
Cavitation and Solid-State Post-Condensation of Polyethylene Terephthalate: Literature Review



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Microstructure of PET Material
2.1. A Three-Phase PET Model
2.2. The Crystallization Process Based on the Three-Phase PET Model
2.3. The Crystal Nucleation Process of PET Material Based on the Three-Phase Model
2.4. PET Microstructure Analysis by the Use of Positron Annihilation Lifetime Spectroscopy (PALS)
3. Cavitation and Solid-State Post-Condensation of PET Material
3.1. Cavitation and Microcavitation Effect in PET Material
- During film-sample deformation, mesophase transformations to higher-density structures lead to the sample’s volume contraction, possibly forming voids in the sample.
- The porosity of stretched films, driven by the presence of voids, increased as the drawing ratio rose.
- Cavitation occurs sooner with a higher number of tangential lamellae within a spherulite. Conversely, reducing the number of tangential daughter lamellae accelerates void formation.
- Cavitation emerged at the spherulite boundaries or in their equatorial regions, where the lamellar crystals are oriented perpendicularly to the tensile direction.
- Annealing can accelerate the onset of cavitation [75]. In annealed samples, cavitation behavior was greatly intensified due to an increase in crystal thickness and a rise in stress concentration sites. This behavior may explain the reduction in pressure resistance observed in PET bottles produced in a hot mold during the SBM process, as noted in another work by the authors [6]).
- The presence of thinner lamellae generally inhibit void formation due to their higher density of tie-molecules. This denser network facilitates more effective load transfer to the lamellae, promoting their plastic deformation over cavitation within the amorphous phase, as observed in research on polybutene [81].
- Samples with lower molecular weight exhibited stronger cavitation due to fewer entanglements in the amorphous phase.
- When crystallite movement in the amorphous phase is restricted at relatively low temperatures, cavitation increases as the sample is stretched parallel to the lamella orientation (based on research of PP) and void forms before fragmentation and reorientation of microstructure—with rising temperature during stretching, void size diminishes.
- Introducing a low molecular weight modifier into the free volume pores of the amorphous phase results in reduced intensity (“the type of liquid is not relevant, except that it should not dissolve polymer crystals” [85]), or complete elimination, of the cavitation phenomenon. SEM microphotographs of cavitating (a) and saturated with chloroform non-cavitating polypropylene samples (b) are shown in Figure 3 [86].
- Initially, during stretching, voids elongate perpendicular to the stretching direction and subsequently reorient along the stretch axis. In the early stages, incorporating a nucleating agent (NJS) has minimal impact on void size. However, at later stages, voids along the stretching direction in iPP/NJS composites grow rapidly.
- Cavitation occurs in various semicrystalline polymers when they are stretched uniaxially above their glass transition temperatures. Void formation is typically affected by the polymer’s morphology, including lamellae thickness, orientation, and the microstructure of the amorphous phase. During stretching, void sizes vary according to local strain levels [68].
3.2. Solid-State Post-Condensation of PET
4. General Conclusions from Literature Review
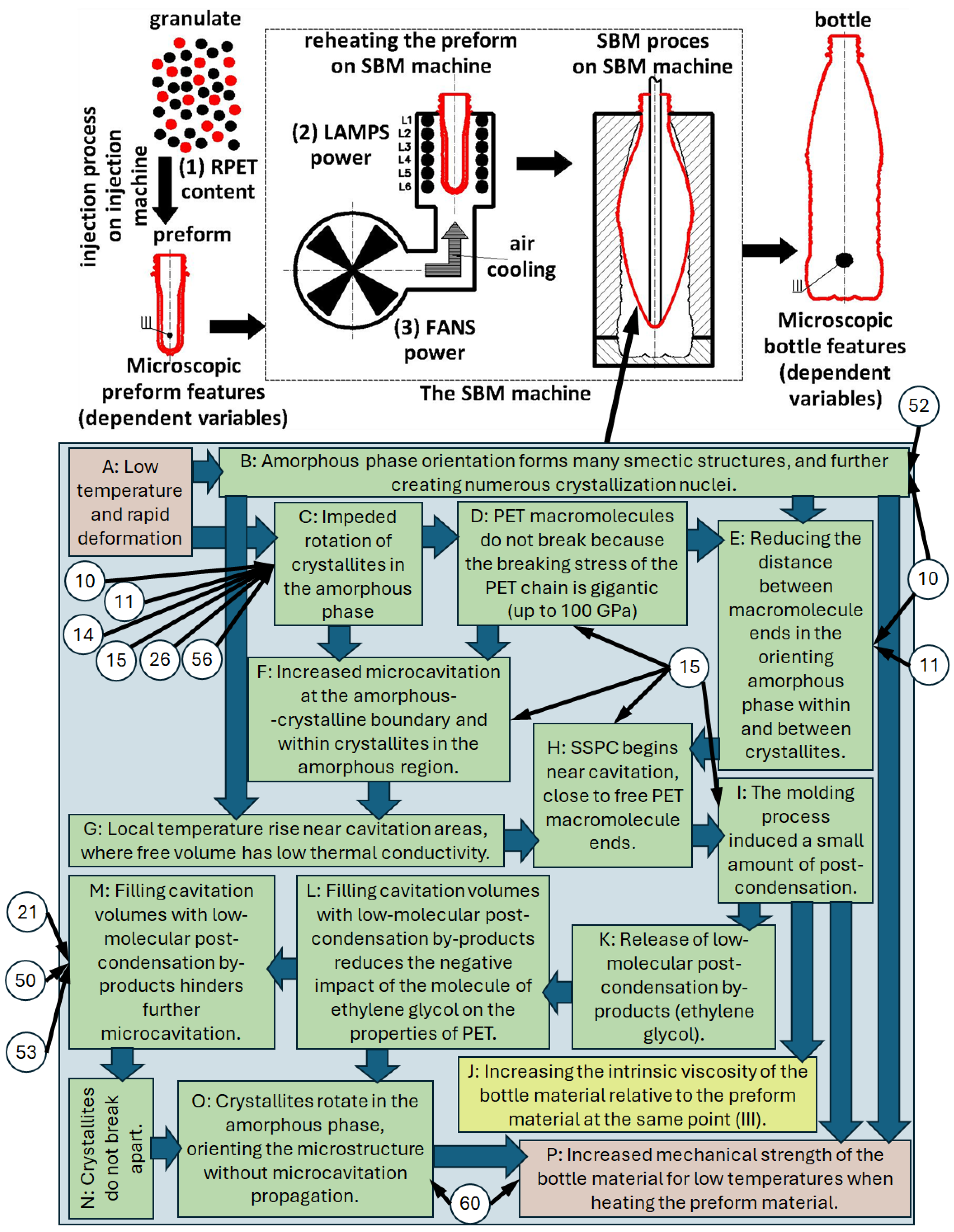
- A:
- Relatively low temperature and very quick deformation of the PET material during the SBM process [97] (stiffer amorphous phase)—the temperature of the preform, depending on the shape and thickness of the side wall of the preform and the shape of the container intended for cold fill, ranges from 105 °C to 128 °C [1].
