
First-Principles Study of Doped CdX(X = Te, Se) Compounds: Enhancing Thermoelectric Properties



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
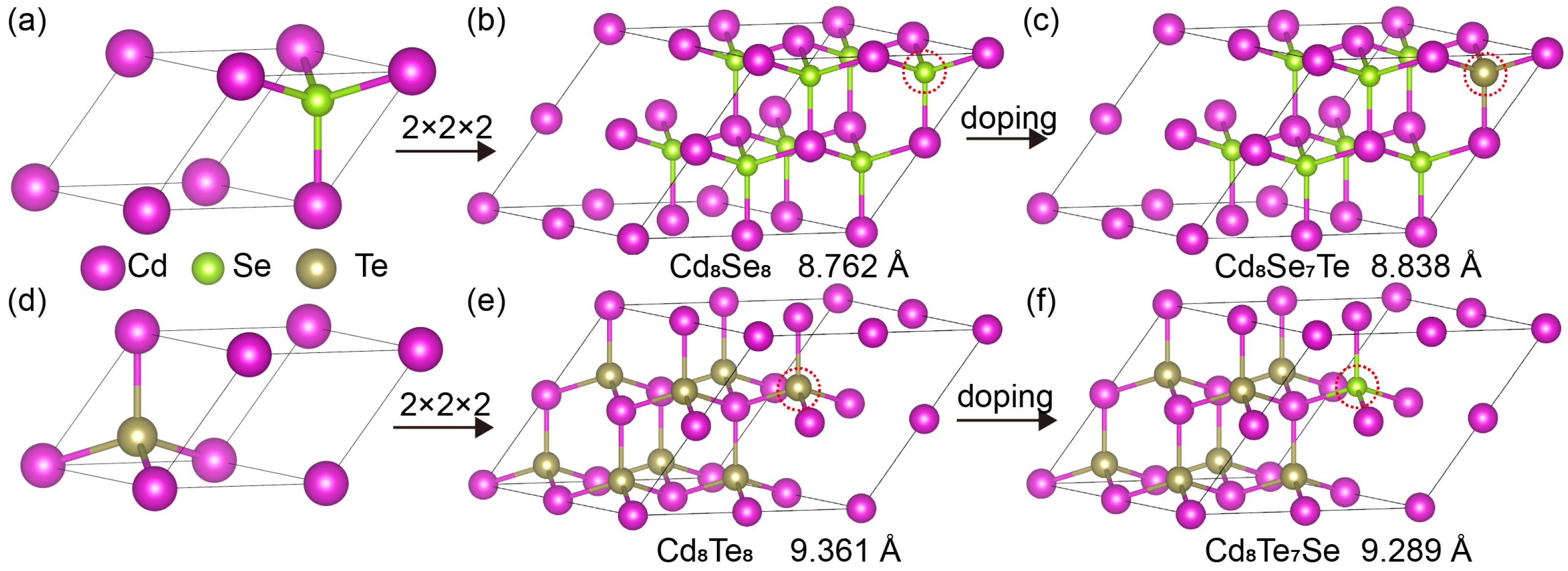
2. Computational Methods
3. Results and Discussion
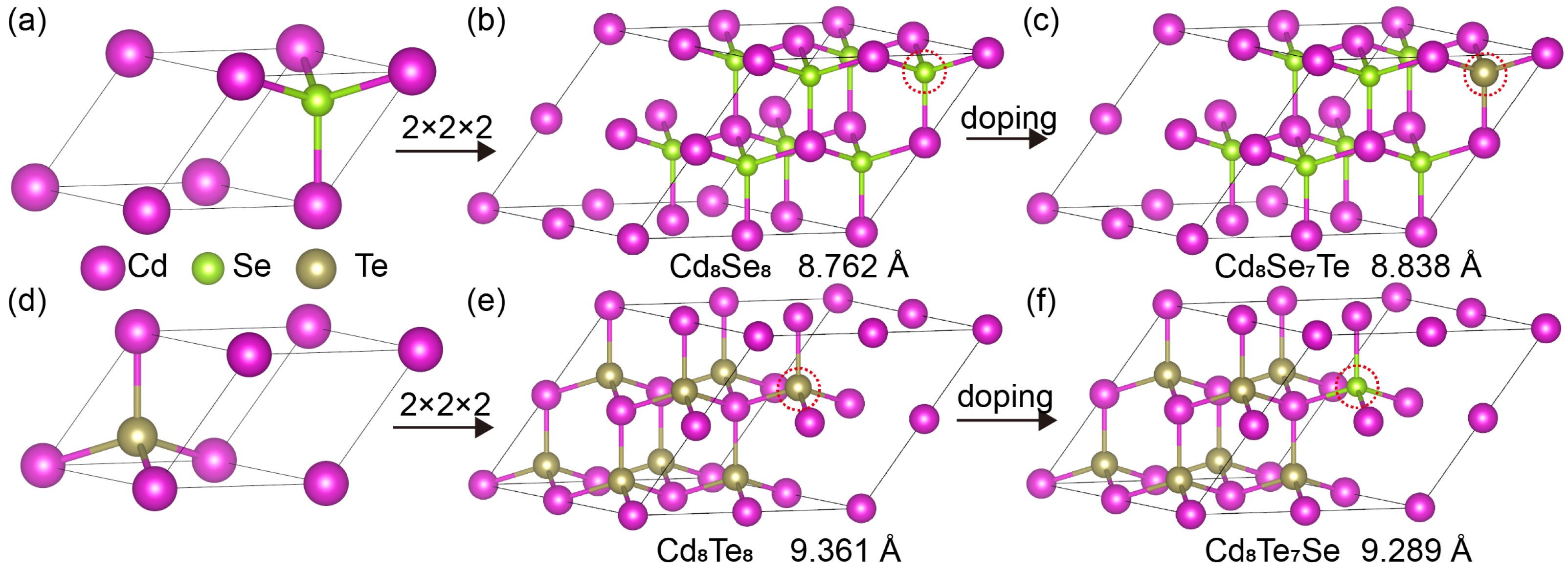
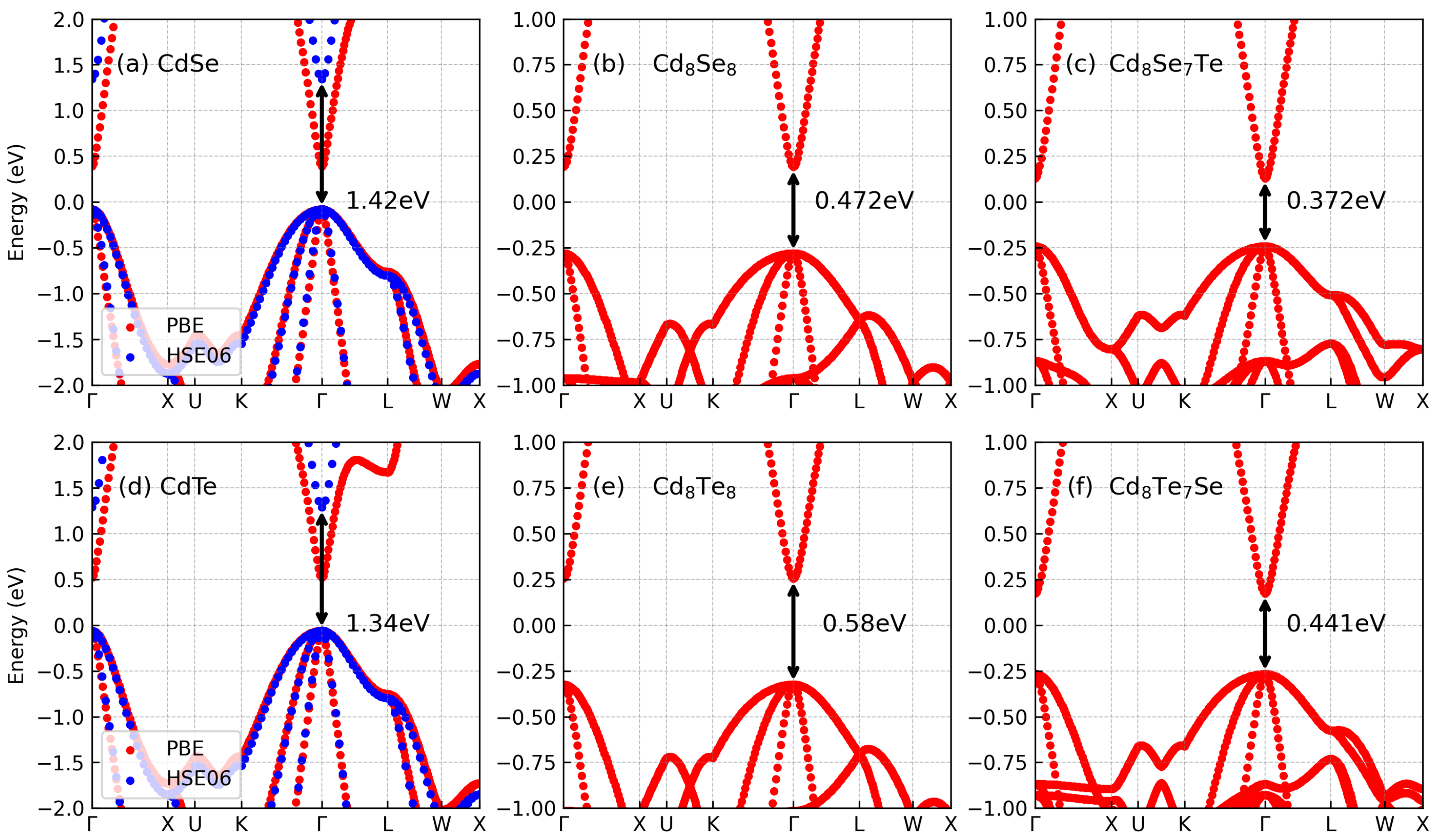
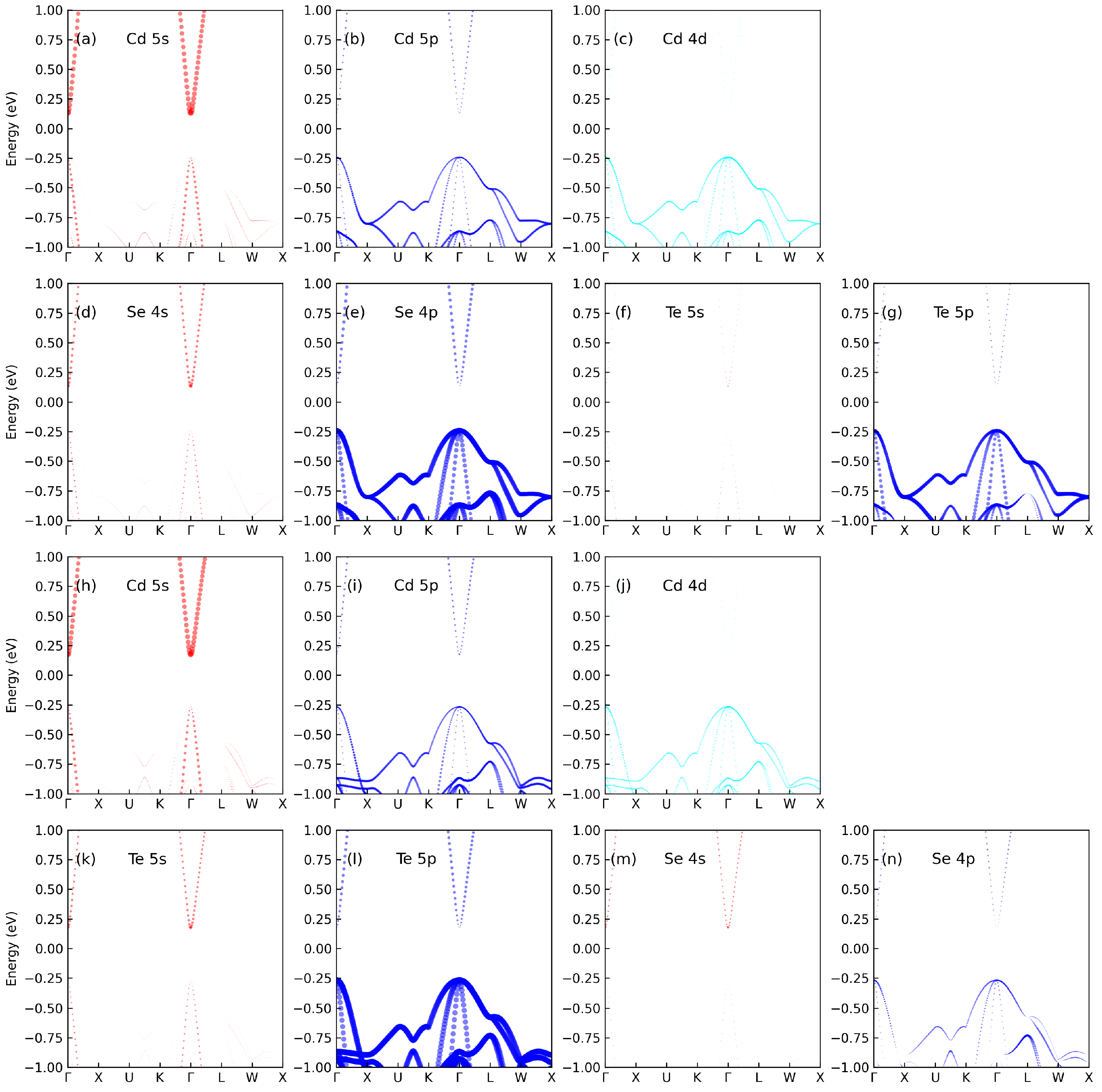
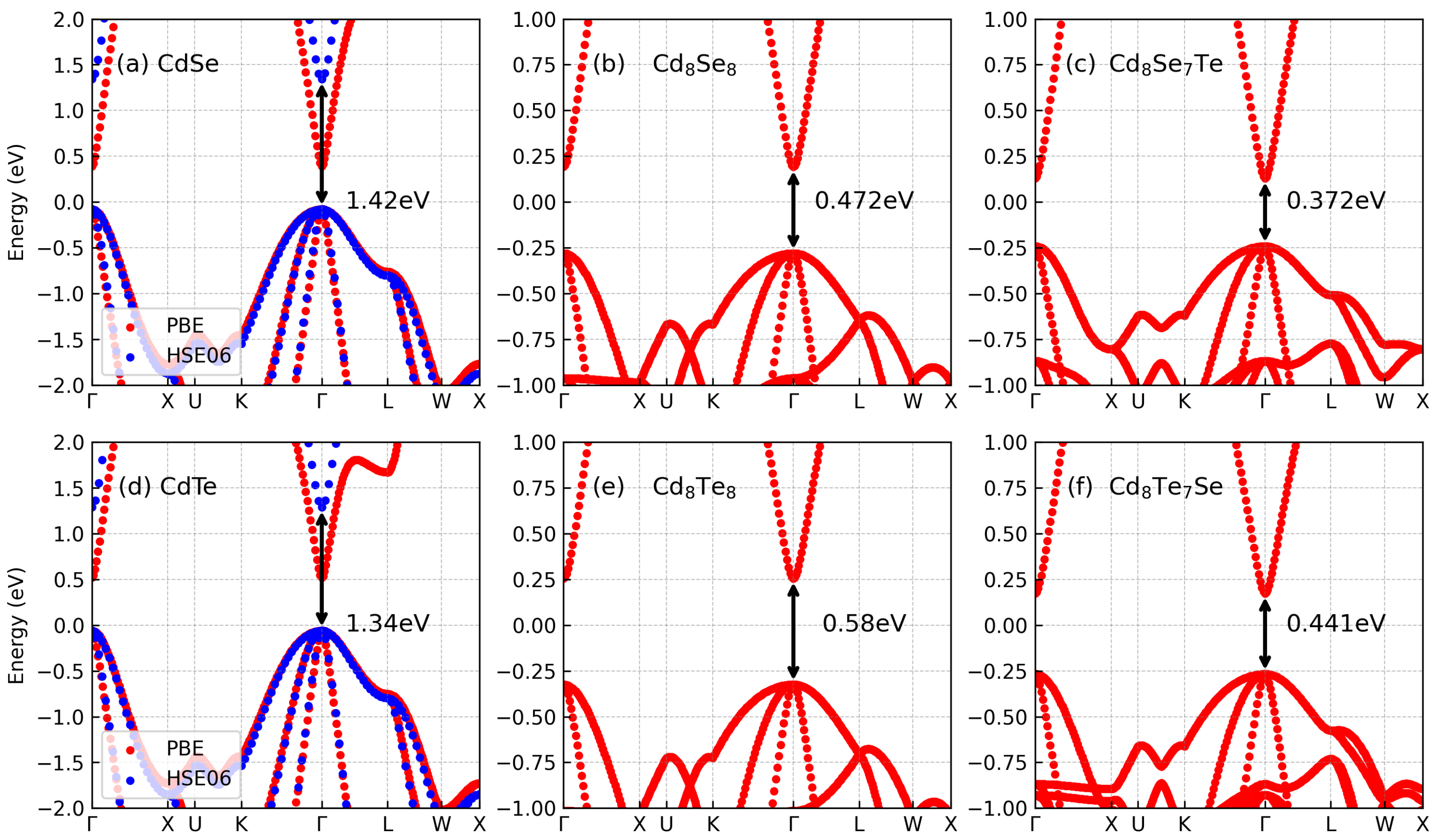
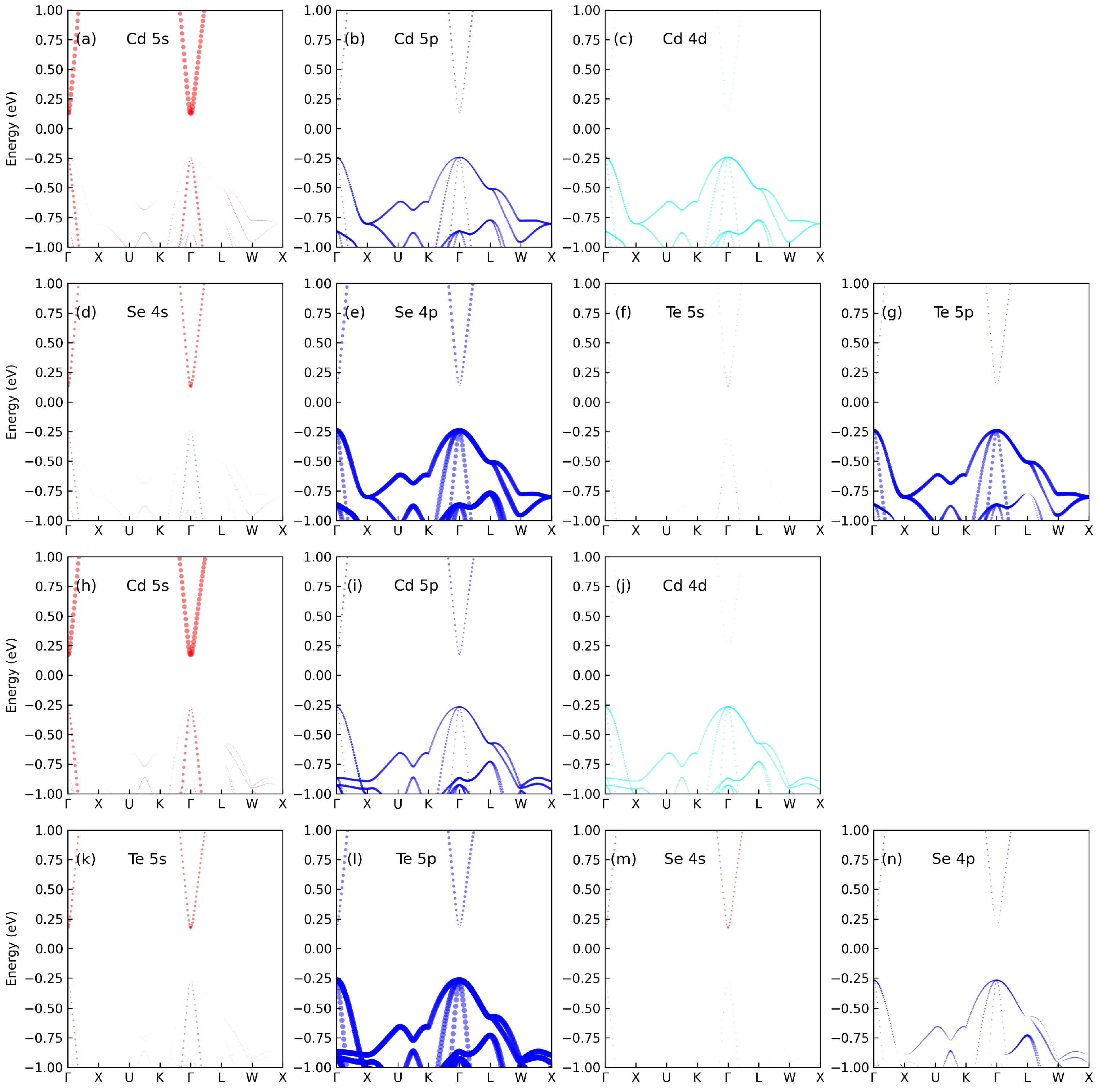
3.1. Electronic Properties
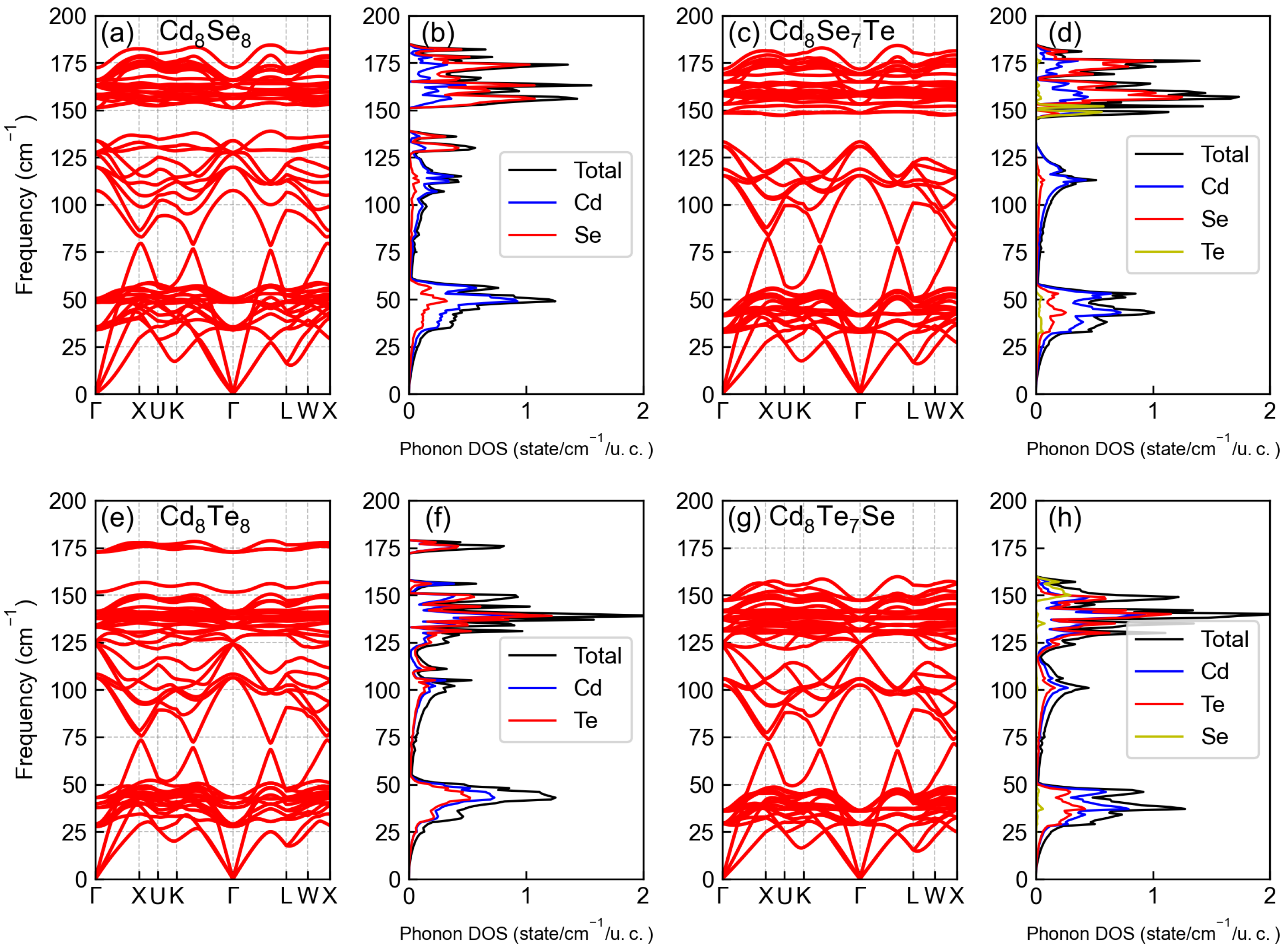
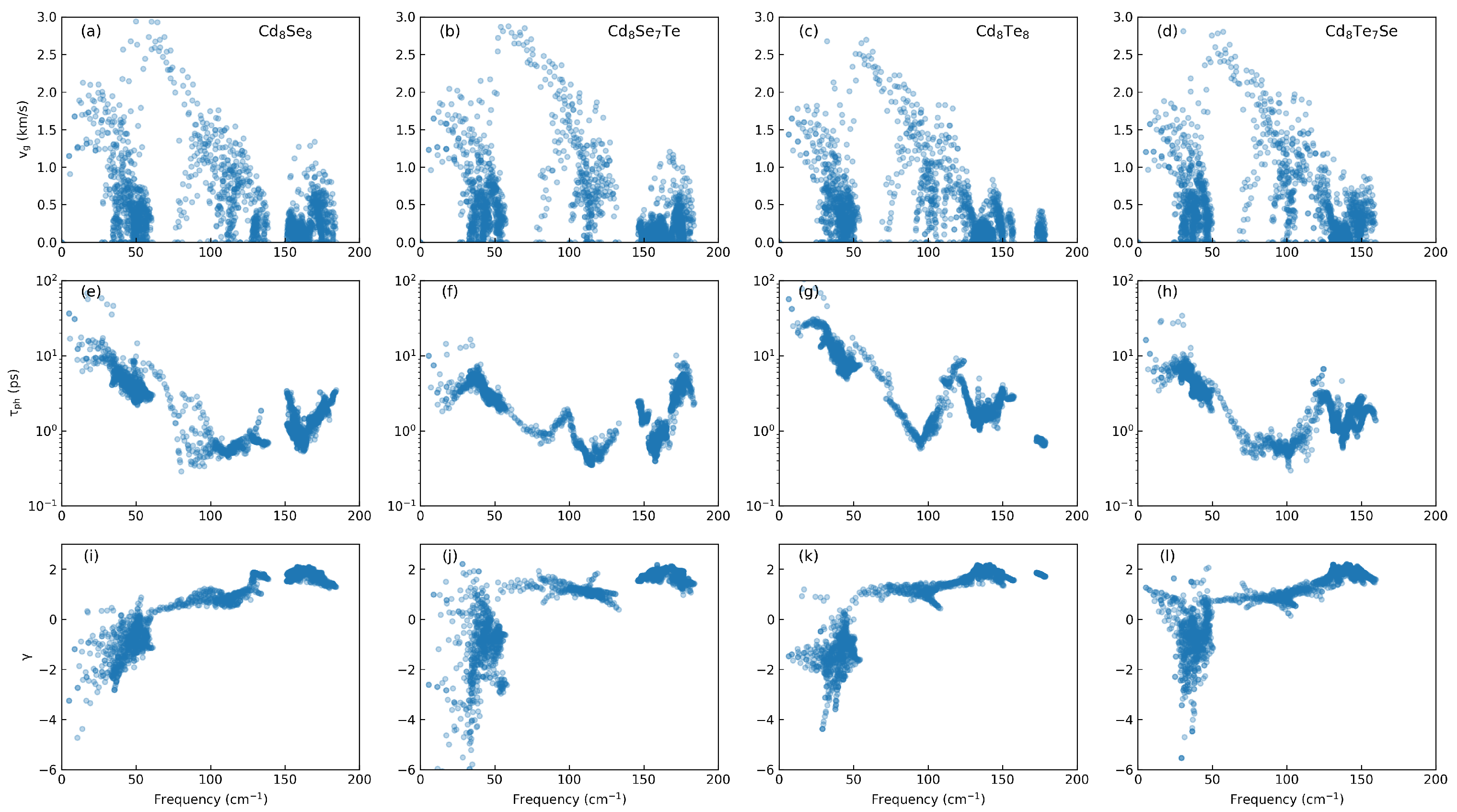
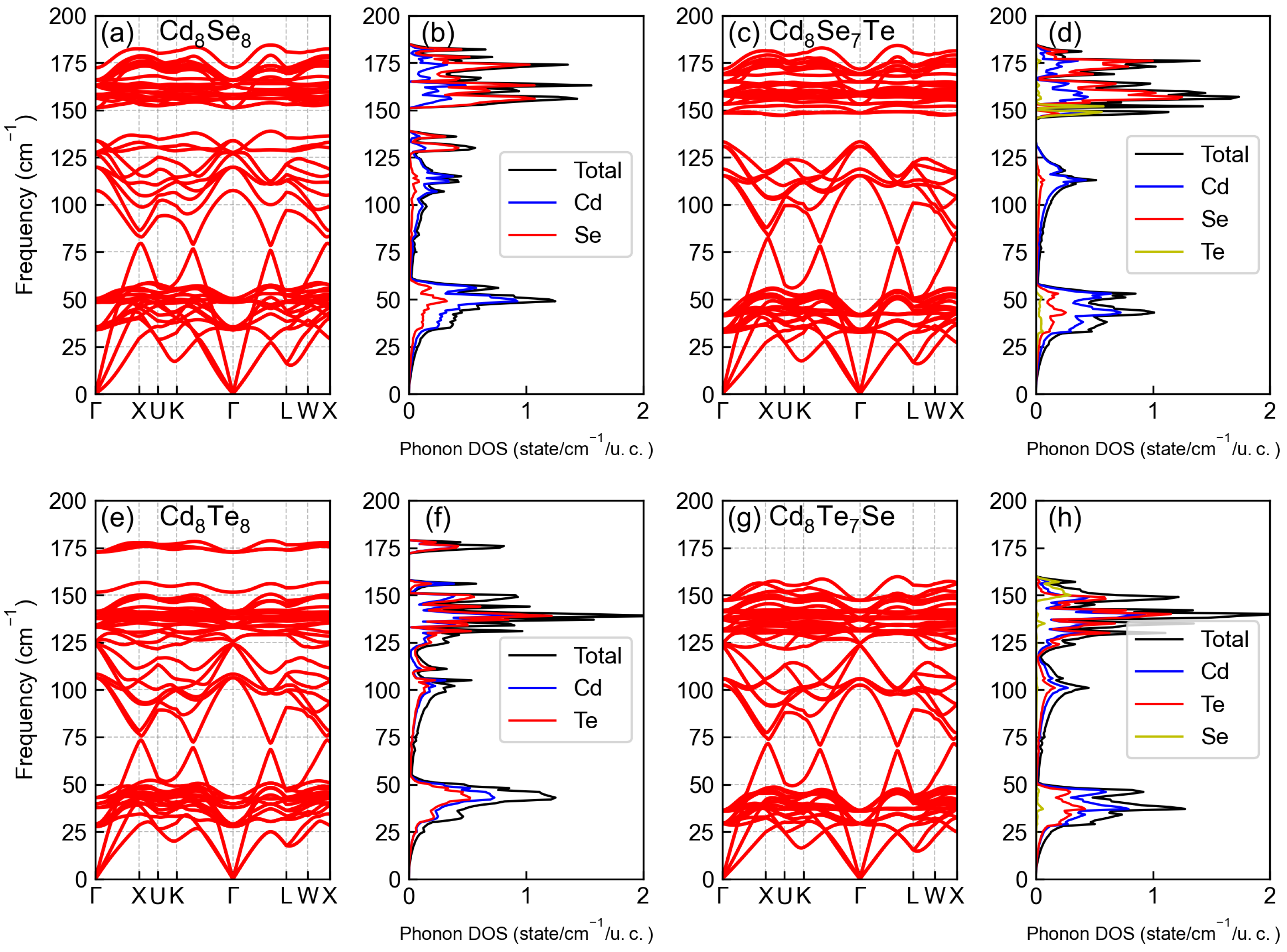
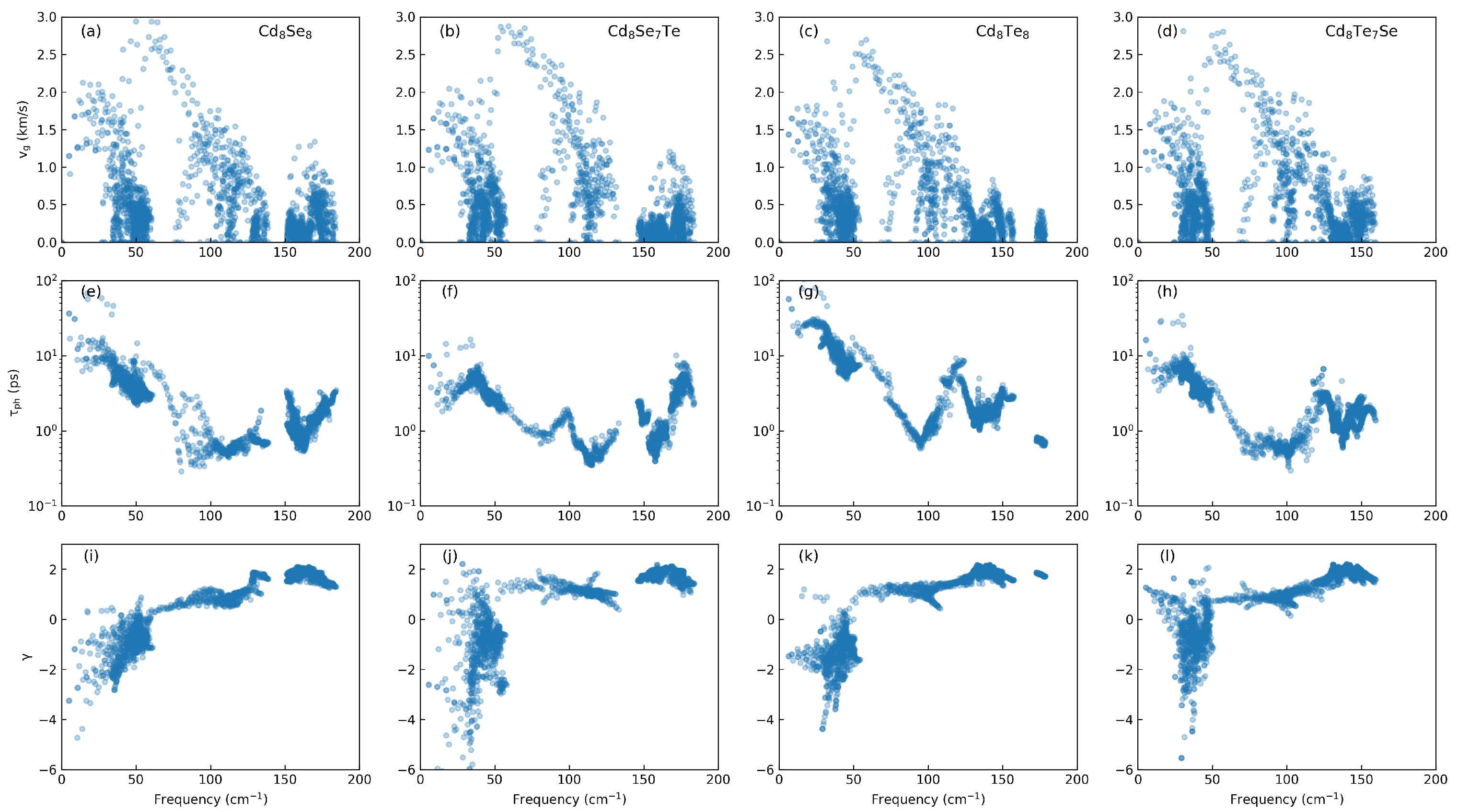
3.2. Phononic Properties
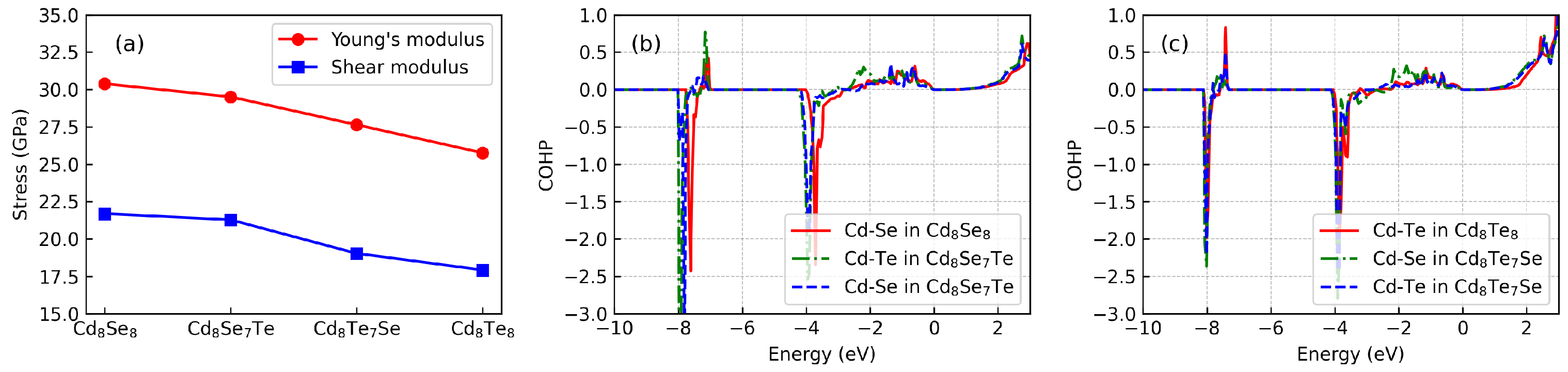
3.3. Mechanical Properties and Bonding Analysis
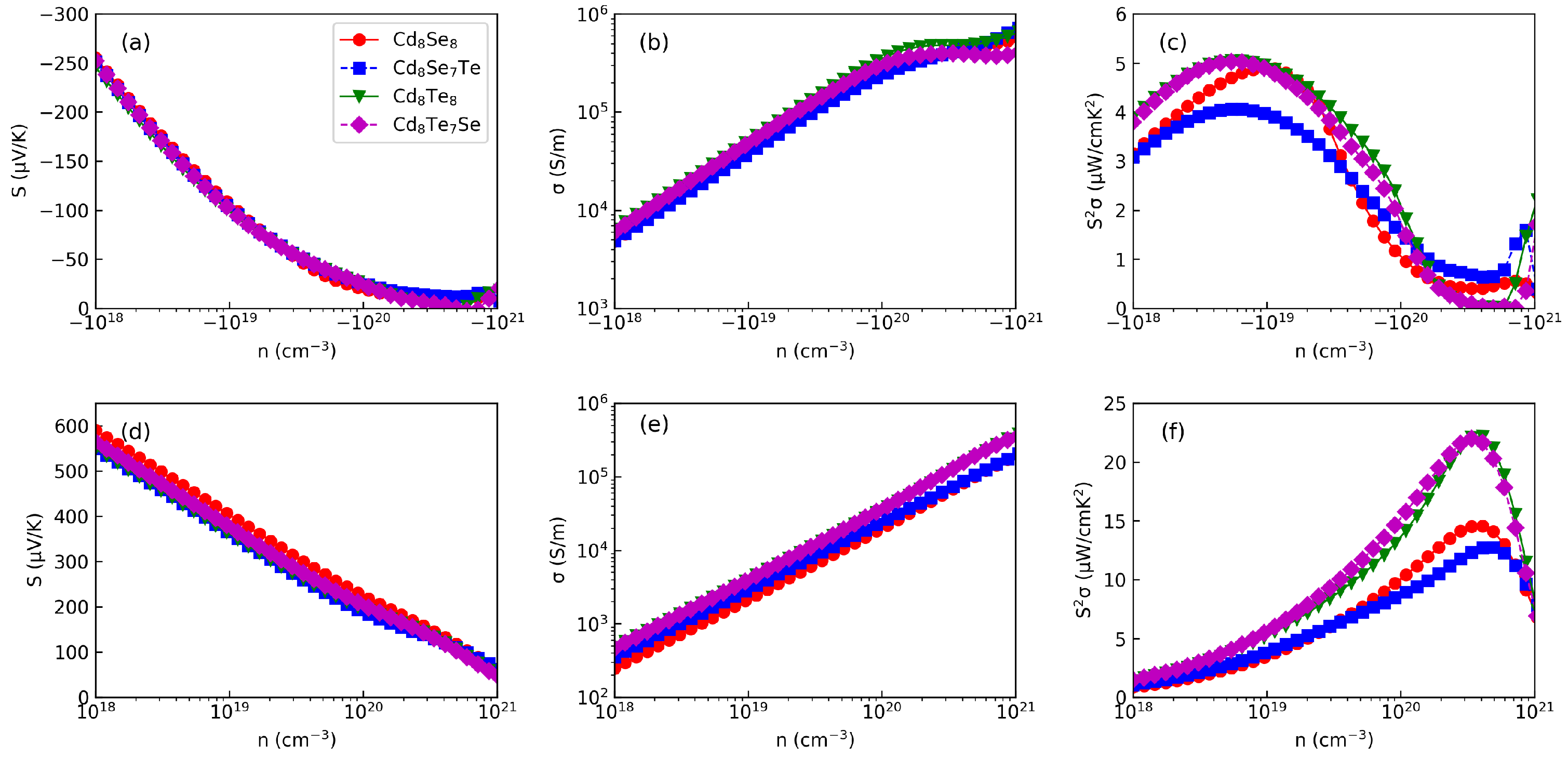
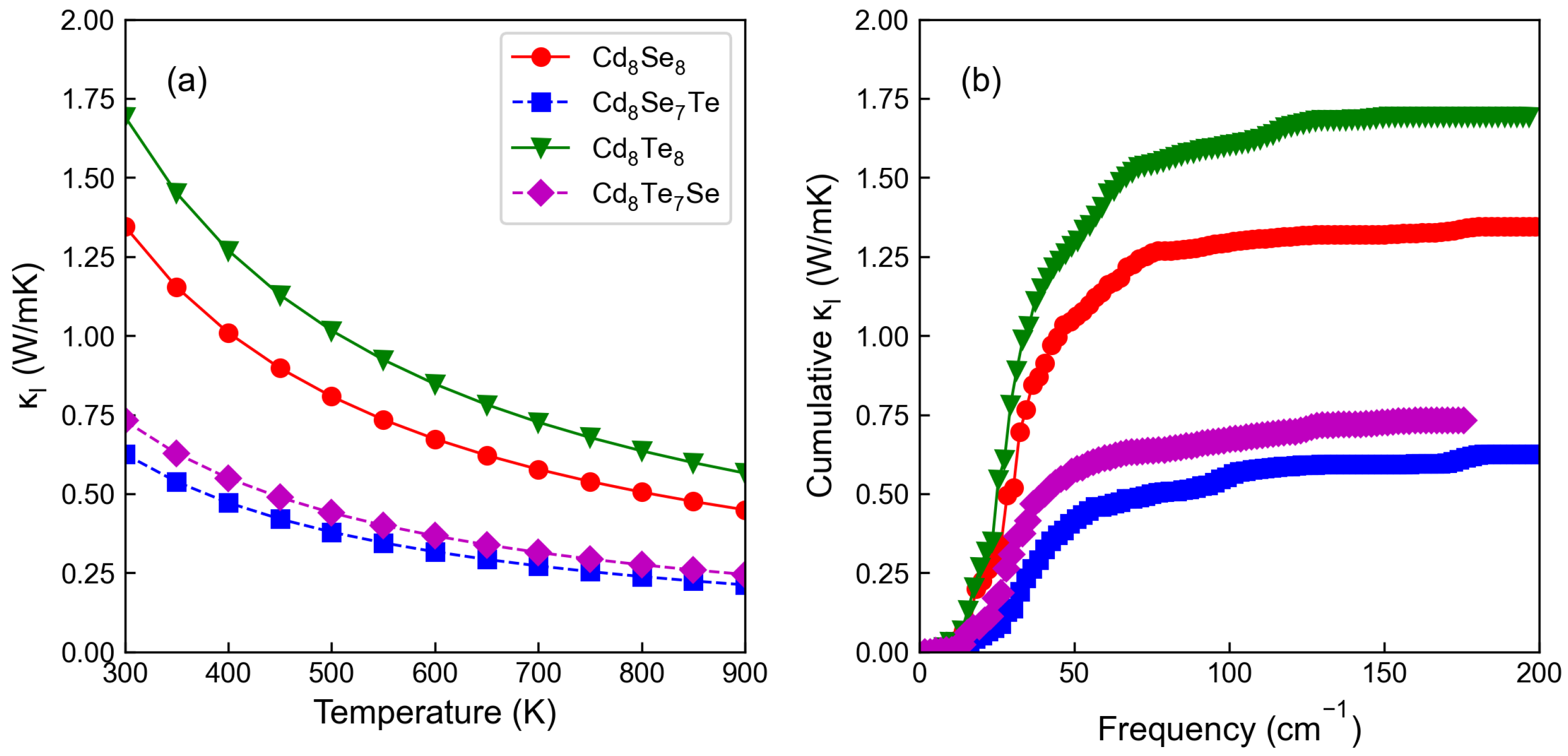
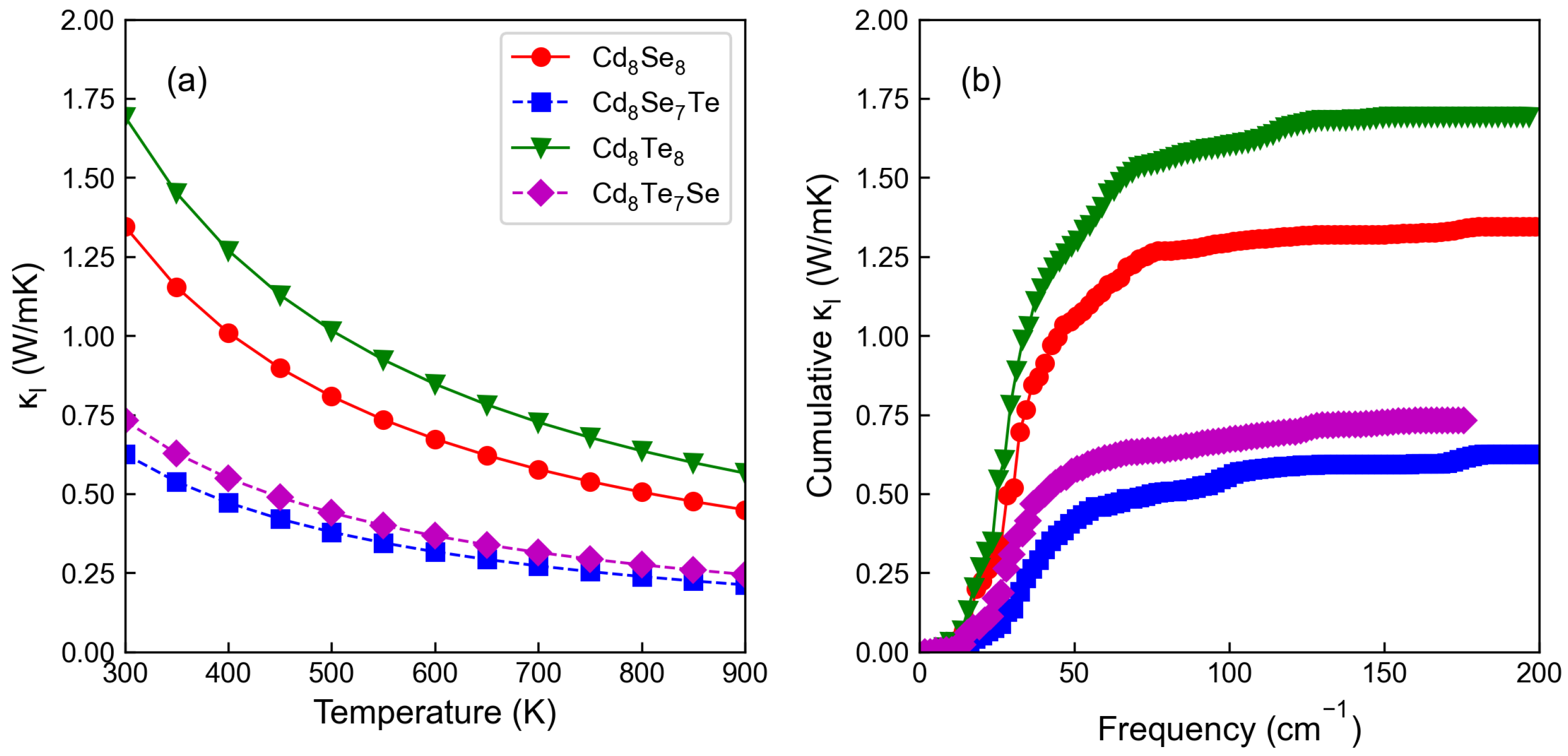
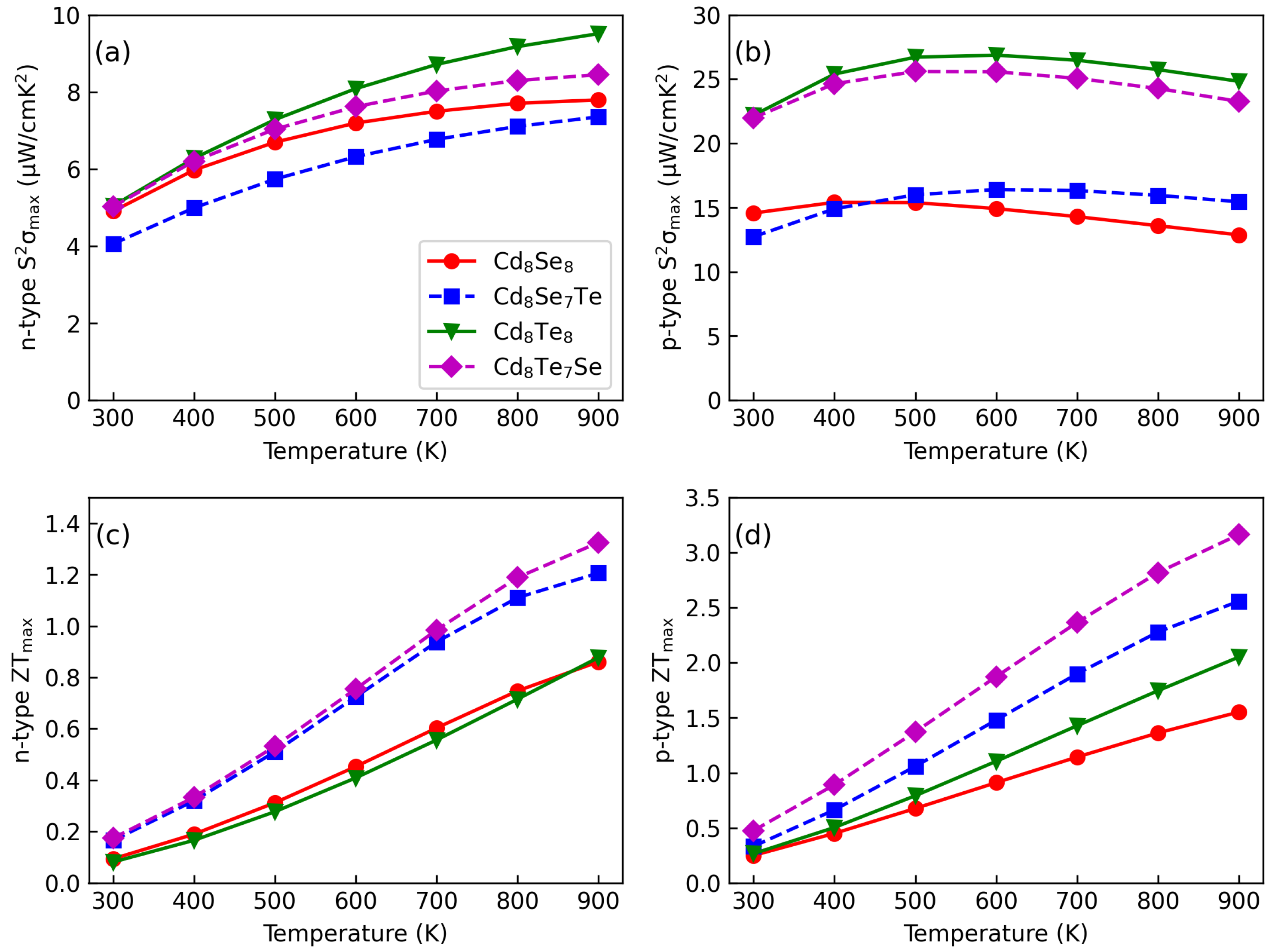
3.4. Thermoelectric Properties
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zoui, M.A.; Bentouba, S.; Stocholm, J.G.; Bourouis, M. A Review on Thermoelectric Generators: Progress and Applications. Energies 2020, 13, 3606. [Google Scholar] [CrossRef]
- Sootsman, J.R.; Chung, D.Y.; Kanatzidis, M.G. New and Old Concepts in Thermoelectric Materials. Angew. Chem. Int. Ed. 2009, 48, 8616–8639. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.L.; Zou, J.; Chen, Z.G. Advanced Thermoelectric Design: From Materials and Structures to Devices. Chem. Rev. 2020, 120, 7399–7515. