
Waste for Product—Synthesis and Electrocatalytic Properties of Palladium Nanopyramid Layer Enriched with PtNPs
 , , , , , and
, , , , , and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Method of Analysis
3. Results
3.1. Process of Metal Recovery from Synthetic Waste Solution and Catalyst Synthesis
3.1.1. Pd(II) and Pt(IV) Adsorption on Active Carbon Pellets (ROY 0.8)—Kinetic Study
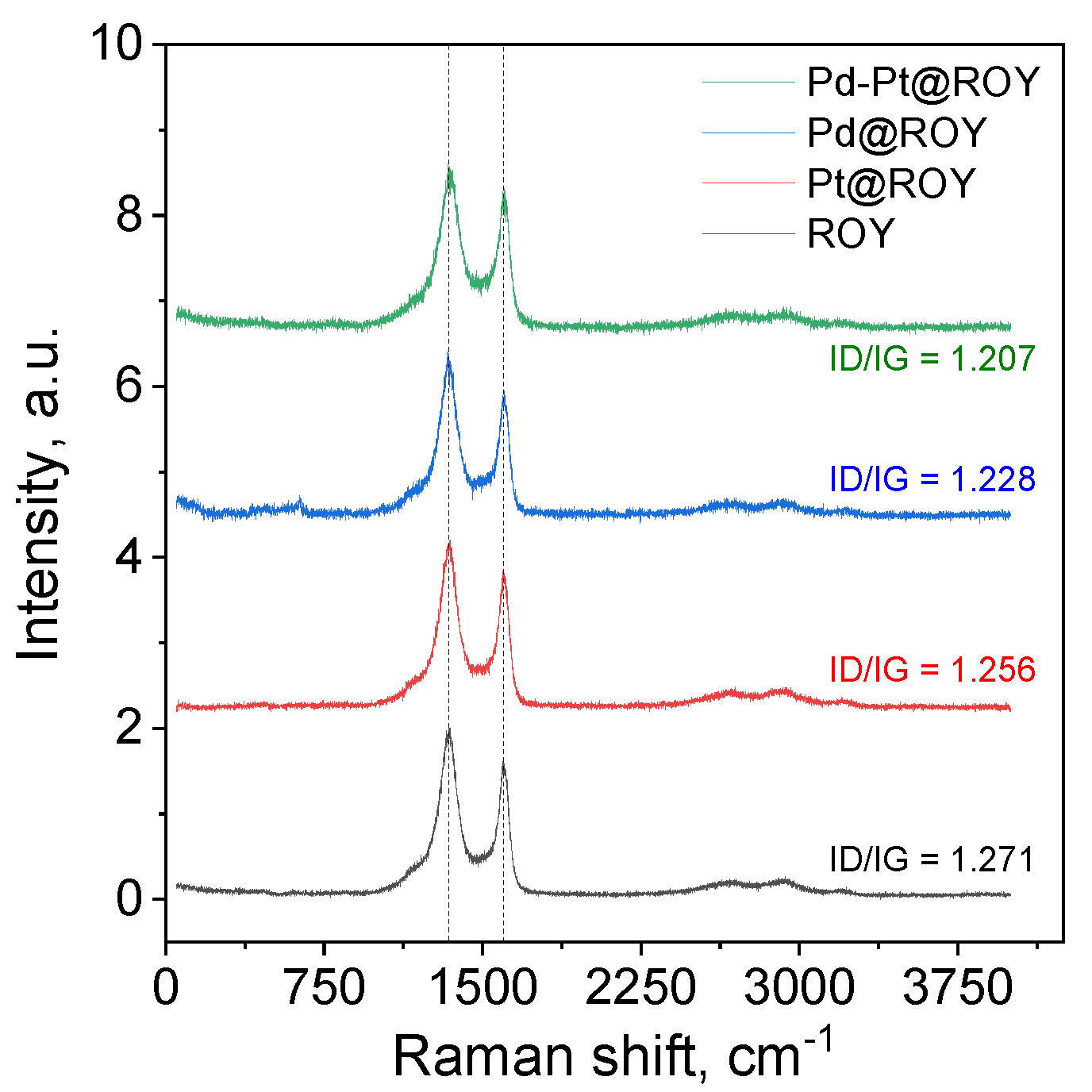
3.1.2. Raman Analysis of ROY, Pd@ROY, Pt@ROY, and Pd-Pt@ROY
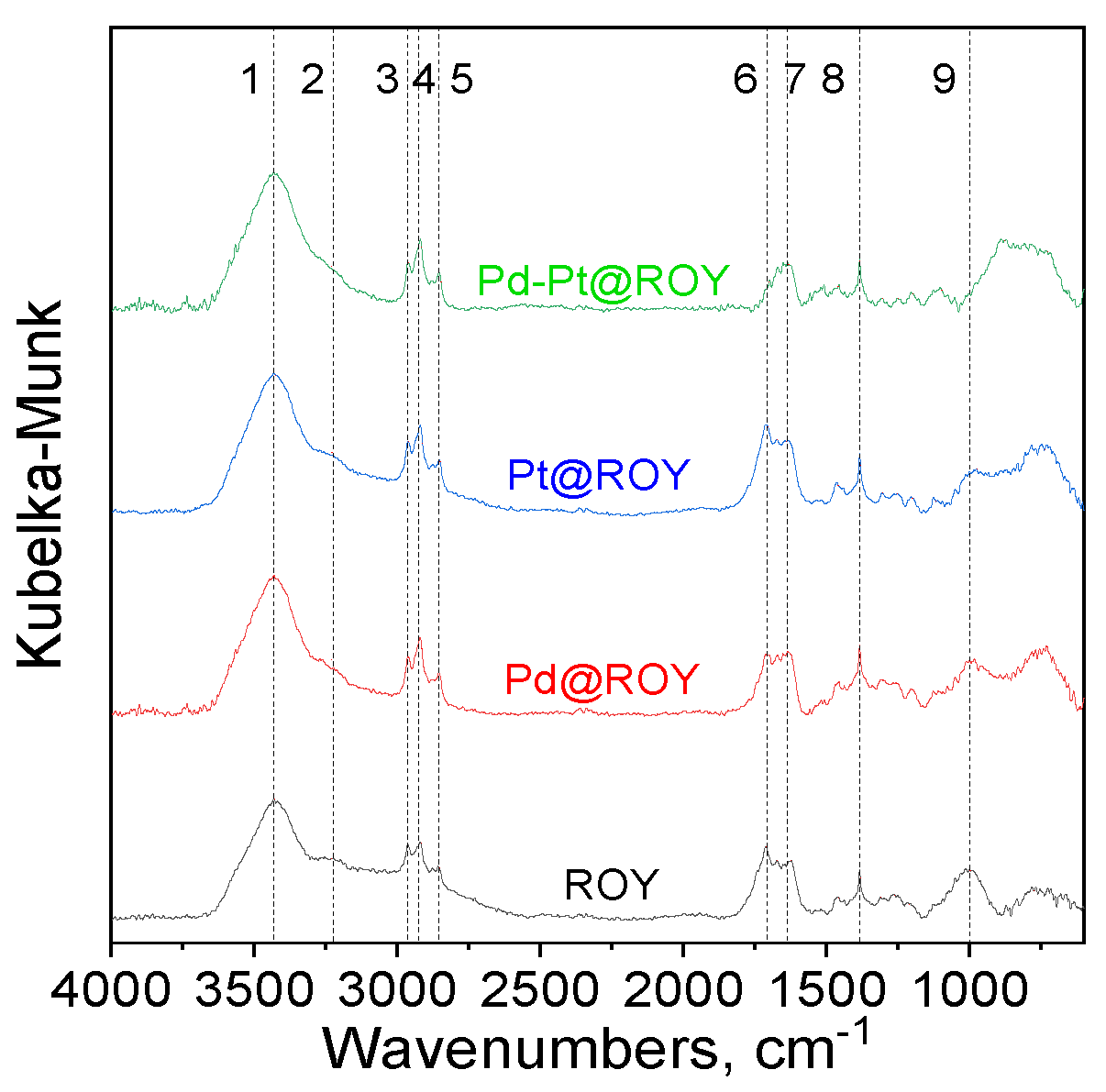
3.1.3. IR Analysis
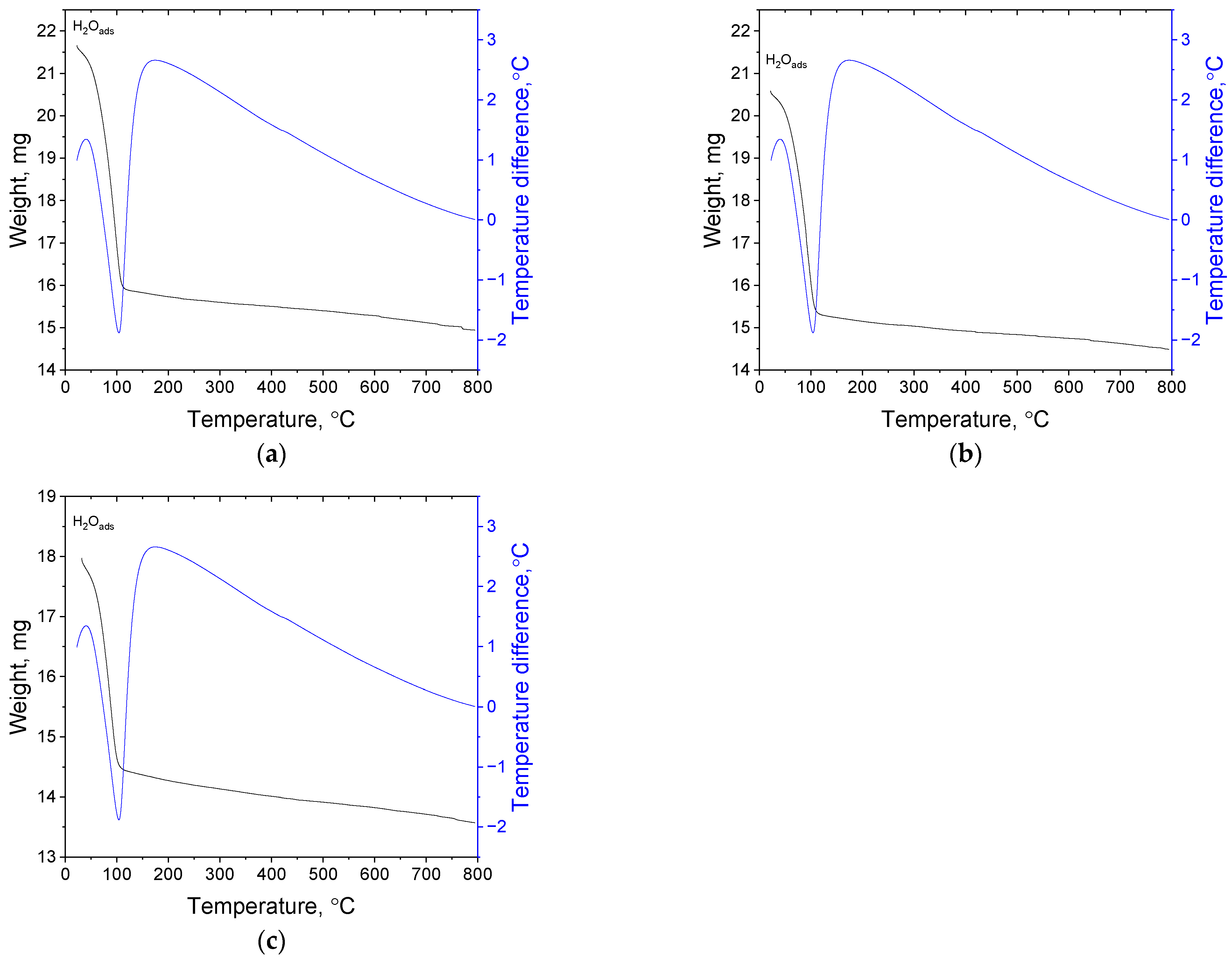
3.1.4. Thermal Gravimetric Analysis of the Obtained Catalyst
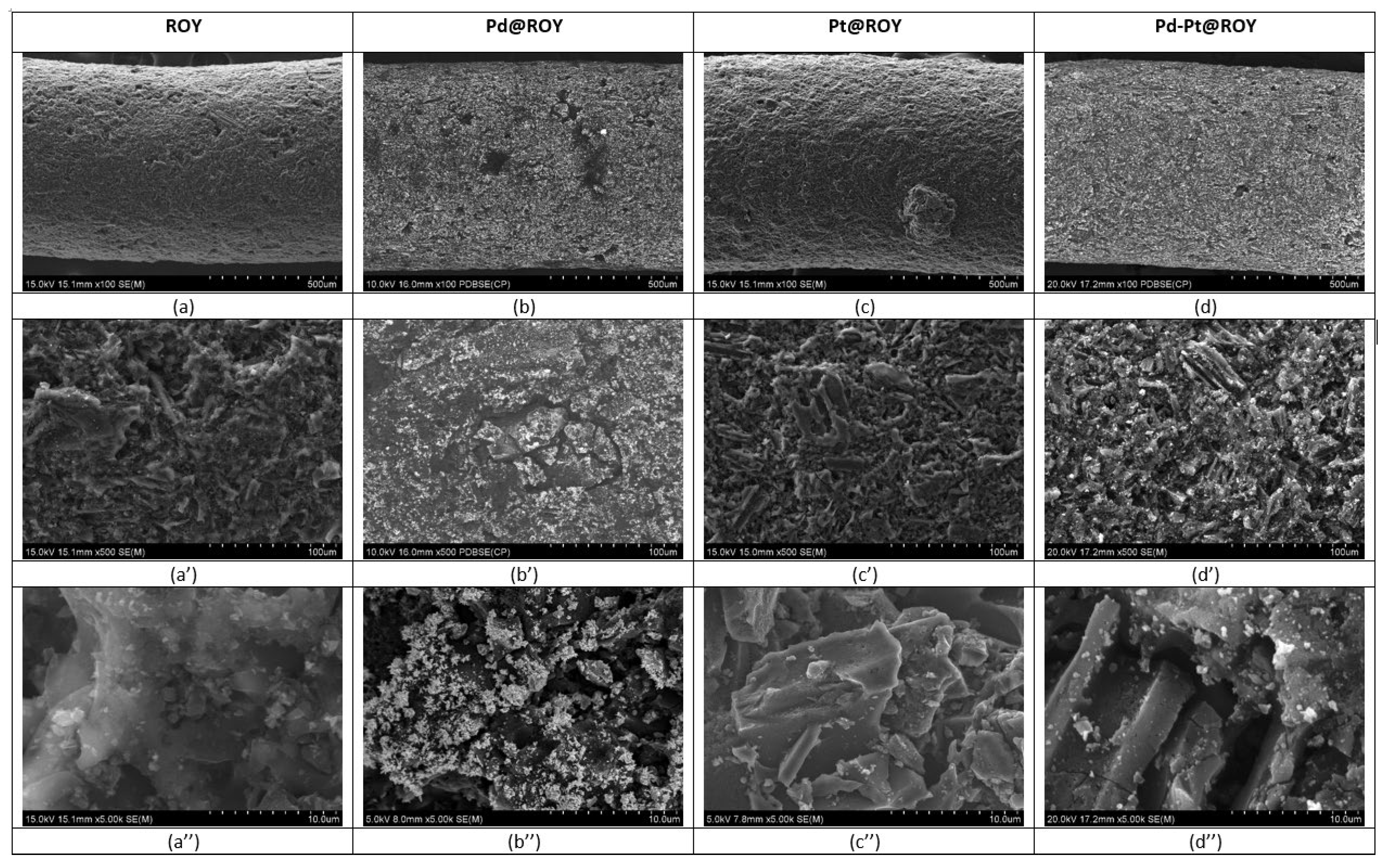
3.1.5. SEM/EDS Analysis
3.1.6. Wettability Tests
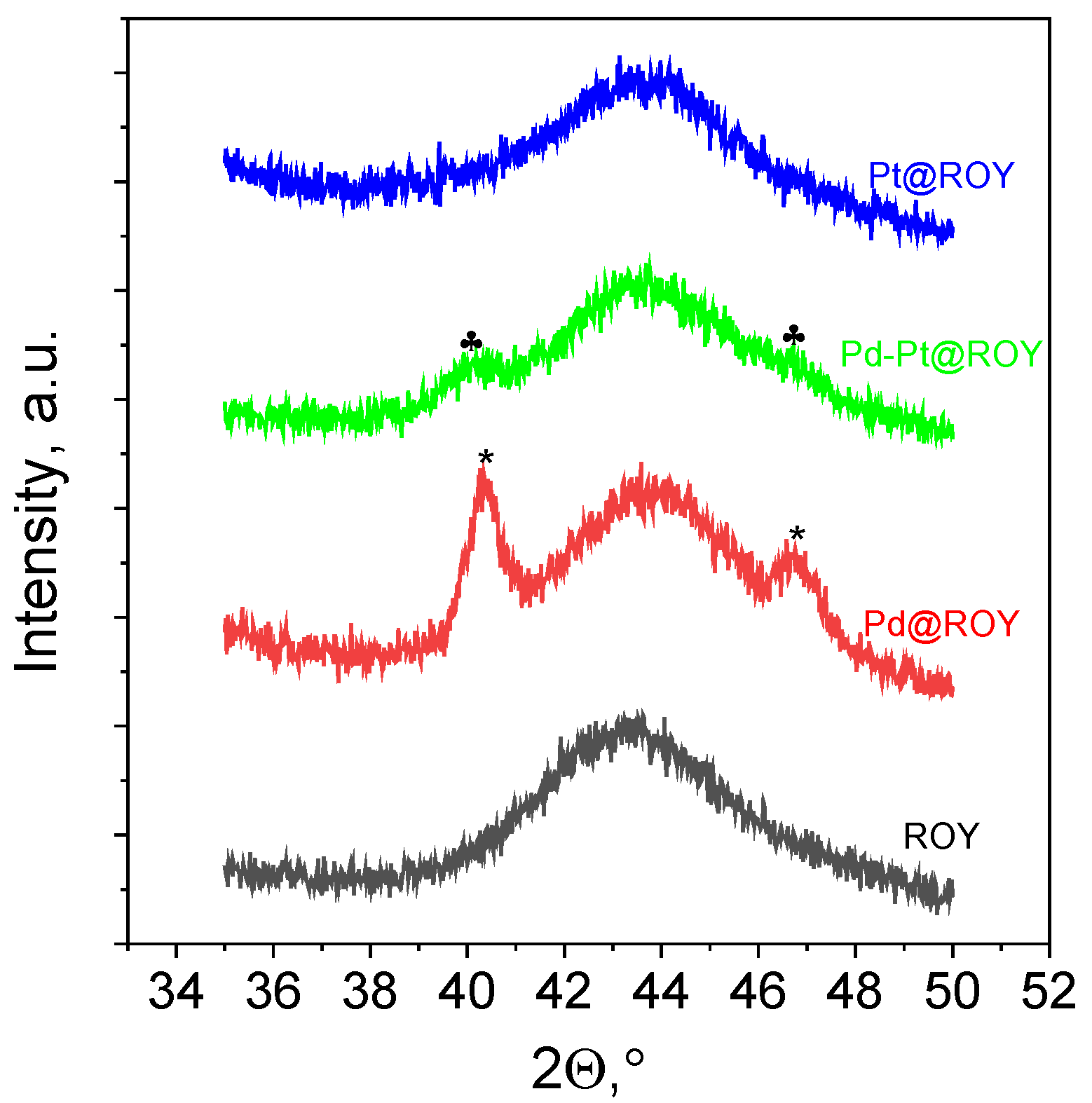
3.1.7. XRD Analysis
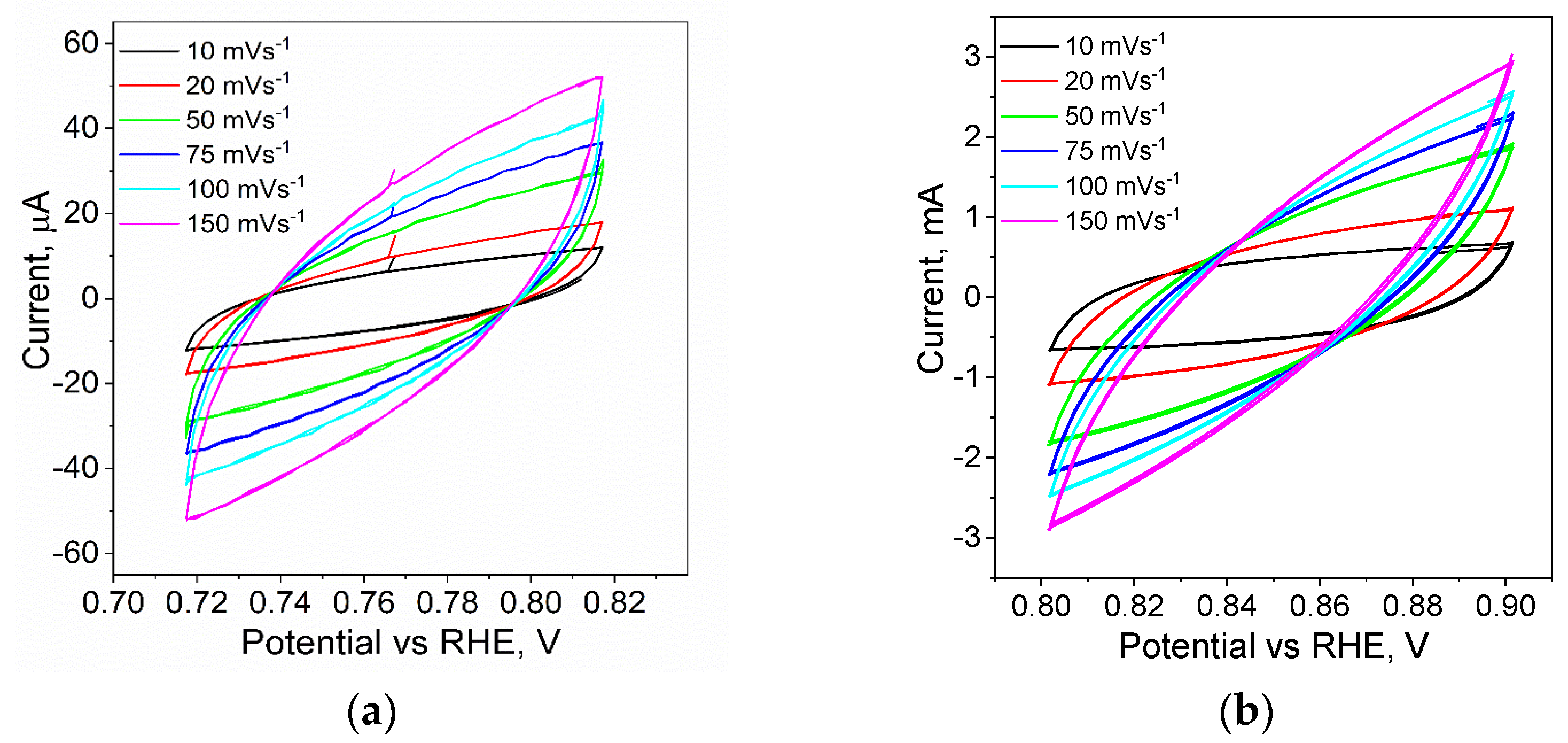
4. Electrocatalytic Properties of Obtained Materials for the Hydrogen Evolution Reaction
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Briffa, J.; Sinagra, E.; Blundell, R. Heavy metal pollution in the environment and their toxicological effects on humans. Heliyon 2020, 6, e04691. [Google Scholar] [CrossRef] [PubMed]
- Myriam El, A.-H.; Fayçal, H. Heavy Metals in the Environment and Health Impact. In Environmental Health; Takemi, O., Ed.; IntechOpen: Rijeka, Croatia, 2021; Chapter 4. [Google Scholar]
- Tchounwou, P.B.; Yedjou, C.G.; Patlolla, A.K.; Sutton, D.J. Heavy metal toxicity and the environment. In Molecular, Clinical and Environmental Toxicology; Springer: Basel, Switzerland, 2012. [Google Scholar]
- Pach, A.; Zaryczny, A.; Michałek, T.; Kamiński, H.; Kutyła, D.; Tokarski, T.; Chat-Wilk, K.; Hessel, V.; Luty-Błocho, M. One-Step Synthesis of Pt–Pd@ACF Catalyst in the Microreactor System for the Hydrogen Evolution Reaction. Ind. Eng. Chem. Res. 2024, 63, 7018–7030. [Google Scholar] [CrossRef]
- Xiang, H.; Min, X.; Tang, C.-J.; Sillanpää, M.; Zhao, F. Recent advances in membrane filtration for heavy metal removal from wastewater: A mini review. J. Water Process Eng. 2022, 49, 103023. [Google Scholar] [CrossRef]