- B:
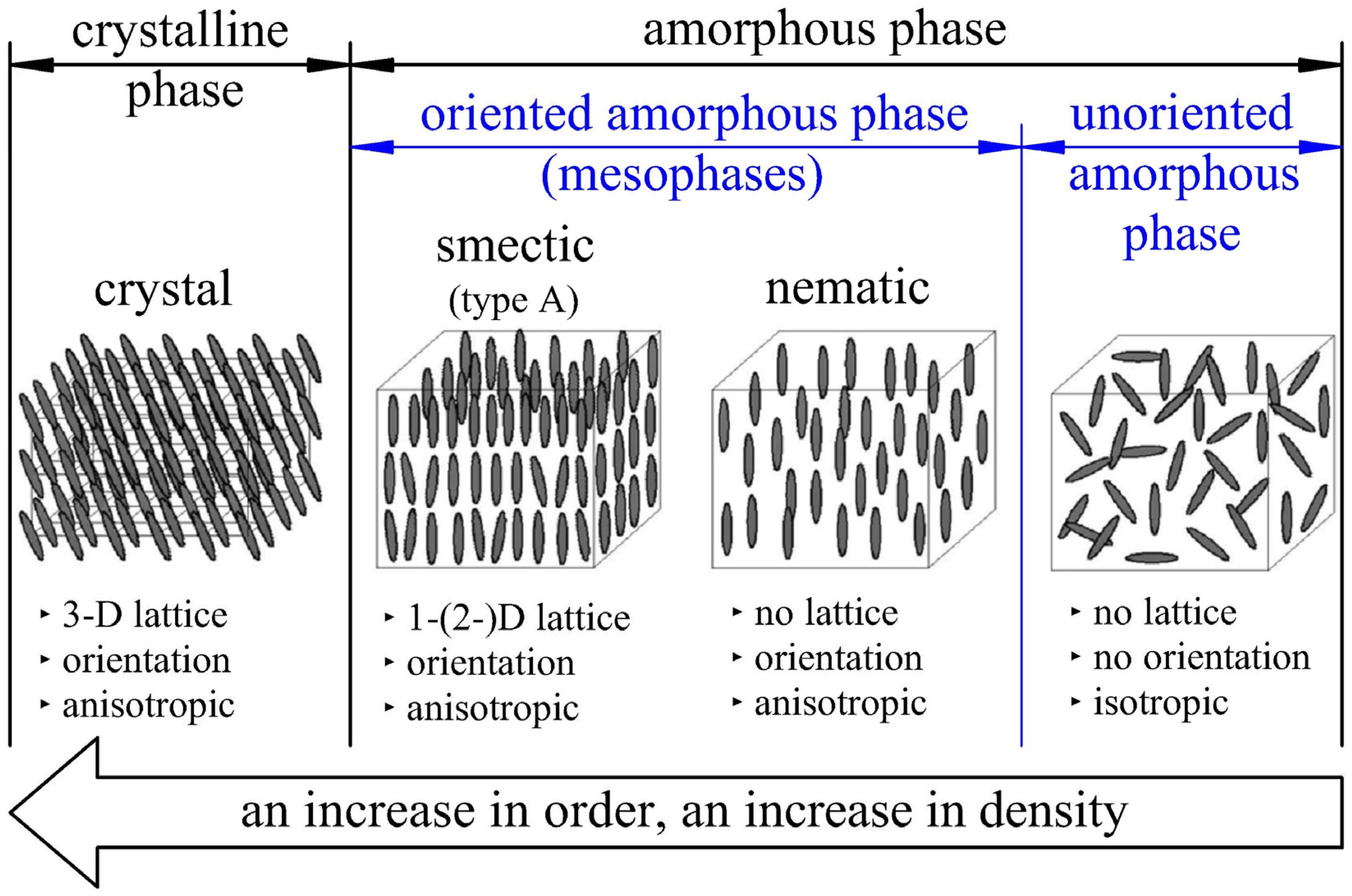
- Occurrence of the orientation of the amorphous phase with the formation of a large number of smectic structures and thus a very large number of crystallization nuclei [25] (no crystallization of the PET material during rapid deformation at relatively low temperatures), especially in biaxial stretching [42]—immediate crystallization induced by the deformation of the material occurs after the fast deformation process is completed [43,44].