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Ding, J.; Zou, Y.; Di, C.a.; Zhu, D. Chemical doping of organic semiconductors for thermoelectric applications. Chem. Soc. Rev. 2020, 49, 7210–7228. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.H.; Shi, X.L.; Ao, D.W.; Liu, W.D.; Chen, Y.X.; Li, F.; Chen, S.; Tian, X.Q.; Li, X.R.; Duan, J.Y.; et al. Rational band engineering and structural manipulations inducing high thermoelectric performance in n-type CoSb3 thin films. Nano Energy 2021, 81, 105683. [Google Scholar] [CrossRef]
- Hooshmand Zaferani, S.; Ghomashchi, R.; Vashaee, D. Strategies for engineering phonon transport in Heusler thermoelectric compounds. Renew. Sustain. Energy Rev. 2019, 112, 158–169. [Google Scholar] [CrossRef]
- Novak, T.G.; Kim, K.; Jeon, S. 2D and 3D nanostructuring strategies for thermoelectric materials. Nanoscale 2019, 11, 19684–19699. [Google Scholar] [CrossRef] [PubMed]
- Slade, T.J.; Pal, K.; Grovogui, J.A.; Bailey, T.P.; Male, J.; Khoury, J.F.; Zhou, X.; Chung, D.Y.; Snyder, G.J.; Uher, C.; et al. Contrasting SnTe-NaSbTe2 and SnTe-NaBiTe2 Thermoelectric Alloys: High Performance Facilitated by Increased Cation Vacancies and Lattice Softening. J. Am. Chem. Soc. 2020, 142, 12524–12535. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Cheng, H.; Le, Q.; Chen, R.; Li, J.; Ouyang, J. Giant Thermoelectric Properties of Ionogels with Cationic Doping. Adv. Energy Mater. 2022, 12, 2200858. [Google Scholar] [CrossRef]