- Silva, J.E.; Paiva, A.P.; Soares, D.; Labrincha, A.; Castro, F. Solvent extraction applied to the recovery of heavy metals from galvanic sludge. J. Hazard. Mater. 2005, 120, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Gulliani, S.; Volpe, M.; Messineo, A.; Volpe, R. Recovery of metals and valuable chemicals from waste electric and electronic materials: A critical review of existing technologies. RSC Sustain. 2023, 1, 1085–1108. [Google Scholar] [CrossRef]
- Qasem, N.A.A.; Mohammed, R.H.; Lawal, D.U. Removal of heavy metal ions from wastewater: A comprehensive and critical review. NPJ Clean Water 2021, 4, 36. [Google Scholar] [CrossRef]
- Fei, Y.; Hu, Y.H. Design, synthesis, and performance of adsorbents for heavy metal removal from wastewater: A review. J. Mater. Chem. A 2022, 10, 1047–1085. [Google Scholar] [CrossRef]
- Mariana, M.; H.P.S, A.K.; Mistar, E.M.; Yahya, E.B.; Alfatah, T.; Danish, M.; Amayreh, M. Recent advances in activated carbon modification techniques for enhanced heavy metal adsorption. J. Water Process Eng. 2021, 43, 102221. [Google Scholar] [CrossRef]
- George, R.; Bahadur, N.; Singh, N.; Singh, R.; Verma, A.; Shukla, A.K. Environmentally Benign TiO2 Nanomaterials for Removal of Heavy Metal Ions with Interfering Ions Present in Tap Water. Mater. Today Proc. 2016, 3, 162–166. [Google Scholar] [CrossRef]
- Yang, J.; Liu, S.; Xu, X.; Pan, B.; Long, Q.; Cheng, L.; Deng, J.; Yao, Q.; Lu, Z.; Wang, Z.; et al. Mechanism of Pd(II) adsorption by nanoscale titanium dioxide loaded bamboo shoot shell biomass. Environ. Sci. Pollut. Res. Int. 2023, 30, 113394–113408. [Google Scholar] [CrossRef]
- Chen, B.; Li, L.; Liu, L.; Cao, J. Effective adsorption of heavy metal ions in water by sulfhydryl modified nano titanium dioxide. Front. Chem. 2023, 10, 1072139. [Google Scholar] [CrossRef]
- Zhao, X.; Jia, Q.; Song, N.; Zhou, W.; Li, Y. Adsorption of Pb(II) from an Aqueous Solution by Titanium Dioxide/Carbon Nanotube Nanocomposites: Kinetics, Thermodynamics, and Isotherms. J. Chem. Eng. Data 2010, 55, 4428–4433. [Google Scholar] [CrossRef]
- Akpomie, K.G.; Conradie, J.; Adegoke, K.A.; Oyedotun, K.O.; Ighalo, J.O.; Amaku, J.F.; Olisah, C.; Adeola, A.O.; Iwuozor, K.O. Adsorption mechanism and modeling of radionuclides and heavy metals onto ZnO nanoparticles: A review. Appl. Water Sci. 2022, 13, 20. [Google Scholar] [CrossRef]
