1. Introduction
A primary way in which proteins evolve is through rearrangement of their functional/structural units, known as
protein domains [
1,
2]. The domains are independent folding and functional modules, and thus they exhibit conserved sequence segments. Prediction algorithms exploited this information and used, as input features, the domain composition of a protein for various tasks. For example, [
3] classified the cellular location, and [
4,
5] predicted the associated Gene Ontology (GO) terms. There exist two ways to represent domains; either by the linear order in a protein,
domain architectures [
6], or by a graph where nodes are domains and edges connect domains that co-exist in a protein [
1,
2].
Moreover, [
7] investigated whether the domain architectures had grammar as a natural spoken language. They compared the bi-gram entropy of
domain architectures for Pfam domains [
8] to the respective entropy of the English language, showing that although it was lower than the English language, it was significantly different from a language produced after shuffling the domains. Prior to this result, methods had exploited the
domain architecture representation to various applications, such as fast homology search [
9] and retrieval of similar proteins [
10].
Word embeddings are unsupervised learning methods which have, as input, large corpora, and where they output a dense vector representation of words contained in the sentences of these documents based on the distributional semantic hypothesis, that is, the meaning of a word can be understood by its context. Thus, a word vector represents local linguistic features, such as lexical or semantical information, of the respective word. Several methods to train word embeddings have been established, for example, [
11,
12,
13]. These representations have been shown to hold several properties, such as analogy and grouping of semantically similar words [
14,
15]. Importantly, these properties are learned without the need of a labeled data set. Word embeddings are currently the mainstream input for neural networks in the Natural Language Processing (NLP) field, as firstly, they reduce the feature space, compared to 1-hot representation, and secondly, they provide word features that encapsulate relations between words based on linguistic features. The use of word embeddings improved the performance on most of the tasks, such as sentiment analysis or Named Entity Recognition (NER) [
16].
Various methods used to create embeddings for proteins have been proposed [
17,
18,
19,
20,
21,
22,
23]. ProtVec fragmented the protein sequence in 3-mers for all possible starting shifts, then learned embeddings for each 3-mer and represented the respective protein as the average of its constituting 3-mer vectors [
17]. SeqVec utilized and extended the Embeddings from Language Models (ELMo) [
24] to learn a dense representation per amino acid residue, resulting in matrix representations of proteins, created by concatenating their learned residue vectors [
21].
Focusing on their word segmentation, we note that learning embeddings for each amino acid or 3-mer may not always reflect evolutionary signals. That is, a pair of proteins with low sequence similarity is still a member of the same protein super-family, preserving similar function [
25].
The previous embedding approaches evaluated the learned representations intrinsically, in a qualitative manner. They averaged out the whole protein amino acid embeddings to compute the aggregated vector. Then, known biological characteristics of proteins are used, such as biophysical, chemical, structural, enzymatic, and taxonomic, as distinct colors in a reduced 2-D embedding space. In such visualizations, previous embedding approaches reported the appearance of distinct clusters of proteins, each consisting of proteins with similar properties. For downstream evaluation, they measured the improvement of performance in downstream tasks.
Concerning the qualitative intrinsic evaluation, two caveats exist. First, researchers averaged out the protein amino acid vectors, where consequently, this qualitative evaluation is not related in a straightforward way with each learned embedding vector trained per amino-acid. In addition, this averaging-out operation may not reveal the function of the most important sites of a protein, meaning the comparative result holds a low degree of biological significance. Second, we argue that the presented qualitative evaluations lack the ability to assess different learned embeddings in a sophisticated manner. This is because there is no systematic way to quantitatively compare 2-D plots of reduced embedding spaces, each produced by a protein-embedding method in investigation.
Indeed for word embeddings, there has been an increase in methods to evaluate word representations intrinsically and in a quantitative manner, such as [
26,
27]. Having such evaluation metrics allows us to validate the knowledge acquired
per each word vector and use the best-performing space for downstream tasks. However, intrinsic evaluations of current amino acid embedding representations are prevented by incomplete biological metadata at amino acid level, for all disposed proteins, in the UniProtKnowledgeBase (UniProtKB) [
28].