- Perumal, S.; Samanta, M.; Ghosh, T.; Shenoy, U.S.; Bohra, A.K.; Bhattacharya, S.; Singh, A.; Waghmare, U.V.; Biswas, K. Realization of High Thermoelectric Figure of Merit in GeTe by Complementary Co-doping of Bi and In. Joule 2019, 3, 2565–2580. [Google Scholar] [CrossRef]
- Chen, C.; Jacobs, I.E.; Kang, K.; Lin, Y.; Jellett, C.; Kang, B.; Lee, S.B.; Huang, Y.; BaloochQarai, M.; Ghosh, R.; et al. Observation of Weak Counterion Size Dependence of Thermoelectric Transport in Ion Exchange Doped Conducting Polymers Across a Wide Range of Conductivities. Adv. Energy Mater. 2023, 13, 2202797. [Google Scholar] [CrossRef]
- Zhao, W.; Jin, K.; Fu, L.; Shi, Z.; Xu, B. Mass Production of Pt Single-Atom-Decorated Bismuth Sulfide for n-Type Environmentally Friendly Thermoelectrics. Nano Lett. 2022, 22, 4750–4757. [Google Scholar] [CrossRef]
- Van Du, N.; Rahman, J.U.; Huy, P.T.; Shin, W.H.; Seo, W.S.; Kim, M.H.; Lee, S. X-site aliovalent substitution decoupled charge and phonon transports in XYZ half-Heusler thermoelectrics. Acta Mater. 2019, 166, 650–657. [Google Scholar] [CrossRef]
- Han, S.; Dai, S.; Ma, J.; Ren, Q.; Hu, C.; Gao, Z.; Duc Le, M.; Sheptyakov, D.; Miao, P.; Torii, S.; et al. Strong phonon softening and avoided crossing in aliovalence-doped heavy-band thermoelectrics. Nat. Phys. 2023, 19, 1649–1657. [Google Scholar] [CrossRef]
- Baranets, S.; Balvanz, A.; Darone, G.M.; Bobev, S. On the Effects of Aliovalent Substitutions in Thermoelectric Zintl Pnictides. Varied Polyanionic Dimensionality and Complex Structural Transformations—The Case of Sr3ZnP3 vs. Sr3AlxZn1-xP3. Chem. Mater. 2022, 34, 4172–4185. [Google Scholar] [CrossRef]