- Le, A.T.; Pung, S.-Y.; Sreekantan, S.; Matsuda, A.; Huynh, D.P. Mechanisms of removal of heavy metal ions by ZnO particles. Heliyon 2019, 5, e01440. [Google Scholar] [CrossRef] [PubMed]
- Saad, A.H.A.; Azzam, A.M.; El-Wakeel, S.T.; Mostafa, B.B.; Abd El-latif, M.B. Removal of toxic metal ions from wastewater using ZnO@Chitosan core-shell nanocomposite. Environ. Nanotechnol. Monit. Manag. 2018, 9, 67–75. [Google Scholar] [CrossRef]
- Alanazi, A.G.; Habila, M.A.; Alothman, Z.A.; Badjah-Hadj-Ahmed, A.-Y. Synthesis and Characterization of Zinc Oxide Nanoparticle Anchored Carbon as Hybrid Adsorbent Materials for Effective Heavy Metals Uptake from Wastewater. Crystals 2024, 14, 447. [Google Scholar] [CrossRef]
- Haripriyan, U.; Gopinath, K.P.; Arun, J. Chitosan based nano adsorbents and its types for heavy metal removal: A mini review. Mater. Lett. 2022, 312, 131670. [Google Scholar] [CrossRef]
- Boukhlifi, F. Sustainable Treatment of Heavy Metals by Adsorption on Raw Chitin/Chitosan. In Trace Metals in the Environment; Mario Alfonso, M.-T., Hugo, S.-N., Agnieszka, S., Eds.; IntechOpen: Rijeka, Croatia, 2021; Chapter 13. [Google Scholar]
- Trikkaliotis, D.G.; Ainali, N.M.; Tolkou, A.K.; Mitropoulos, A.C.; Lambropoulou, D.A.; Bikiaris, D.N.; Kyzas, G.Z. Removal of Heavy Metal Ions from Wastewaters by Using Chitosan/Poly(Vinyl Alcohol) Adsorbents: A Review. Macromol 2022, 2, 403–425. [Google Scholar] [CrossRef]
- Wang, K.; Zhang, F.; Xu, K.; Che, Y.; Qi, M.; Song, C. Modified magnetic chitosan materials for heavy metal adsorption: A review. RSC Adv. 2023, 13, 6713–6736. [Google Scholar] [CrossRef]
- Duan, C.; Ma, T.; Wang, J.; Zhou, Y. Removal of heavy metals from aqueous solution using carbon-based adsorbents: A review. J. Water Process Eng. 2020, 37, 101339. [Google Scholar] [CrossRef]
- Wang, B.; Lan, J.; Bo, C.; Gong, B.; Ou, J. Adsorption of heavy metal onto biomass-derived activated carbon: Review. RSC Adv. 2023, 13, 4275–4302. [Google Scholar] [CrossRef]