To address this limitation in quantitative intrinsic evaluations of protein sequence embeddings, we present our approach with five major contributions:
Our dom2vec approach is developed, in which words are InterPro annotations and sentences are the domain architectures. Then, we use the word2vec method to learn the embedding vector representation for each InterPro annotation.
A quantitative intrinsic evaluation method is established based on the most significant biological information for a domain—its structure and function. First, we evaluated the learned embedding space for domain hierarchy comparing known domain parent–children relations to cosine similarity of the parent domain. Then, we investigated the performance of a nearest neighbor classifier,
, to predict the secondary structure class provided by SCOPe secondary structure class [
29] and the Enzyme Commission (EC) primary class. Finally, we equally examined the performance of the
classifier to predict the GO molecular function class for three example model organisms and one human pathogen.
Strikingly, we observed that reaches adequate accuracy, compared to on randomized domains vectors, for secondary structure, enzymatic function, and GO molecular function. Thus, we hypothesized an analogy between word embedding clustering by local linguistic features and protein domains clustering by domain structure and function.
To evaluate our embeddings extrinsically, we inputted the learned domains embeddings to simple neural networks and compared their performance with state-of-the-art protein sequence embeddings in three full-protein tasks. We surpassed both SeqVec and ProtVec for the toxin presence and enzymatic primary function prediction task, and we reported comparable results in the cellular location prediction task.
The remainder of the paper is organized as follows: related work on protein embeddings is reviewed in
Section 2. The methodology used to train and evaluate dom2vec embeddings is described in
Section 3. The intrinsic and extrinsic evaluation results are presented in
Section 4. In
Section 5, we conclude.
2. Background
Current studies on protein embeddings are evaluated intrinsically and extrinsically. In extrinsic evaluation, prediction measures, like performance on a supervised learning task, are most commonly used to evaluate the quality of embeddings. For example, the ProtVec work [
17] evaluated their proposed embeddings extrinsically by measuring their performance in predicting protein family and disorder. SeqVec [
21] assessed their embeddings extrinsically by measuring performance on protein-level tasks, prediction of sub-cellular localization and water solubility, and residue-level tasks, and prediction of the functional effect of single amino acid mutations. However, extrinsic evaluation methods are based on a downstream prediction task, thus not measuring the biological information captured by each learned subsequence vector separately.
Previous studies evaluated the quality of their sequence embeddings
intrinsically, by averaging the amino acid embedding vectors per protein and then drawing t-SNE visualizations [
30] using distinct biological labels of a protein as colors, such as taxonomy, SCOPe, and EC primary class. However, this qualitative assessment hinders the selection of the best-performing embeddings, irrespective of the downstream task, because there is not a sophisticated method to rank 2-D visualizations.
Nevertheless, in NLP, the quality of a learned word embedding space is often evaluated intrinsically in a quantitative manner by considering relationships among words, such as analogies. Compared to qualitative evaluation, quantitative intrinsic evaluation enables assessment of the degree of biological information captured by the embeddings. This advantage allows us to choose the best set of parameters to create the embeddings that contain the highest degree of meaningful information without choosing a specific downstream task.
From all discussed protein embeddings studies, only [
23] developed quantitative intrinsic evaluation methods. To benchmark their Pfam domain embeddings, they used the following three experiments. First, they benchmarked the performance of the nearest neighbor classifier predicting the three main GO ontologies of a Pfam using its embedding vector. Second, they assessed the Matthew’s correlation coefficient [
31] between Pfam embedding and first-order Markov encodings. They also assessed the vector arithmetic to compare GO conceptual binary assignment—for example, one pair was intracellular (GO:0005622) vs. extracellular (GO:0005615).