- Musah, J.D.; Ilyas, A.M.; Venkatesh, S.; Mensah, S.; Kwofie, S.; Roy, V.A.L.; Wu, C.M.L. Isovalent substitution in metal chalcogenide materials for improving thermoelectric power generation—A critical review. Nano Res. Energy 2022, 1, e9120034. [Google Scholar] [CrossRef]
- Lee, K.H.; Kim, H.S.; Kim, M.; Roh, J.W.; Lim, J.H.; Kim, W.J.; Kim, S.-i.; Lee, W. Isovalent sulfur substitution to induce a simultaneous increase in the effective mass and weighted mobility of a p-type Bi–Sb–Te alloy: An approach to enhance the thermoelectric performance over a wide temperature range. Acta Mater. 2021, 205, 116578. [Google Scholar] [CrossRef]
- He, X.; Zhang, H.; Nose, T.; Katase, T.; Tadano, T.; Ide, K.; Ueda, S.; Hiramatsu, H.; Hosono, H.; Kamiya, T. Degenerated Hole Doping and Ultra-Low Lattice Thermal Conductivity in Polycrystalline SnSe by Nonequilibrium Isovalent Te Substitution. Adv. Sci. 2022, 9, 2105958. [Google Scholar] [CrossRef]
- Gao, B.; Tang, J.; Meng, F.; Li, W. Band manipulation for high thermoelectric performance in SnTe through heavy CdSe-alloying. J. Mater. 2019, 5, 111–117. [Google Scholar] [CrossRef]
- Tao, Q.; Deng, R.; Li, J.; Yan, Y.; Su, X.; Poudeu, P.F.P.; Tang, X. Enhanced Thermoelectric Performance of Bi0.46Sb1.54Te3 Nanostructured with CdTe. ACS Appl. Mater. Interfaces 2020, 12, 26330–26341. [Google Scholar] [CrossRef]