- C:
- D:
- PET macromolecules do not break because the breaking stress of the covalent carbon-carbon bond in PET chain is gigantic (the strength of a polymer chain depends on the strength of –C–C– bonds, practically 60–100 GPa [96]).
- E:
- Shortening the distance between the free ends of PET macromolecules in the orienting amorphous phase inside the crystallites and at the boundary of the crystalline and amorphous phases [25]. Free ends of PET macromolecules emerging from the crystallites are not entangled with each other [12], which occurs only in the amorphous phase, which may favor post-condensation of PET macromolecules in free volumes (vacuum) created as a result of microcavitation between the lamellas in the crystallite, as well as at the boundary of the crystalline and amorphous phases.
- F:
- G:
- Local increase in temperature (e.g., as a result of intermolecular friction [46,89] or as a result of a change in the conformation of the macromolecule from gauche to trans [100]), whereas for very rapid deformation the heat transfer conditions are almost adiabatic, so all the released heat of crystallization (which occurs after completion of deformation at high deformation rates) will increase the sample temperature [101]) in the vicinity of microcavitation areas (the free volume is characterized by a very low thermal conductivity coefficient) [89]
- H:
- Initiation of the post-condensation process in the solid state near the microcavitation area in the vicinity of the free ends of PET macromolecules near crystallite boundaries [12].
- I:
- The molding process induced a small amount of post-condensation, which can be determined by (J).
- J:
- Increasing the intrinsic viscosity of the bottle material relative to the preform material at the same point (Figure 4b in [7]).
- K:
- The release of low-molecular by-products of the post-condensation process (molecule of ethylene glycol) [12].
- L:
- Filling free microcavitation volumes with low-molecular products of the post-condensation process, reducing the negative impact of the molecule of ethylene glycol on the properties of PET [12].
- M:
- The process of filling empty microcavitation volumes with low-molecular products of the post-condensation process hinders further microcavitation processes [88], as a result of which the microcavitation area does not propagate in the amorphous phase inside the crystallite, and at the border of the crystalline and amorphous phases [77] (the reduction of the dimensions of the free volumes results in a decrease in their susceptibility to ellipsoidization during polymer deformation as a consequence of what microcavitation volumes do not propagate into cavitation volumes [9]—the mechanism of the influence of deformation on the ellipsoidization of the free volumes, their approaching each other, and consequently their propagation into the cavitation volumes is schematically shown in Figure 5 [9]), increasing the strength of the amorphous phase inside the crystalline phase (by filling free volume pores, their average size decreases, increasing the strength of the amorphous phase, which in turn hinders the process of creating cavitation pores—higher stresses are required to create them and stabilize such pores) [77].
- N:
- Crystallites do not break apart (filling the free volume pores and reducing their average size increases the strength of the amorphous phase [77]).
- O:
- The crystallites rotate in the amorphous phase, orienting the microstructure without the propagation of microcavitation due to the free volumes being filled with low-molecular products of the post-condensation process.
- P:

- Is it possible to control the post-condensation process and the microcavitation phe-nomenon during the blow molding process?
- What effect do post-condensation and microcavitation have on the mechanical strength of blow molding packages?
- Do the described phenomena also occur in other blow molding plastics, with particular emphasis on PP (Polypropylene)?
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wawrzyniak, P.; Datta, J. Characteristics of the blowing stages of poly (ethylene terephthalate) preforms in the blowing process with simultaneous stretching. Przem. Chem. 2015, 94, 1114–1118. [Google Scholar] [CrossRef]
- Wawrzyniak, P.; Datta, J. Stretch blow molding machines used for manufacturing PET bottles. Przem. Chem. 2015, 94, 1110–1113. [Google Scholar] [CrossRef]
- Wawrzyniak, P.; Karaszewski, W. A literature survey of the influence of preform reheating and stretch blow molding with hot mold process parameters on the properties of PET containers. Part I. Polimery 2020, 65, 346–356. [Google Scholar] [CrossRef]
- Wawrzyniak, P.; Karaszewski, W. A literature survey of the influence of preform reheating and stretch blow molding with hot mold process parameters on the properties of PET containers. Part II. Polimery 2020, 65, 437–448. [Google Scholar] [CrossRef]