- Sheraz, N.; Shah, A.; Haleem, A.; Iftikhar, F.J. Comprehensive assessment of carbon-, biomaterial- and inorganic-based adsorbents for the removal of the most hazardous heavy metal ions from wastewater. RSC Adv. 2024, 14, 11284–11310. [Google Scholar] [CrossRef] [PubMed]
- Jüntgen, H. Activated carbon as catalyst support: A review of new research results. Fuel 1986, 65, 1436–1446. [Google Scholar] [CrossRef]
- Iwanow, M.; Gärtner, T.; Sieber, V.; König, B. Activated carbon as catalyst support: Precursors, preparation, modification and characterization. Beilstein J. Org. Chem. 2020, 16, 1188–1202. [Google Scholar] [CrossRef] [PubMed]
- Wojnicki, M.; Socha, R.P.; Pędzich, Z.; Mech, K.; Tokarski, T.; Fitzner, K. Palladium(II) Chloride Complex Ion Recovery from Aqueous Solutions Using Adsorption on Activated Carbon. J. Chem. Eng. Data 2018, 63, 702–711. [Google Scholar] [CrossRef]
- Wojnicki, M.; Fitzner, K. Kinetic modeling of the adsorption process of Pd(II) complex ions onto activated carbon. React. Kinet. Mech. Catal. 2018, 124, 453–468. [Google Scholar] [CrossRef]
- Wojnicki, M.; PacŁAwski, K.; Socha, R.P.; Fitzner, K. Adsorption and reduction of platinum(IV) chloride complex ions on activated carbon. Trans. Nonferrous Met. Soc. China 2013, 23, 1147–1156. [Google Scholar] [CrossRef]
- Wojnicki, M.; Rudnik, E.; Socha, R.P.; Fitzner, K. Platinum(IV) Chloride Complex Ions Adsorption on Activated Carbon Organosorb 10CO. Aust. J. Chem. 2017, 70, 769–775. [Google Scholar] [CrossRef]
- Wojnicki, M.; Socha, R.P.; Luty-Błocho, M.; Fitzner, K. Kinetic studies of the removal of Pt(IV) chloride complex ions from acidic aqueous solutions using activated carbon. React. Kinet. Mech. Catal. 2017, 120, 715–734. [Google Scholar] [CrossRef]
- Michałek, T.; Wojtaszek, K.; Małecki, S.; Kornaus, K.; Wandor, S.; Druciarek, J.; Fitzner, K.; Wojnicki, M. Recovery of Pd(II) Ions from Aqueous Solutions Using Activated Carbon Obtained in a Single-Stage Synthesis from Cherry Seeds. C 2023, 9, 46. [Google Scholar] [CrossRef]
- Gurrath, M.; Kuretzky, T.; Boehm, H.P.; Okhlopkova, L.B.; Lisitsyn, A.S.; Likholobov, V.A. Palladium catalysts on activated carbon supports: Influence of reduction temperature, origin of the support and pretreatments of the carbon surface. Carbon 2000, 38, 1241–1255. [Google Scholar] [CrossRef]