- Giannozzi, P.; Baroni, S.; Bonini, N.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Chiarotti, G.L.; Cococcioni, M.; Dabo, I.; et al. Quantum ESPRESSO: A modular and open-source software project for quantum simulations of materials. J. Phys. Condens. Matter 2009, 21, 395502. [Google Scholar] [CrossRef] [PubMed]
- Giannozzi, P.; Andreussi, O.; Brumme, T.; Bunau, O.; Nardelli, M.B.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Cococcioni, M.; et al. Advanced capabilities for materials modelling with Quantum ESPRESSO. J. Phys. Condens. Matter 2017, 29, 465901. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Prandini, G.; Marrazzo, A.; Castelli, I.E.; Mounet, N.; Marzari, N. Precision and efficiency in solid-state pseudopotential calculations. NPJ Comput. Mater. 2018, 4, 72. [Google Scholar] [CrossRef]
- Heyd, J.; Scuseria, G.E.; Ernzerhof, M. Hybrid functionals based on a screened Coulomb potential. J. Chem. Phys. 2003, 118, 8207–8215. [Google Scholar] [CrossRef]
- Krukau, A.V.; Vydrov, O.A.; Izmaylov, A.F.; Scuseria, G.E. Influence of the exchange screening parameter on the performance of screened hybrid functionals. J. Chem. Phys. 2006, 125, 224106. [Google Scholar] [CrossRef] [PubMed]
- Maintz, S.; Deringer, V.L.; Tchougréeff, A.L.; Dronskowski, R. Analytic projection from plane-wave and PAW wavefunctions and application to chemical-bonding analysis in solids. J. Comput. Chem. 2013, 34, 2557–2567. [Google Scholar] [CrossRef] [PubMed]