- Wawrzyniak, P.; Karaszewski, W. Blowing kinetics, pressure resistance, thermal stability, and relaxation of the amorphous phase of the PET container in the SBM process with hot and cold mold. Part I: Research methodology and results. Polymers 2020, 12, 1749. [Google Scholar] [CrossRef] [PubMed]
- Wawrzyniak, P.; Karaszewski, W. Blowing kinetics, pressure resistance, thermal stability, and relaxation of the amorphous phase of the PET container in the SBM process with hot and cold mold. Part II: Statistical Analysis and Interpretation of Tests. Polymers 2020, 12, 1761. [Google Scholar] [CrossRef]
- Wawrzyniak, P.; Karaszewski, W.; Różański, A. Effect of rPET Content and Preform Heating/Cooling Conditions in the Stretch Blow Molding Process on Microcavitation and Solid-State Post-Condensation of vPET-rPET Blend: Part I—Research Methodology and Results. Materials 2024, 17, 5233. [Google Scholar] [CrossRef]
- Wawrzyniak, P.; Karaszewski, W.; Safandowska, M.; Idczak, R. Effect of rPET Content and Preform Heating/Cooling Conditions in the SBM Process on Microcavitation and Solid State Post-condensation of vPET-rPET Blend. Part II: Statistical Analysis and Interpretation of Tests. Materials, 2024; in review. [Google Scholar]
- Makarewicz, C.; Safandowska, M.; Idczak, R.; Rozanski, A. Plastic Deformation of Polypropylene Studied by Positron Annihilation Lifetime Spectroscopy. Macromolecules 2022, 55, 10062–10076. [Google Scholar] [CrossRef]
- Fakirov, S. Condensation Polymers: Their Chemical Peculiarities Offer Great Opportunities. Prog. Polym. Sci. 2019, 89, 1–18. [Google Scholar] [CrossRef]
- Fakirov, S. Chemical healing in poly(ethylene terephthalate). J. Polym. Sci. Polym. Phys. Ed. 1984, 22, 2095–2104. [Google Scholar] [CrossRef]
- Gantillon, B.; Spitz, R.; McKenna, T.F. The Solid State Postcondensation of PET, 1. Macromol. Mater. Eng. 2004, 289, 88–105. [Google Scholar] [CrossRef]
- Ge, M.; Sun, C.; Zhang, G.; Coutier-Delgosha, O.; Fan, D. Combined suppression effects on hydrodynamic cavitation performance in Venturi-type reactor for process intensification. Ultrason. Sonochemistry 2022, 86, 106035. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.Z.D.; Cao, M.-Y.; Wunderlich, B. Glass Transition and Melting Behavior of Poly (oxy-1,4-phenyleneoxy-1,4-phenylenecarbonyl-1,4-phenylene). Macromolecules 1986, 19, 1868–1876. [Google Scholar] [CrossRef]
- Cheng, S.Z.D.; Wu, Z.Q.; Wunderlich, B. Glass Transition and Melting Behavior of Poly(thio-1,4-phenylene). Macromolecules 1987, 20, 2802–2810. [Google Scholar] [CrossRef]
- Schlosser, E.; Schönhals, A. Recent development in dielectric relaxation spectroscopy of polymers. Colloid Polym. Sci. 1989, 267, 963–969. [Google Scholar] [CrossRef]
- Shantarovich, V.P. Positron annihilation and free volume studies in polymer glasses. J. Polym. Sci. B Polym. Phys. 2008, 46, 2485–2503. [Google Scholar] [CrossRef]
- Lin, J.; Shenogin, S.; Nazarenko, S. Oxygen solubility and specific volume of rigid amorphous fraction in semicrystalline poly(ethylene terephthalate). Polymer 2002, 43, 4733–4743. [Google Scholar] [CrossRef]
- Michaels, A.S.; Vieth, W.R.; Barrie, J.A. Solution of Gases in Polyethylene Terephthalate. J. Appl. Phys. 1963, 34, 1–12. [Google Scholar] [CrossRef]
- Olson, B.G.; Lin, J.; Nazarenko, S.; Jamieson, A.M. Positron Annihilation Lifetime Spectroscopy of Poly(ethylene terephthalate): Contributions from Rigid and Mobile Amorphous Fractions. Macromolecules 2003, 36, 7618–7623. [Google Scholar] [CrossRef]
- Rabek, J.F. Współczesna Wiedza o Polimerach (ang. Modern Knowledge of Polymers); PWN: Warszawa, Poland, 2008. (In Polish) [Google Scholar]
- Rastogi, R.; Vellinga, W.P.; Rastogi, S.; Schick, C.; Meijer, H.E.H. The Three-Phase Structure and Mechanical Properties of Poly(ethylene terephthalate). J. Polym. Sci. Part B Polym. Phys. 2004, 42, 2092–2106. [Google Scholar] [CrossRef]
- Awaja, F.; Pavel, D. Recycling of PET. Eur. Polym. J. 2005, 41, 1453–1477. [Google Scholar] [CrossRef]
- Dong, W.; Zhao, J.; Li, C.; Guo, M.; Zhao, D.; Fan, Q. Study of the amorphous phase in semicrystalline poIy(ethyIene terephthalate) via dynamie mechanical thermal analysis. Polym. Bull. 2002, 49, 197–203. [Google Scholar] [CrossRef]
- Kawakami, D.; Hsiao, B.S.; Burger, C.; Ran, S.; Avila-Orta, C.; Sics, I.; Kikutani, T.; Jacob, K.I.; Chu, B. Deformation-Induced Phase Transition and Superstructure Formation in Poly(ethylene terephthalate). Macromolecules 2005, 38, 91–103. [Google Scholar] [CrossRef]
- Boyd, T.J. Transient Crystallization of Poly(ethylene terephthalate) Bottles. Ph.D. Thesis, The University of Toledo, Toledo, OH, USA, 2004. Available online: http://rave.ohiolink.edu/etdc/view?acc_num=toledo1091111913 (accessed on 20 September 2024).
- Ivanov, D.A.; Amalou, Z.; Koch, M.H.J. Aromatic Semirigid Chain Polymers: Studies of Semicrystalline Morphology and its Reorganization upon Annealing. Annual Reports. 2002. Available online: https://hasyweb.desy.de/science/annual_reports/2002_report/part2/contrib/73/7701.pdf (accessed on 20 September 2024).