- Luty-Błocho, M.; Wojnicki, M.; Włoch, G.; Fitzner, K. Green method for efficient PdNPs deposition on carbon carrier in the microreactor system. J. Nanopart. Res. 2018, 20, 239. [Google Scholar] [CrossRef] [PubMed]
- Luty-Błocho, M.; Wojnicki, M.; Pacławski, K.; Fitzner, K. The synthesis of platinum nanoparticles and their deposition on the active carbon fibers in one microreactor cycle. Chem. Eng. J. 2013, 226, 46–51. [Google Scholar] [CrossRef]
- Pikna, Ľ.; Milkovič, O.; Saksl, K.; Heželová, M.; Smrčová, M.; Puliš, P.; Michalik, Š.; Gamcová, J. The structure of nano-palladium deposited on carbon-based supports. J. Solid State Chem. 2014, 212, 197–204. [Google Scholar] [CrossRef]
- Luty-Błocho, M. The influence of steric stabilization on process of Au, Pt nanoparticles formation. Arch. Metall. Mater. 2023, 64, 55–63. [Google Scholar] [CrossRef]
- Sharififard, H.; Soleimani, M.; Zokaee Ashtiani, F. Application of nanoscale iron oxide-hydroxide-impregnated activated carbon (Fe-AC) as an adsorbent for vanadium recovery from aqueous solutions. Desalination Water Treat. 2016, 57, 15714–15723. [Google Scholar] [CrossRef]
- Ho Kim, Y.; Nakano, Y. Adsorption mechanism of palladium by redox within condensed-tannin gel. Water Res. 2005, 39, 1324–1330. [Google Scholar] [CrossRef]
- Wojnicki, M.; Podborska, A. The Mechanism of Redox Reaction between Palladium(II) Complex Ions and Potassium Formate in Acidic Aqueous Solution. Arch. Metall. Mater. 2017, 62, 737–745. [Google Scholar] [CrossRef]
- Sun, Z.; Chai, L.; Shu, Y.; Li, Q.; Liu, M.; Qiu, D. Chemical bond between chloride ions and surface carboxyl groups on activated carbon. Colloids Surf. A Physicochem. Eng. Asp. 2017, 530, 53–59. [Google Scholar] [CrossRef]
- Simonov, P.A.; Moroz, E.M.; Chuvilin, A.L.; Kolomiichuk, V.N.; Boronin, A.I.; Likholobov, V.A. Influence of an interaction of PdCl2 with carbon support on state and catalytic properties of Pd/C catalysts. In Studies in Surface Science and Catalysis; Poncelet, G., Martens, J., Delmon, B., Jacobs, P.A., Grange, P., Eds.; Elsevier: Amsterdam, The Netherlands, 1995; Volume 91, pp. 977–987. [Google Scholar]