- Maintz, S.; Deringer, V.L.; Tchougréeff, A.L.; Dronskowski, R. LOBSTER: A tool to extract chemical bonding from plane-wave based DFT. J. Comput. Chem. 2016, 37, 1030–1035. [Google Scholar] [CrossRef] [PubMed]
- Hill, R. The Elastic Behaviour of a Crystalline Aggregate. Proc. Phys. Soc. Sect. 1952, 65, 349. [Google Scholar] [CrossRef]
- Yalameha, S.; Nourbakhsh, Z.; Vashaee, D. ElATools: A tool for analyzing anisotropic elastic properties of the 2D and 3D materials. Comput. Phys. Commun. 2022, 271, 108195. [Google Scholar] [CrossRef]
- Momma, K.; Izumi, F. VESTA: A three-dimensional visualization system for electronic and structural analysis. J. Appl. Crystallogr. 2008, 41, 653–658. [Google Scholar] [CrossRef]
- Madsen, G.K.; Singh, D.J. BoltzTraP. A code for calculating band-structure dependent quantities. Comput. Phys. Commun. 2006, 175, 67–71. [Google Scholar] [CrossRef]
- Samsonidze, G.; Kozinsky, B. Accelerated Screening of Thermoelectric Materials by First-Principles Computations of Electron–Phonon Scattering. Adv. Energy Mater. 2018, 8, 1800246. [Google Scholar] [CrossRef]
- Li, W.; Carrete, J.; Katcho, N.A.; Mingo, N. ShengBTE: A solver of the Boltzmann transport equation for phonons. Comput. Phys. Commun. 2014, 185, 1747–1758. [Google Scholar] [CrossRef]
- Gopal, P.; Fornari, M.; Curtarolo, S.; Agapito, L.A.; Liyanage, L.S.I.; Nardelli, M.B. Improved predictions of the physical properties of Zn- and Cd-based wide band-gap semiconductors: A validation of the ACBN0 functional. Phys. Rev. B 2015, 91, 245202. [Google Scholar] [CrossRef]