- Liu, R.Y.F.; Hu, Y.S.; Schiraldi, D.A.; Hiltner, A.; Baer, E. Crystallinity and Oxygen Transport Properties of PET Bottle Walls. J. Appl. Polym. Sci. 2004, 94, 671–677. [Google Scholar] [CrossRef]
- Mohanty, S. Liquid crystals—The ‘fourth’ phase of matter. Reson 2003, 8, 52–70. [Google Scholar] [CrossRef]
- Wawrzyniak, P. The Influence of Stretch Blow Moulding Process Parameters on the Properties of the Produced PET Containers. Ph.D. Dissertation, Politechnika Gdańska, Gdańsk, Poland, 2016. [Google Scholar]
- Okada, K.; Higashioji, T.; Nakagawa, T.; Uchida, H.; Takahashi, K.; Inoue, R.; Nishida, K.; Kanaya, T. Structural analysis of poly(ethylene terephthalate) during uniaxial drawing above the glass transition temperature. Polym. J. 2013, 45, 50–56. [Google Scholar] [CrossRef]
- Nikolov, S.; Lebensohn, R.A.; Raabe, D. Self-consistent modeling of large plastic deformation, texture and morphology evolution in semi-crystalline polymers. J. Mech. Phys. Solids 2006, 54, 1350–1375. [Google Scholar] [CrossRef]
- Abe, A.; Furuya, H.; Hiejima, T.; Nishiyama, T. On the Stability of the Nematic Order Observed During the Cold-crystallization of PET. Polym. J. 2008, 40, 910–914. [Google Scholar] [CrossRef]
- Huang, J.-M.; Chu, P.P.; Chang, F.-C. Conformational changes and molecular motion of poly(ethylene terephthalate) annealed above glass transition temperature. Polymer 2000, 41, 1741–1748. [Google Scholar] [CrossRef]
- Baldenegro-Perez, L.A.; Navarro-Rodriguez, D.; Medellin-Rodriguez, F.J.; Hsiao, B.; Avila-Orta, C.A.; Sics, I. Molecular Weight and Crystallization Temperature Effects on Poly(ethylene terephthalate) (PET) Homopolymers, an Isothermal Crystallization Analysis. Polymers 2014, 6, 583–600. [Google Scholar] [CrossRef]
- Gouissem, L.; Douibi, A.; Benachour, D. The Evolution of Properties of Recycled Poly(ethylene terephthalate) as Function of Chain Extenders, the Extrusion Cycle and Heat Treatment. Polym. Sci. Ser. A 2014, 56, 844–855. [Google Scholar] [CrossRef]
- Królikowski, B. Effect of poly(ethylene terephthalate) and polyethylene comminution on the mechanical and thermal properties of polymer compositions. Polimery 2007, 52, 752–759. [Google Scholar] [CrossRef]
- Gaonkar, A.A.; Murudkar, V.V.; Deshpande, V.D. Comparison of crystallization kinetics of polyethylene terephthalate (PET) and reorganized PET. Thermochim. Acta 2020, 683, 178472. [Google Scholar] [CrossRef]
- Sylvestre, N.; Bouvard, J.-L.; Derrien, M.; Monnier, X.; Combeaud, C. Effects of mechanical recycling on PET stretchability. Polymer 2024, 307, 127256. [Google Scholar] [CrossRef]
- Gorlier, E.; Haudin, J.M.; Bilion, N. Strain-induced crystallisation in bulk amorphous PET under uni-axial loading. Polymer 2001, 42, 9541–9549. [Google Scholar] [CrossRef]
- Torres, N.; Robin, J.J.; Boutevin, B. Study of thermal and mechanical properties of virgin and recycled poly(ethylene terephthalate) before and after injection molding. Eur. Polym. J. 2000, 36, 2075–2080. [Google Scholar] [CrossRef]
- Shen, Y.; Harkin-Jones, E.; Hornsby, P.; McNally, T.; Abu-Zurayk, R. The effect of temperature and strain rate on the deformation behaviour, structure development and properties of biaxially stretched PET–clay nanocomposites. Compos. Technol. 2011, 71, 758–764. [Google Scholar] [CrossRef]
- Mahendrasingam, A.; Blundell, D.J.; Martin, C.; Fuller, D.H.; MacKerron, W.; Harvie, J.L.; Oldman, R.J.; Riekel, C. Influence of temperature and chain orientation on the crystallization of poly(ethylene terephthalate) during fast drawing. Polymer 2000, 41, 7803–7814. [Google Scholar] [CrossRef]
- Mahendrasingam, A.; Martin, C.; Fuller, W.; Blundell, D.J.; Oldman, R.J.; MacKerron, D.H.; Harvie, J.L.; Riekel, C. Observation of a transient structure prior to strain-induced crystallization in poly(ethylene terephthalate). Polymer 2000, 41, 1217–1221. [Google Scholar] [CrossRef]
- Lu, X.F.; Hay, J.N. Isothermal crystallization kinetics and melting behaviour of poly(ethylene terephthalate). Polymer 2001, 42, 9423–9431. [Google Scholar] [CrossRef]
- Menary, G.H.; Tan, C.W.; Harkin-Jones, E.M.A.; Armstrong, C.G.; Martin, P.J. Biaxial Deformation and Experimental Study of PET at Conditions Applicable to Stretch Blow Molding. Polym. Eng. Sci. 2012, 52, 671–688. [Google Scholar] [CrossRef]
- de Daubeny, R.P.; Bunn, C.W.; Brown, C.J. The crystal structure of polyethylene terephthalate. Proc. R. Soc. Lond. Ser. A Math. Phys. Sci. 1954, A226, 531. [Google Scholar] [CrossRef]
- Ceelen, J.M.J. Microscopic Characterization of PET Foil Used as Flexible Electronics Substrate. Master’s Thesis, Eindhoven University of Technology, Eindhoven, The Netherlands, 2011. Available online: https://pure.tue.nl/ws/portalfiles/portal/207024741/MT_11.16.pdf (accessed on 20 September 2024).