Our approach differs from [
23] in four main points. First, we trained embeddings for all domain annotations of all proteins available in Interpro. That is, we included all available InterPro annotations, consisting of super-family, family, single domains, and functional sites, as “words” input to the
word2vec method. Therefore, we used a broader set of annotations for the whole spectrum of organisms. Besides,
word2vec was developed for sentences of natural languages, which have a moderate number of words. In order to copy with this assumption for the sentence length, we resolved overlapping and redundant annotations in order to increase the number of InterPro annotations, making our input more
suitable for the
word2vec method. Second, we benchmarked over the two
word2vec models (CBOW and SKIP) and their parameters for each experiment of our quantitative intrinsic evaluation step, and consequently, we used our assessment to choose the best embedding space. Third, we established three unique intrinsic evaluation benchmarks for domain hierarchy, SCOPe secondary structure, and EC primary class. Lastly, our approach was also extrinsically evaluated on three downstream tasks in order to show that
dom2vec embeddings can surpass or be comparable to state-of-the-art protein sequence embeddings.
3. Materials and Methods
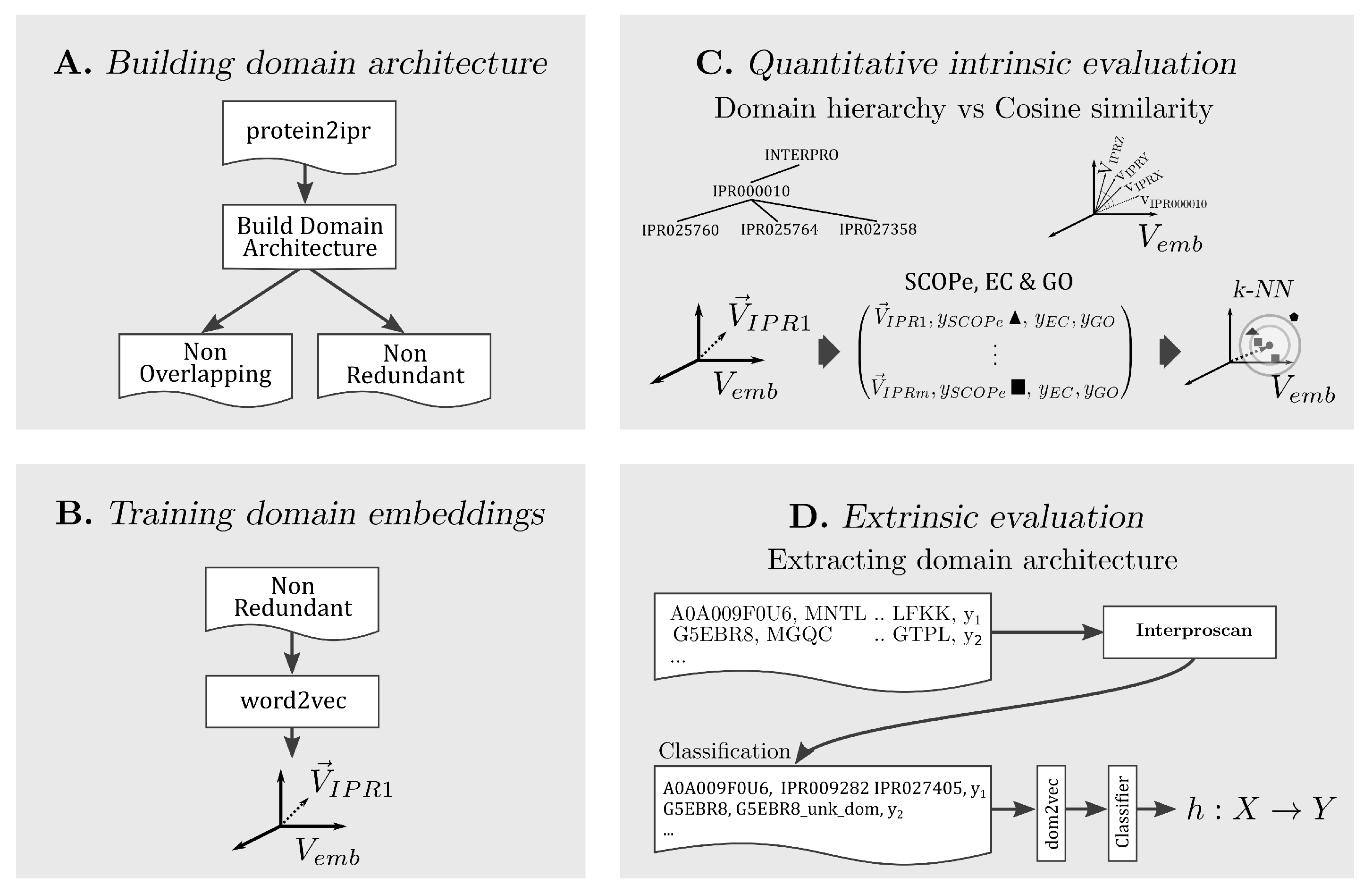
In the following, the methodology for each part of our approach is explained. A conceptual summary is presented in
Figure 1.