- Polak, M.P.; Scharoch, P.; Kudrawiec, R. The effect of isovalent doping on the electronic band structure of group IV semiconductors. J. Phys. Appl. Phys. 2020, 54, 085102. [Google Scholar] [CrossRef]
- Snyder, G.J.; Toberer, E.S. Complex thermoelectric materials. Nat. Mater. 2008, 7, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Yadav, A.; Deshmukh, P.; Roberts, K.; Jisrawi, N.; Valluri, S. An analytic study of the Wiedemann–Franz law and the thermoelectric figure of merit. J. Phys. Commun. 2019, 3, 105001. [Google Scholar] [CrossRef]
- Ahamed, S.T.; Kulsi, C.; Kirti.; Banerjee, D.; Srivastava, D.N.; Mondal, A. Synthesis of multifunctional CdSe and Pd quantum dot decorated CdSe thin films for photocatalytic, electrocatalytic and thermoelectric applications. Surfaces Interfaces 2021, 25, 101149. [Google Scholar] [CrossRef]
- Cao, W.; Wang, Z.; Miao, L.; Shi, J.; Xiong, R. Thermoelectric Properties of Strained β-Cu2Se. ACS Appl. Mater. Interfaces 2021, 13, 34367–34373. [Google Scholar] [CrossRef]
- Cao, W.; Wang, Z.; Miao, L.; Shi, J.; Xiong, R. Extremely Anisotropic Thermoelectric Properties of SnSe Under Pressure. Energy Environ. Mater. 2023, 6, e12361. [Google Scholar] [CrossRef]
- Huang, L.F.; Zeng, Z. Roles of Mass, Structure, and Bond Strength in the Phonon Properties and Lattice Anharmonicity of Single-Layer Mo and W Dichalcogenides. J. Phys. Chem. C 2015, 119, 18779–18789. [Google Scholar] [CrossRef]