- Hagihara, H.; Oishi, A.; Funabashi, M.; Kunioka, M.; Suda, H. Free-volume hole size evaluated by positron annihilation lifetime spectroscopy in the amorphous part of poly(ethylene terephthalate) degraded by a weathering test. Polym. Degrad. Stab. 2014, 110, 389–394. [Google Scholar] [CrossRef]
- Dimonie, D.; Socoteanu, R.; Pop, S.; Fierascu, I.; Fierascu, R.; Petrea, C.; Zaharia, C.; Petrache, M. Overview on Mechanical Recycling by Chain Extension of POSTC-PET Bottles. In Material Recycling; IntechOpen: London, UK, 2012. [Google Scholar] [CrossRef]
- Ilišković, N.; Bravar, M. Correlation of physico-chemical, mechanical and electrical properties of ultraviolet-degraded poly(ethylene terephthalate). Polym. Degrad. Stab. 1986, 15, 173–182. [Google Scholar] [CrossRef]
- Ballara, A.; Verdu, J. Physical aspects of the hydrolysis of polyethylene terephthalate. Polym. Degrad. Stab. 1989, 26, 361–374. [Google Scholar] [CrossRef]
- Mohamed, H.F.M.; Ito, Y.; Imai, M. Change of distribution of free-volume holes during crystallization of poly(ethylene terephthalate) revealed by positron annihilation lifetime spectroscopy. J. Chem. Phys. 1996, 105, 4841–4845. [Google Scholar] [CrossRef]
- Sathyanarayana, P.M.; Shariff, G.; Thimmegowda, M.C.; Ashalatha, M.B.; Ramani, R.; Ranganathaiah, C. Structural relaxation in poly(ethylene terephthalate) studied by positron annihilation lifetime spectroscopy. Polym. Int. 2002, 51, 765–771. [Google Scholar] [CrossRef]
- Buttafava, A.; Consolati, G.; Mariani, M.; Quasso, F.; Ravasio, U. Effects induced by gamma irradiation of different polyesters studied by viscometry, thermal analysis and positron annihilation spectroscopy. Polym. Degrad. Stab. 2005, 89, 133. [Google Scholar] [CrossRef]
- Buttafava, A.; Consolati, G.; Di Landro, L.; Mariani, M. γ-Irradiation effects on polyethylene terephthalate studied by positron annihilation lifetime spectroscopy. Polymer 2002, 43, 7477–7481. [Google Scholar] [CrossRef]
- Siegel, R.W. Positron annihilation spectroscopy. Annu. Rev. Mater. Sci. 1980, 10, 393–425. [Google Scholar] [CrossRef]
- Pethrick, R.A. Positron annihilation—A probe for nanoscale voids and free volume? Prog. Polym. Sci. 1997, 22, 1–47. [Google Scholar] [CrossRef]
- Grafutin, V.I.; Prokop’ev, E.P. Positron annihilation spectroscopy in materials structure studies. Physics-Uspekhi 2002, 45, 59. [Google Scholar] [CrossRef]
- Yampolskii, Y.; Pinnau, I.; Freeman, B.D. Materials Science of Membranes for Gas and Vapor Separation; John Wiley & Sons: West Sussex, UK, 2006. [Google Scholar] [CrossRef]
- Sharma, S.K.; Pujari, P.K. Role of free volume characteristics of polymer matrix in bulk physical properties of polymer nanocomposites: A review of positron annihilation lifetime studies. Prog. Polym. Sci. 2017, 75, 31–47. [Google Scholar] [CrossRef]
- Takahashi, K.Z. Mapping positron annihilation lifetime spectroscopy data of a polymer to classical molecular dynamics simulations without shifting the glass transition temperature. J. Chem. Phys. 2023, 159, 084903. [Google Scholar] [CrossRef]
- Consolati, G.; Nichetti, D.; Quasso, F. Probing the Free Volume in Polymers by Means of Positron Annihilation Lifetime Spectroscopy. Polymers 2023, 15, 3128. [Google Scholar] [CrossRef]
- Shantarovich, V.P.; Bekeshev, V.G.; Pastukhov, A.V.; Davankov, V.A.; Krasil’nikova, O.K.; Belousova, E.V.; Kevdina, I.B.; Filimonov, M.K.; Gustov, V.W. When some elementary free volumes in polymers are not seen by positron annihilation experiments. J. Phys. Conf. Ser. 2015, 618, 012021. [Google Scholar] [CrossRef]
- Pawlak, A.; Galeski, A. Cavitation during tensile deformation of polypropylene. Macromolecules 2008, 41, 2839–2851. [Google Scholar] [CrossRef]
- Makarewicz, C.; Safandowska, M.; Idczak, R.; Kolodziej, S.; Rozanski, A. Strain Rate and Temperature Influence on Micromechanisms of Plastic Deformation of Polyethylenes Investigated by Positron Annihilation Lifetime Spectroscopy. Polymers 2024, 16, 420. [Google Scholar] [CrossRef]
- Makarewicz, C.; Safandowska, M.; Idczak, R.; Rozanski, A. Positron Annihilation Lifetime Spectroscopic Analysis of Plastic Deformation of High-Density Polyethylene. Macromolecules 2021, 54, 9649–9662. [Google Scholar] [CrossRef]
- Chang, P.; Schneider, K.; Kuehnert, I.; Heinrich, G. Cavitation Behavior of Semi-Crystalline Polymers during Uniaxial Stretching Studied by Synchrotron Small-Angle X-Ray Scattering. In Small Angle Scattering and Diffraction; InTech: London, UK, 2018. [Google Scholar] [CrossRef]