3.1. Building Domain Architectures
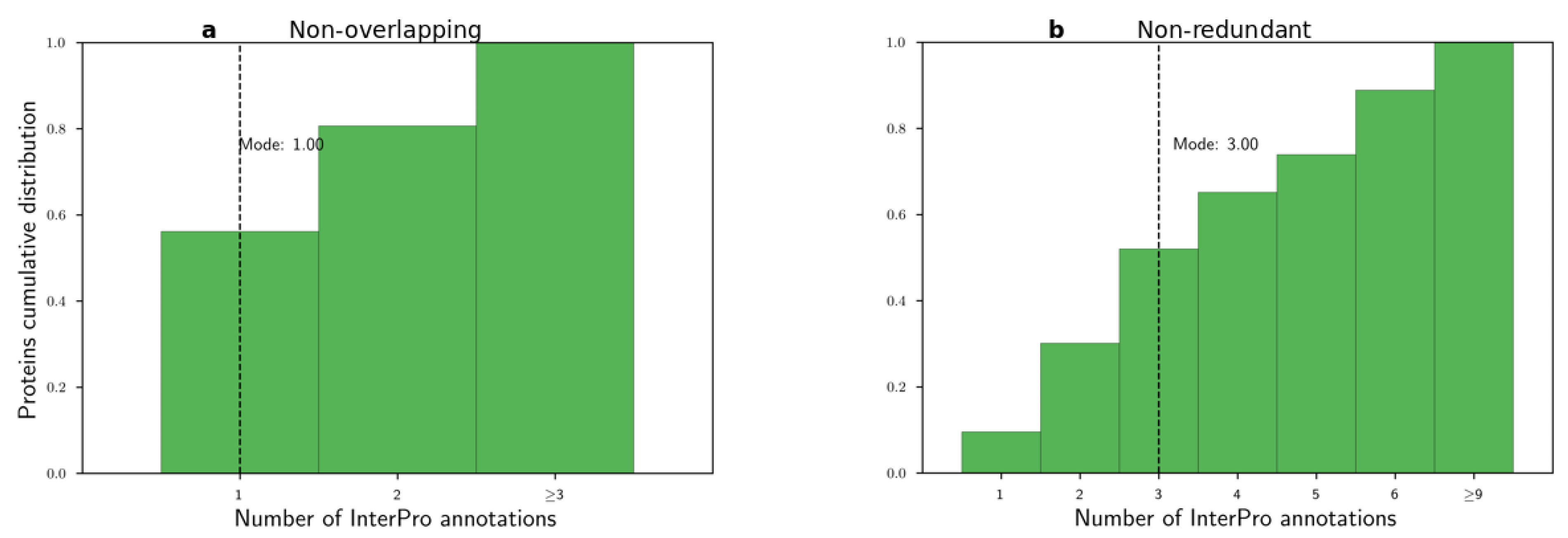
The InterPro database contains functional annotations for super-family, family, and single domains, as well as functional protein sites. Hereafter, we will refer to all such functional annotations as InterPro annotations. Furthermore, we will denote by domain architectures the ordered arrangement of domains in a protein. We consider two distinct strategies to represent a protein based on its domain architecture, consisting of either non-overlapping or non-redundant annotations. For both annotation types, we insert each annotation, based on the annotation’s beginning and end, in an interval tree . For each entry of the , we save the annotation InterPro identifier, significance score, and length. Based on the annotation type, we apply the following two distinct strategies to create the linear domain architectures:
Non-overlapping annotations. For each overlapping region in a protein, we keep the longest annotation out of all overlapping ones. Annotations of non-overlapping regions are included.