- Luty-Błocho, M.; Wojnicki, M.; Csapo, E.; Fitzner, K. On the Rate of Interaction of Sodium Borohydride with Platinum (IV) Chloride Complexes in Alkaline Media. Materials 2021, 14, 3137. [Google Scholar] [CrossRef] [PubMed]
- Simonov, P.A.; Romanenko, A.V.; Prosvirin, I.P.; Moroz, E.M.; Boronin, A.I.; Chuvilin, A.L.; Likholobov, V.A. On the nature of the interaction of H2PdCl4 with the surface of graphite-like carbon materials. Carbon 1997, 35, 73–82. [Google Scholar] [CrossRef]
- Higashi, N.; Ozaki, Y. Potential of far-ultraviolet absorption spectroscopy as a highly sensitive quantitative and qualitative analysis method for aqueous solutions, part I: Determination of hydrogen chloride in aqueous solutions. Appl. Spectrosc. 2004, 58, 910–916. [Google Scholar] [CrossRef] [PubMed]
- Boily, J.-F.; Seward, T.M. Palladium(II) chloride complexation: Spectrophotometric investigation in aqueous solutions from 5 to 125°C and theoretical insight into Pd-Cl and Pd-OH2 interactions. Geochim. Cosmochim. Acta 2005, 69, 3773–3789. [Google Scholar] [CrossRef]
- Saito, S.; Numadate, N.; Teraoka, H.; Enami, S.; Kobayashi, H.; Hama, T. Impurity contribution to ultraviolet absorption of saturated fatty acids. Sci. Adv. 2023, 9, eadj6438. [Google Scholar] [CrossRef] [PubMed]
- Coyle, J.D. Photochemistry of carboxylic acid derivatives. Chem. Rev. 1978, 78, 97–123. [Google Scholar] [CrossRef]
- Kaeswurm, J.A.H.; Scharinger, A.; Teipel, J.; Buchweitz, M. Absorption Coefficients of Phenolic Structures in Different Solvents Routinely Used for Experiments. Molecules 2021, 26, 4656. [Google Scholar] [CrossRef] [PubMed]
- Shimodaira, N.; Masui, A. Raman spectroscopic investigations of activated carbon materials. J. Appl. Phys. 2002, 92, 902–909. [Google Scholar] [CrossRef]
- Xing, Z.; Qi, Y.; Tian, Z.; Xu, J.; Yuan, Y.; Bommier, C.; Lu, J.; Tong, W.; Jiang, D.-E.; Ji, X. Identify the Removable Substructure in Carbon Activation. Chem. Mater. 2017, 29, 7288–7295. [Google Scholar] [CrossRef]