- Benyathiar, P.; Kumar, P.; Carpenter, G.; Brace, J.; Mishra, D.K. Polyethylene Terephthalate (PET) Bottle-to-Bottle Recycling for the Beverage Industry: A Review. Polymers 2022, 14, 2366. [Google Scholar] [CrossRef]
- Pawlak, A. Cavitation during deformation of polymers on the example of polypropylene. J. Appl. Polym. Sci. 2012, 125, 4177–4187. [Google Scholar] [CrossRef]
- Aboulfaraj, M.; G’Sell, C.; Ulrich, B.; Dahoun, A. In situ observation of the plastic deformation of polypropylene spherulites under uniaxial tension and simple shear in the scanning electron microscope. Polymer 1995, 36, 731–742. [Google Scholar] [CrossRef]
- Nitta, K.H.; Takayanagi, M. Direct observation of the deformation of isolated huge spherulites in isotactic polypropylene. J. Mater. Sci. 2003, 38, 4889–4894. [Google Scholar] [CrossRef]
- Pawlak, A. Cavitation during tensile deformation of isothermally crystallized polypropylene and high-density polyethylene. Colloid Polym. Sci. 2013, 291, 773–787. [Google Scholar] [CrossRef] [PubMed]
- Na, B.; Lv, R. Effect of cavitation on the plastic deformation and failure of isotactic polypropylene. J. Appl. Polym. Sci. 2007, 105, 3274–3279. [Google Scholar] [CrossRef]
- Bai, H.; Luo, F.; Zhou, T.; Deng, H.; Wang, K.; Fu, Q. New insight on the annealing induced microstructural changes and their roles in the toughening of beta-form polypropylene. Polymer 2011, 52, 2351–2360. [Google Scholar] [CrossRef]
- Peterlin, A. Plastic deformation of polymers with fibrous structure. Colloid Polym. Sci. 1975, 253, 809–823. [Google Scholar] [CrossRef]
- Rozanski, A.; Galeski, A. Modification of amorphous phase of semi-crystalline polymers. Polimery 2012, 57, 433–440. [Google Scholar] [CrossRef]
- Schneider, K.; Trabelsi, S.; Zafeiropoulos, N.E.; Davies, R.; Riekel, C.; Stamm, M. The study of cavitation in HDPE using time resolved synchrotron X-ray scattering during tensile deformation. Macromol. Symp. 2006, 236, 241–248. [Google Scholar] [CrossRef]
- Pawlak, A.; Galeski, A. Cavitation during tensile drawing of annealed high density polyethylene. Polymer 2010, 51, 5771–5779. [Google Scholar] [CrossRef]
- Wang, Y.; Jiang, Z.; Fu, L.; Lu, Y.; Men, Y. Lamellar thickness and stretching temperature dependency of cavitation in semi-crystalline polymers. PLoS ONE 2014, 9, e97234. [Google Scholar] [CrossRef]
- Thomas, C.; Seguela, R.; Detrez, F.; Miri, V.; Vanmansart, C. Plastic deformation of spherulitic semi-crystalline polymers: An in situ AFM study of polybutene under tensile drawing. Polymer 2009, 50, 3714–3723. [Google Scholar] [CrossRef]
- Pantani, R.; Coccorullo, I.; Speranza, V.; Titomanlio, G. Modeling of morphology evolution in the injection molding process of thermoplastic polymers. Prog. Polym. Sci. 2005, 30, 1185–1222. [Google Scholar] [CrossRef]
- Rhoades, A.M.; Williams, J.L.; Wonderling, N.; Androsch, R.; Guo, J. Skin/core crystallinity of injection-molded poly (butylene terephthalate) as revealed by microfocus X-ray diffraction and fast scanning chip calorimetry. J. Therm. Anal. Calorim. 2017, 127, 939–946. [Google Scholar] [CrossRef]
- van Erp, T.B.; Govaert, L.E.; Gerrit, W.M.P. Mechanical performance of injection-molded poly (propylene): Characterization and modeling. Macromol. Mater. Eng. 2013, 29, 8348–8358. [Google Scholar] [CrossRef]
- Rozanski, A.; Galeski, A. Controlling cavitation of semi-crystalline polymers during tensile drawing. Macromolecules 2011, 44, 7273–7287. [Google Scholar] [CrossRef]
- Rozanski, A.; Galeski, A. Crystalline Lamellae Fragmentation during Drawing of Polypropylene. Macromolecules 2015, 48, 5310–5322. [Google Scholar] [CrossRef]
- Zhang, Y.; Ben Jar, P.Y.; Xue, S.; Li, L. Quantification of strain-induced damage in semi-crystalline polymers: A review. J. Mater. Sci. 2019, 54, 62–82. [Google Scholar] [CrossRef]
- Galeski, A.; Rozanski, A. Cavitation during Drawing of Crystalline Polymers. Macromol. Symp. 2011, 298, 1–9. [Google Scholar] [CrossRef]
- Ronkay, F.; Czigany, T. Cavity formation and stress-oscillation during the tensile test of injection molded specimens made of PET. Polym. Bull. 2006, 57, 989–998. [Google Scholar] [CrossRef]
- Viana, M.E.; Riul, A.; Carvalho, G.M.; Rubira, A.F.; Muniz, E.C. Chemical recycling of PET by catalyzed glycolysis: Kinetics of the heterogeneous reaction. J. Chem. Eng. 2011, 173, 210–219. [Google Scholar] [CrossRef]
- Coates, G.W.; Getzler, Y.D.Y.L. Chemical recycling to monomer for an ideal, circular polymer economy. Nat. Rev. Mater. 2020, 5, 501–516. [Google Scholar] [CrossRef]
- Guan, Q.; Deng, X.; Zhang, H.; Zhong, S.; Liu, P.; Zhuang, Y.; Hu, X.; Yao, M.; Neisiany, R.E.; You, Z. Intrinsic flame retarding and non-dripping liquid crystal polyethylene terephthalate copolyesters for fire safety system. Chem. Eng. J. 2023, 453, 139329. [Google Scholar] [CrossRef]