- Yang, X.X.; Li, J.W.; Zhou, Z.F.; Wang, Y.; Yang, L.W.; Zheng, W.T.; Sun, C.Q. Raman spectroscopic determination of the length, strength, compressibility, Debye temperature, elasticity, and force constant of the C-C bond in graphene. Nanoscale 2012, 4, 502–510. [Google Scholar] [CrossRef] [PubMed]
- Delaire, O.; Ma, J.; Marty, K.; May, A.F.; McGuire, M.A.; Du, M.H.; Singh, D.J.; Podlesnyak, A.; Ehlers, G.; Lumsden, M.D.; et al. Giant anharmonic phonon scattering in PbTe. Nat. Mater. 2011, 10, 614–619. [Google Scholar] [CrossRef]
- Kundu, A.; Ma, J.; Carrete, J.; Madsen, G.K.H.; Li, W. Anomalously large lattice thermal conductivity in metallic tungsten carbide and its origin in the electronic structure. Mater. Today Phys. 2020, 13, 100214. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jin, J.; Lv, F.; Cao, W.; Wang, Z. First-Principles Study of Doped CdX(X = Te, Se) Compounds: Enhancing Thermoelectric Properties. Materials 2024, 17, 1797. https://doi.org/10.3390/ma17081797
Jin J, Lv F, Cao W, Wang Z. First-Principles Study of Doped CdX(X = Te, Se) Compounds: Enhancing Thermoelectric Properties. Materials. 2024; 17(8):1797. https://doi.org/10.3390/ma17081797
Chicago/Turabian StyleJin, Junfeng, Fang Lv, Wei Cao, and Ziyu Wang. 2024. "First-Principles Study of Doped CdX(X = Te, Se) Compounds: Enhancing Thermoelectric Properties" Materials 17, no. 8: 1797. https://doi.org/10.3390/ma17081797
APA StyleJin, J., Lv, F., Cao, W., & Wang, Z. (2024). First-Principles Study of Doped CdX(X = Te, Se) Compounds: Enhancing Thermoelectric Properties. Materials, 17(8), 1797. https://doi.org/10.3390/ma17081797