- Bokobza, L.; Bruneel, J.-L.; Couzi, M. Raman Spectra of Carbon-Based Materials (from Graphite to Carbon Black) and of Some Silicone Composites. C 2015, 1, 77–94. [Google Scholar] [CrossRef]
- Zambrzycki, M.; Jeleń, P.; Fraczek-Szczypta, A. Structure and electrical transport properties of electrospun carbon nanofibers/carbon nanotubes 3D hierarchical nanocomposites: Effect of the CCVD synthesis conditions. J. Mater. Sci. 2022, 57, 9334–9356. [Google Scholar] [CrossRef]
- Cançado, L.G.; Takai, K.; Enoki, T.; Endo, M.; Kim, Y.A.; Mizusaki, H.; Jorio, A.; Coelho, L.N.; Magalhães-Paniago, R.; Pimenta, M.A. General equation for the determination of the crystallite size La of nanographite by Raman spectroscopy. Appl. Phys. Lett. 2006, 88, 163106. [Google Scholar] [CrossRef]
- Cansado, I.P.; Mourão, P.A.; Castanheiro, J.E. Performance of Regenerated Activated Carbons on Pesticides Removal from the Aqueous Phase. Processes 2023, 11, 2496. [Google Scholar] [CrossRef]
- Azhagapillai, P.; Al Shoaibi, A.; Chandrasekar, S. Surface functionalization methodologies on activated carbons and their benzene adsorption. Carbon Lett. 2021, 31, 419–426. [Google Scholar] [CrossRef]
- Rashidi, N.A.; Bokhari, A.; Yusup, S. Evaluation of kinetics and mechanism properties of CO2 adsorption onto the palm kernel shell activated carbon. Environ. Sci. Pollut. Res. 2021, 28, 33967–33979. [Google Scholar] [CrossRef] [PubMed]
- Pudney, P.D.A.; Mutch, K.J.; Zhu, S. Characterising the phase behaviour of stearic acid and its triethanolamine soap and acid–soap by infrared spectroscopy. Phys. Chem. Chem. Phys. 2009, 11, 5010–5018. [Google Scholar] [CrossRef] [PubMed]
- Squeo, G.; Grassi, S.; Paradiso, V.M.; Alamprese, C.; Caponio, F. FT-IR extra virgin olive oil classification based on ethyl ester content. Food Control 2019, 102, 149–156. [Google Scholar] [CrossRef]
- De la Puente, G.; Pis, J.J.; Menéndez, J.A.; Grange, P. Thermal stability of oxygenated functions in activated carbons. J. Anal. Appl. Pyrolysis 1997, 43, 125–138. [Google Scholar] [CrossRef]