Non-redundant annotations. For each overlapping region in a protein, we keep all annotations with a distinct InterPro identifier. We break ties for annotations with the equal InterPro identifier by filtering in the longest one. Similarly, we keep annotations of non-overlapping regions.
For both annotation types, we sort the filtered-in annotations by their starting position. Finally, following the approach of [
5], we also added the “GAP” domain to annotate more than 30 amino acid sub-sequences, which does not match any InterPro annotation entry.
An example of the resulting domain architectures for the
Diphthine synthase protein is shown in
Figure 2. All domains are overlapping, with the largest one colored in blue, and the non-overlapping annotation is the single longest domain (IPR035966). All other domains have a unique InterProID; therefore, the set of non-redundant InterPro annotations includes all presented domains which are sorted with respect to their starting position, and colored in green.
Applying the previous steps for all annotated proteins produces the domain architectures, constituting the input corpus to the following embedding module.
3.2. Training Domain Embeddings
Given a protein, we assumed that words were its resolved InterPro annotations and sentences were the protein domain architectures. By this assumption, we learned task-independent embeddings for each InterPro annotation using two variants of
word2vec: a continuous bag of words and skip-gram model, hereafter denoted as CBOW and SKIP respectively. See [
12] for technical details on the difference between these approaches. Through this training, each InterPro annotation is associated with a task-independent embedding vector.
3.3. Quantitative Intrinsic Evaluation
In the following, we use the metadata for the most characteristic properties of domains, in order to evaluate the learned embedding space for various hyper-parameters of word2vec. We propose four intrinsic evaluation approaches for domain embeddings: domain hierarchy based on the family/subfamily relation, SCOPe secondary structure class, EC primary class, and GO molecular function annotation.
We refer to the embedding space learned by word2vec for a particular set of hyperparameters as . The k nearest neighbors of a domain d is found by using the Euclidean distance, and it is denoted as .
To inspect the relative performance of on each of the following evaluations, we randomized all domain vectors and ran each evaluation task. That is, we assigned to each domain vector a newly created random vector, for each unique dimensionality of embedding space, irrespective of all other embedding method parameters.
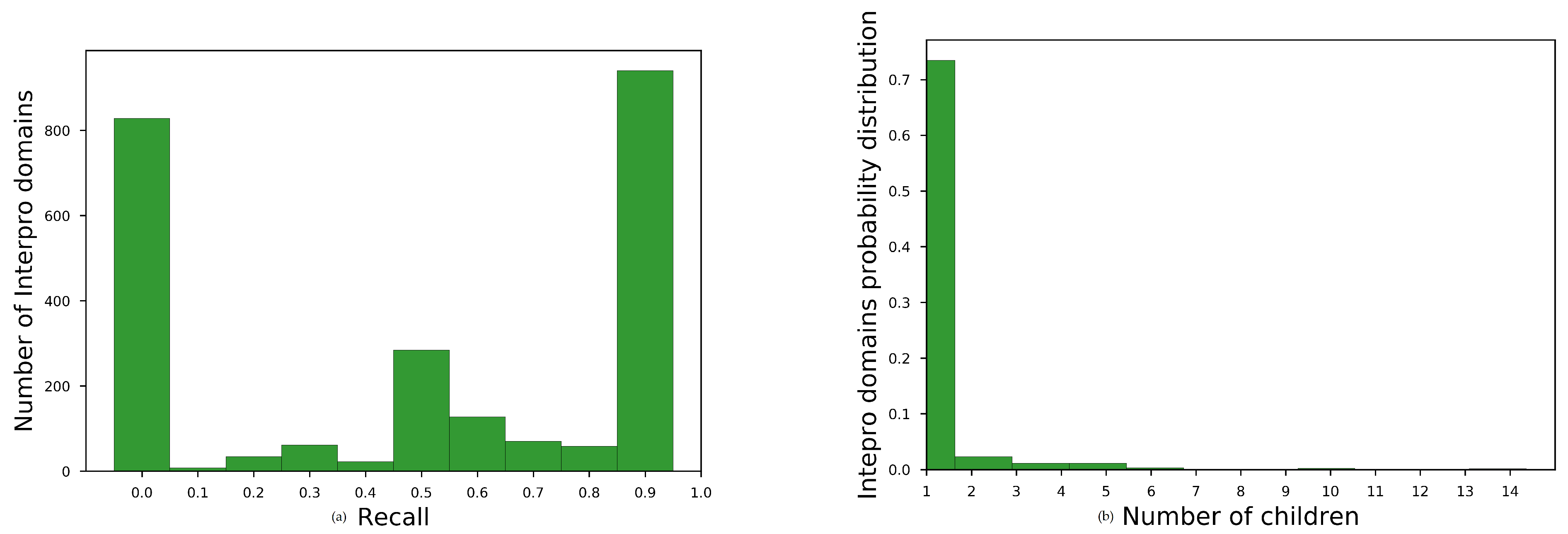
Domain Hierarchy
InterPro defines a strict family–subfamily relationship among domains. This relationship is based on sequence similarity of the domain signatures. We refer to the children of domain p as . We use these relationships to evaluate an embedding space, posing the following research question,
: Did vectors of hierarchically close domains form clusters in the ?
