yu, S.; Ren, J.; Zhang, Z.; Liu, S.; Liu, X.; Wang, Z.; Wu, Y.; Zhang, Y. Lychee seed-derived microporous carbon for high-performance sodium-sulfur batteries. Carbon 2023, 201, 864–870. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, Z.; Yu, S.; Johnson, H.M.; Zhao, D.C.; Tan, S.C.; Pan, Z.D.; Wang, Z.L.; Wu, Y.T.; Liu, X. Three-dimensional nanostructured Co2VO4-decorated carbon nanotubes for sodium-ion battery anode materials. Rare Met. 2023, 42, 4060–4069. [Google Scholar] [CrossRef]
- Fan, K.; Huang, H. Two-dimensional host materials for lithium-sulfur batteries: A review and perspective. Energy Storage Mater. 2022, 50, 696–717. [Google Scholar] [CrossRef]
- Forouzandeh, P.; Pillai, S.C. Two-dimensional (2D) electrode materials for supercapacitors. Mater. Today Proc. 2021, 41, 498–505. [Google Scholar] [CrossRef]
- Naguib, M.; Mochalin, V.N.; Barsoum, M.W.; Gogotsi, Y. 25th anniversary article: MXenes: A new family of two-dimensional materials. Adv. Mater. 2014, 19, 992–1005. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, Z.; Zhou, Z. MXene-based materials for electrochemical energy storage. J. Energy Chem. 2018, 27, 73–85. [Google Scholar] [CrossRef]
- Shetti, N.P.; Mishra, A.; Basu, S.; Aminabhavi, T.M.; Alodhayb, A.; Pandiaraj, S. MXenes as Li-ion battery electrodes: Progress and outlook. Energy Fuels 2023, 37, 12541–12557. [Google Scholar] [CrossRef]
- Wu, Y.; Sun, Y.; Zheng, J.; Rong, J.; Li, H.; Niu, L. MXenes: Advanced materials in potassium ion batteries. Chem. Eng. J. 2021, 404, 126565. [Google Scholar] [CrossRef]
- Xiao, M.; Li, M.; Song, E.; Song, H.; Li, Z.; Bi, J. Halogenated Ti3C2 MXene as high capacity electrode material for Li-ion batteries. J. Inorg. Mater. 2022, 37, 660–668. [Google Scholar] [CrossRef]
- Li, Z.; Xiao, X.; Zhang, D.; Niu, H.; Shu, Q.; Yan, C.; Wang, X.; Guan, P.; Hu, X. Electrostatics and chemistry combination divalent cobalt ions and alkali-treated MXene for high-performance lithium-ion batteries. Energy Fuels 2022, 36, 13266–13277. [Google Scholar] [CrossRef]
- Li, J.; Liu, H.; Shi, X.; Li, X.; Li, W.; Guan, E.; Lu, T.; Pan, L. MXene-based anode materials for high performance sodium-ion batteries. J. Colloid Interf. Sci. 2024, 658, 425–440. [Google Scholar] [CrossRef]
- Wu, Y.; Nie, P.; Wang, J.; Dou, H.; Zhang, X. Few-layer MXenes delaminated via high-energy mechanical milling for enhanced sodium-ion batteries performance. ACS Appl. Mater. Interfaces 2017, 9, 39610–39617. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Wang, M.; Lyu, M.; Jiao, Y.; Du, A.; Luo, B.; Gentle, I.; Wang, L. Two-dimensional titanium carbonitride Mxene for high-performance sodium ion batteries. ACS Appl. Nano Mater. 2018, 1, 6854–6863. [Google Scholar] [CrossRef]
- Fan, K.; Ying, Y.; Li, X.; Luo, X.; Huang, H. Theoretical investigation of V3C2 MXene as prospective high-capacity anode material for metal-ion (Li, Na, K, and Ca) batteries. J. Phys. Chem. C 2019, 123, 18207–18214. [Google Scholar] [CrossRef]
- Xu, J.; Wang, D.; Lian, R.; Gao, X.; Liu, Y.; Yury, G.; Chen, G.; Wei, Y. Structural prediction and multilayer Li+ storage in two-dimensional VC2 carbide studied by first-principles calculations. J. Mater. Chem. A 2019, 7, 8873–8881. [Google Scholar] [CrossRef]