- Skibińska, K.; Żabiński, P. Nanocones: A Compressive Review of Their Electrochemical Synthesis and Applications. Materials 2024, 17, 3089. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, G.; He, Z.; Chen, J.; Gao, W.; Cao, P. Superhydrophobic Ni nanocone surface prepared by electrodeposition and its overall performance. Surf. Coat. Technol. 2023, 464, 129548. [Google Scholar] [CrossRef]
- Penki, T.R.; Shanmughasundaram, D.; Kishore, B.; Munichandraiah, N. High rate capability of coconut kernel derived carbon as an anode material for lithium-ion batteries. Adv. Mat. Lett. 2014, 5, 184–190. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | |||
|---|---|---|---|
| Pd@ROY | Pt@ROY | Pd-Pt@ROY | |
| Rate constant, min−1 | 0.195 | 0.238 | 0.278 |
| Sample | ID/IG | ID2/IG2 | La ID/IG | La ID2/IG2 |
|---|---|---|---|---|
| a.u. | a.u. | nm | nm | |
| ROY | 1.271 | 1.013 | 15.13 | 18.98 |
| Pt@ROY | 1.256 | 1.013 | 15.31 | 19.22 |
| Pd@ROY | 1.228 | 0.953 | 15.66 | 20.18 |
| Pd-Pt@ROY | 1.207 | 0.827 | 15.93 | 23.25 |
| Plane | Sample | 2 Θ, ° | |
|---|---|---|---|
| Experimental | ICDD Cards/Ref. | ||
| Pt | 39.956 1 | ||
| (1 1 1) | Pt-Pd@ACF | 40.035 | [4] |
| Pd-Pt@ROY | 40.042 | This work | |
| Pd | 40.47 2 | ||
| Pd@ACF | 40.110 | [4] | |
| Pd@ROY | 40.333 | This work | |
| Pt | 46.472 1 | ||
| (2 0 0) | Pt-Pd@ACF | 46.629 | [4] |
| Pd-Pt@ROY | 46.621 | This work | |
| Pd | 46.88 2 | ||
| Pd@ACF | 46.684 | [4] | |
| Pd@ROY | 46.855 | This work | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luty-Błocho, M.; Pach, A.; Kutyła, D.; Kula, A.; Małecki, S.; Jeleń, P.; Hessel, V. Waste for Product—Synthesis and Electrocatalytic Properties of Palladium Nanopyramid Layer Enriched with PtNPs. Materials 2024, 17, 4165. https://doi.org/10.3390/ma17164165
Luty-Błocho M, Pach A, Kutyła D, Kula A, Małecki S, Jeleń P, Hessel V. Waste for Product—Synthesis and Electrocatalytic Properties of Palladium Nanopyramid Layer Enriched with PtNPs. Materials. 2024; 17(16):4165. https://doi.org/10.3390/ma17164165
Chicago/Turabian StyleLuty-Błocho, Magdalena, Adrianna Pach, Dawid Kutyła, Anna Kula, Stanisław Małecki, Piotr Jeleń, and Volker Hessel. 2024. "Waste for Product—Synthesis and Electrocatalytic Properties of Palladium Nanopyramid Layer Enriched with PtNPs" Materials 17, no. 16: 4165. https://doi.org/10.3390/ma17164165