Evaluation We predicted the closest
domains on cosine similarity of their vector to the parent vector, and we denote this predicted set as
. For all learned embedding spaces, we measured their recall performance,
, defined as follows:
SCOPe Secondary Structure Class
We extracted the secondary structure of Interpro domains from the SCOPe database and formed the following research question,
: Did vectors of domains, with same secondary structure class, form clusters in the ?
Evaluation We evaluated by retrieving of each domain. Then, we applied stratified 5-fold cross-validation and measured the performance of a k-nearest neighbor classifier to predict the structure class of each domain. The intrinsic evaluation performance metric is the average accuracy across all folds, .
EC Primary Class
The enzymatic activity of each domain is given by its primary EC class [
32] and we pose the following research question,
: Did vectors of domains, with the same enzymatic primary class, form clusters in the ?
Evaluation We again evaluate using k nearest neighbors in a stratified 5-fold cross-validation setting. The average accuracy across all folds, , is again used to quantify the intrinsic quality of the embedding space.
GO Molecular Function
For our last intrinsic evaluation, we aimed to assess using the molecular function GO annotation. We extracted all molecular function GO annotations associated with each domain. In order to account for differences in specificity of different GO annotations, we always used the depth-1 ancestor of each annotation, that is, children of the root molecular function term, GO:0003674.
Since model organisms have the most-annotated proteins, we created GO molecular function data sets for one example of prokaryote (Escherichia coli, denoted E. coli); one example of a simple eukaryote (Saccharomyces cerevisiae, denoted S.cerevisiae); and one complex eukaryote (Homo sapiens, denoted Human). To also assess our embeddings for not highly annotated organisms, we included a molecular function data set for an example of a human pathogen (Plasmodium falciparum, denoted as Malaria). Finally, we pose the following research question,
: Did vectors of domains, with the same GO molecular function, form clusters in the ?
Evaluation Similarly, k nearest neighbors is used here in a stratified 5-cross-validation setting. Average accuracy across all folds, , is again used to quantify performance.