- Mendes, L.C.; Mallet, I.A.; Cestari, S.P.; de Albuquerque Dias, F.G.; da Costa Pereira, P.S. Solid state polymerization of pet/pc extruded blend: Effect of reaction temperature on thermal, morphological and viscosity properties. Polímeros 2014, 24, 422–427. [Google Scholar] [CrossRef]
- Pandey, A.; Toda, A.; Rastogi, S. Influence of Amorphous Component on Melting of Semicrystalline Polymers. Macromolecules 2011, 44, 8042–8055. [Google Scholar] [CrossRef]
- Steinborn-Rogulska, I.; Rokicki, G. Solid-state polycondensation (SSP) as a method to obtain high molecular weight polymers. Part II. Synthesis of polylactide and polyglycolide via SSP. Polimery 2013, 2, 83–92. [Google Scholar] [CrossRef]
- Galeski, A. Strength and toughness of crystalline polymer systems. Prog. Polym. Sci. 2003, 28, 1643–1699. [Google Scholar] [CrossRef]
- Teng, F.; Menary, G.; Malinov, S.; Yan, S.; Boyet Stevens, J. Predicting the multiaxial stress-strain behavior of polyethylene terephthalate (PET) at different strain rates and temperatures above Tg by using an Artificial Neural Network. Mech. Mater. 2022, 165, 104175. [Google Scholar] [CrossRef]
- Pawlak, A.; Galeski, A.; Rozanski, A. Cavitation during deformation of semicrystalline polymers. Prog. Polym. Sci. 2014, 39, 921–958. [Google Scholar] [CrossRef]
- Rozanski, A. Initiation of Cavitation During Drawing of Crystalline. Ph.D. Dissertation, Centre of Molecular and Macromolucular Studies, Polish Academy of Sciences, Łódź, Poland, 2010. Available online: https://www.cbmm.lodz.pl/pliki/2019/04/PhD_A_Rozanski_2010.pdf (accessed on 20 September 2024).
- Tarazona, A.; Koglin, E.; Pijpers, A.P.; Meier, R.J. On the high-resolution X-ray photoelectron spectrum of poly(ethylene terephthalate): The sensitivity to trans-gauche conformation. Polymer 1997, 38, 2615–2616. [Google Scholar] [CrossRef]
- Blundell, D.J.; MacKerron, D.H.; Fuller, W.; Mahendrasingam, A.; Martin, C.; Oldman, R.J.; Rule, R.J.; Riekel, C. Characterization of strain-induced crystallization of poly(ethylene terephthalate) at fast draw rates using synchrotron radiation. Polymer 1996, 37, 3303–3311. [Google Scholar] [CrossRef]
- Nistico, R. Polyethylene terephthalate (PET) in the packaging industry. Polym. Test. 2020, 90, 106707. [Google Scholar] [CrossRef]
- Ignacio, M.D.; Tumu, K.N.; Munshi, M.; Vorst, K.L.; Curtzwiler, G.W. Suitability of MRF Recovered Post-Consumer Polypropylene Applications in Extrusion Blow Molded Bottle Food Packaging. Polymers 2023, 15, 3471. [Google Scholar] [CrossRef] [PubMed]
- Plastics Europe. Plastics—The Facts 2022. Report. October 2022. Available online: https://plasticseurope.org/wp-content/uploads/2023/03/PE-PLASTICS-THE-FACTS_FINAL_DIGITAL-1.pdf (accessed on 20 September 2024).
- Ge-Zhang, S.; Liu, H.; Song, M.; Wang, Y.; Yang, H.; Fan, H.; Ding, Y.; Mu, L. Advances in Polyethylene Terephthalate Beverage Bottle Optimization: A Mini Review. Polymers 2022, 14, 3364. [Google Scholar] [CrossRef] [PubMed]
- ALPLA Improves PET Bottle Performance with Agr Process Pilot. 23 April 2020. Available online: https://www.agrintl.com/alpla-improves-pet-bottle-performance-with-agr-process-pilot/ (accessed on 20 September 2024).
- Thibault, F.; Lim, L.-T. Modeling and Simulation of Stretch Blow Molding of Polyethylene Terephthalate. Polym. Eng. Sci. 2004, 44, 1460–1472. [Google Scholar] [CrossRef]
- Tsironi, T.N.; Chatzidakis, S.M.; Stoforos, N.G. The future of polyethylene terephthalate bottles: Challenges and sustainability. Packag. Technol. Sci. 2022, 35, 317–325. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wawrzyniak, P.; Karaszewski, W.; Różański, A. Cavitation and Solid-State Post-Condensation of Polyethylene Terephthalate: Literature Review. Materials 2024, 17, 5637. https://doi.org/10.3390/ma17225637
Wawrzyniak P, Karaszewski W, Różański A. Cavitation and Solid-State Post-Condensation of Polyethylene Terephthalate: Literature Review. Materials. 2024; 17(22):5637. https://doi.org/10.3390/ma17225637
Chicago/Turabian StyleWawrzyniak, Paweł, Waldemar Karaszewski, and Artur Różański. 2024. "Cavitation and Solid-State Post-Condensation of Polyethylene Terephthalate: Literature Review" Materials 17, no. 22: 5637. https://doi.org/10.3390/ma17225637
APA StyleWawrzyniak, P., Karaszewski, W., & Różański, A. (2024). Cavitation and Solid-State Post-Condensation of Polyethylene Terephthalate: Literature Review. Materials, 17(22), 5637. https://doi.org/10.3390/ma17225637