- Yu, T.; Zhang, S.; Li, F.; Zhao, Z.; Liu, L.; Xu, H.; Yang, G. Stable and metallic two-dimensional TaC2 as an anode material for lithium-ion battery. J. Mater. Chem. A 2017, 5, 18698–18706. [Google Scholar] [CrossRef]
- Wu, Y.Y.; Bo, T.; Zhang, J.; Lu, Z.; Wang, Z.; Li, Y.; Wang, B.T. Novel two-dimensional tetragonal vanadium carbides and nitrides as promising materials for Li-ion batteries. Phys. Chem. Chem. Phys. 2019, 21, 19513–19520. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Chen, X.; Ong, W.J.; MacFarlane, D.R.; Zhao, X.; Cheetham, A.K.; Sun, C. Understanding of electrochemical mechanisms for CO2 capture and conversion into hydrocarbon fuels in transition-metal carbides (MXenes). ACS Nano 2017, 11, 10825–10833. [Google Scholar] [CrossRef]
- Oganov, A.R.; Glass, C.W. Crystal structure prediction using ab initio evolutionary techniques: Principles and applications. J. Chem. Phys. 2006, 124, 244704. [Google Scholar] [CrossRef]
- Oganov, A.R.; Ma, Y.; Lyakhov, A.O.; Valle, M.; Gatti, C. Evolutionary crystal structure prediction as a method for the discovery of minerals and materials. Rev. Miner. Geochem. 2010, 71, 271–298. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Ernzerhof, M.; Burke, K. Rationale for mixing exact exchange with density functional approximations. J. Chem. Phys. 1996, 105, 9982–9985. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Tang, W.; Sanville, E.; Henkelman, G. A grid-based Bader analysis algorithm without lattice bias. J. Phys. Condens. Matter. 2009, 21, 084204. [Google Scholar] [CrossRef] [PubMed]
- Henkelman, G.; Jónsson, H. Improved tangent estimate in the nudged elastic band method for finding minimum energy paths and saddle points. J. Chem. Phys. 2000, 113, 9978–9985. [Google Scholar] [CrossRef]
- Togo, A.; Tanaka, I. First principles phonon calculations in materials science. Scr. Mater. 2015, 108, 1–5. [Google Scholar] [CrossRef]
- Andersen, H.C. Molecular dynamics simulations at constant pressure and/or temperature. J. Chem. Phys. 1980, 72, 2384–2393. [Google Scholar] [CrossRef]
- Zhang, F.; Jing, T.; Cai, S.; Deng, M.; Liang, D.; Qi, X. Two-dimensional ZrC2 as a novel anode material with high capacity for sodium ion battery. Phys. Chem. Chem. Phys. 2021, 23, 12731–12738. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.; Phantsi, T.D. Bond theory model to study cohesive energy, thermal expansion coefficient and specific heat of nanosolids. Chin. J. Phys. 2018, 56, 2948–2957. [Google Scholar] [CrossRef]
- Mardiyah, R.U.; Arkundato, A.; Misto; Purwandari, E. Energy cohesive calculation for some pure metals using the Lennard-Jones potential in Lammps Molecular Dynamics. J. Phys. Conf. Ser. 2020, 1491, 012020. [Google Scholar] [CrossRef]
- Andrew, R.C.; Mapasha, R.E.; Ukpong, A.M.; Chetty, N. Mechanical properties of graphene and boronitrene. Phys. Rev. B 2012, 85, 125428. [Google Scholar] [CrossRef]
- Mouhat, F.; Coudert, F.X. Necessary and sufficient elastic stability conditions in various crystal systems. Phys. Rev. B 2014, 90, 224104. [Google Scholar] [CrossRef]
- Fan, X.; Zheng, W.T.; Kuo, J.L. Adsorption and diffusion of Li on pristine and defective graphene. ACS Appl. Mater. Interfaces 2012, 4, 2432–2438. [Google Scholar] [CrossRef]
- Singh, T.; Choudhuri, J.R.; Rana, M.K. α-graphyne as a promising anode material for Na-ion batteries: A first-principles study. Nanotechnology 2023, 34, 045404. [Google Scholar] [CrossRef] [PubMed]
- Setiadi, J.; Arnold, M.D.; Ford, M.J. Li-Ion adsorption and diffusion on two-dimensional silicon with defects: A first principles study. ACS Appl. Mater. Interfaces 2013, 5, 10690–10695. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.X.; Guo, P.; Zheng, J.M.; Cao, L.K.; Zhao, P.J. First-principles calculations study of Na adsorbed on silicene. Appl. Surf. Sci. 2015, 341, 69–74. [Google Scholar] [CrossRef]
- Zhao, L.; Ding, B.; Qin, X.Y.; Wang, Z.; Lv, W.; He, Y.B.; Yang, Q.H.; Kang, F. Revisiting the roles of natural graphite in ongoing lithium-ion batteries. Adv. Mater. 2022, 34, 2106704. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yu, Z.; Wang, S.S.; Guan, S.; Yang, H.Y.; Yao, Y.; Yang, S.A. Theoretical prediction of MoN2 monolayer as a high capacity electrode material for metal ion batteries. J. Mater. Chem. A 2016, 4, 15224–15231. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| C11 | C22 | C12 | C66 | |||
|---|---|---|---|---|---|---|
| Our predicted Mo3C2 | 432.3 | 432.3 | 168.4 | 195.6 | 366.7 | 366.7 |
| Mo3C2 MXene | 293.3 | 293.3 | 59.7 | 116.8 | 281.1 | 281.1 |
| Graphene | 352.7 | 352.7 | 60.9 | 145.9 | 342.2 | 342.2 |
| BN | 289.8 | 289.8 | 63.7 | 113.1 | 275.8 | 275.8 |
| SiC | 179.7 | 179.7 | 53.9 | 62.9 | 163.5 | 163.5 |
| Ead (eV) | h (Å) | |||||
|---|---|---|---|---|---|---|
| C1 | H | Mo1 | C1 | H | Mo1 | |
| Li@(3×3)-Mo3C2 | −0.467 | −0.646 | −0.148 | 1.96 | 1.66 | 2.47 |
| Na@(3×3)-Mo3C2 | −0.503 | −0.617 | −0.453 | 2.35 | 2.14 | 2.76 |
| Average Charge | ||||
|---|---|---|---|---|
| Mo | C | Li | Na | |
| Mo3C2 | +0.7 | −1.1 | ||
| Li@(3×3)-Mo3C2 for C1 | +0.7 | −1.1 | +0.9 | |
| Li@(3×3)-Mo3C2 for H | +0.7 | −1.2 | +0.9 | |
| Li@(3×3)-Mo3C2 for Mo1 | +0.7 | −1.1 | +0.9 | |
| Na@(3×3)-Mo3C2 for C1 | +0.7 | −1.1 | +0.8 | |
| Na@(3×3)-Mo3C2 for H | +0.7 | −1.1 | +0.8 | |
| Na@(3×3)-Mo3C2 for Mo1 | +0.7 | −1.1 | +0.8 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xue, B.; Zeng, Q.; Yu, S.; Su, K. Theoretical Investigation of a Novel Two-Dimensional Non-MXene Mo3C2 as a Prospective Anode Material for Li- and Na-Ion Batteries. Materials 2024, 17, 3819. https://doi.org/10.3390/ma17153819
Xue B, Zeng Q, Yu S, Su K. Theoretical Investigation of a Novel Two-Dimensional Non-MXene Mo3C2 as a Prospective Anode Material for Li- and Na-Ion Batteries. Materials. 2024; 17(15):3819. https://doi.org/10.3390/ma17153819
Chicago/Turabian StyleXue, Bo, Qingfeng Zeng, Shuyin Yu, and Kehe Su. 2024. "Theoretical Investigation of a Novel Two-Dimensional Non-MXene Mo3C2 as a Prospective Anode Material for Li- and Na-Ion Batteries" Materials 17, no. 15: 3819. https://doi.org/10.3390/ma17153819