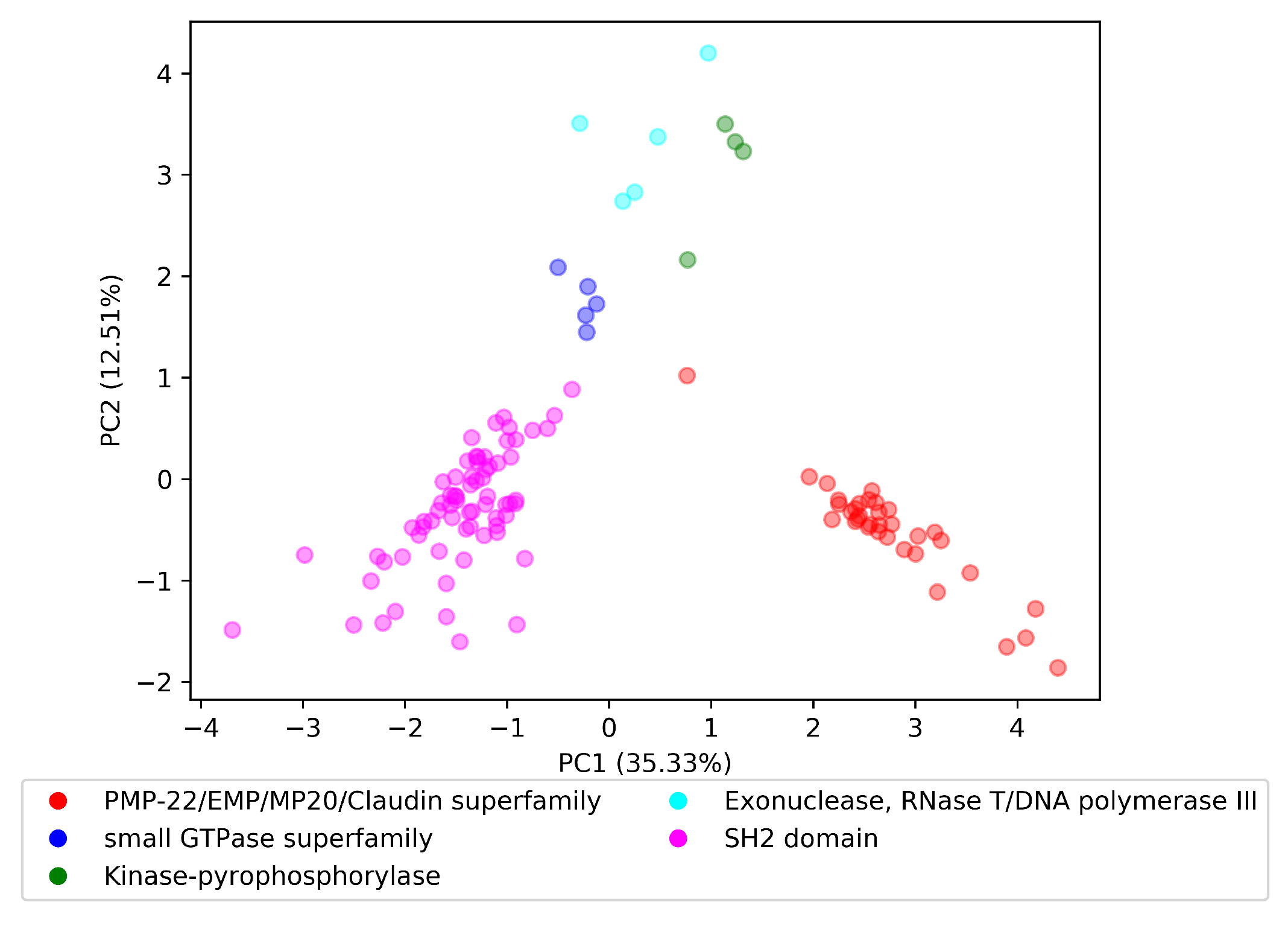
3.4. Qualitative Evaluation
As a preliminary evaluation strategy, we used qualitative evaluation approaches adopted in an existing work. To follow the qualitative approach of ProtVec and SeqVec we also visualized the embedding space for selected domain superfamilies, to answer the following research question,
: Did vectors of each domain superfamily form a cluster in the ?
Evaluation First, we added the vector of each domain in a randomly chosen domain superfamily to an empty space. Then, we performed principle component analysis (PCA) [
33] to reduce the space in two dimensions, and observed the formed clusters.
3.5. Extrinsic Evaluation
In addition, we assessed the learned by examining the performance change in downstream tasks. For the three supervised tasks, TargetP, Toxin, and NEW, the domain representations were used as input in simple neural networks. Next, our model performance was compared to the state-of-the-art protein embeddings, ProtVec and SeqVec.
TargetP
This data set is about predicting the cellular location of a given protein. We downloaded the TargetP data set provided by [
34], and we also used the non-plant data set. This data set consists of 2738 proteins accompanied by their uniprot ID, sequence, and cellular location label, which can be nuclear, cytosol, pathway, or signal and mitochondrial. Finally, we removed all instances with a duplicate set of domains, resulting in a total of 2418. This is a multi-class task, and its class distribution is summarized in
Appendix E.
Evaluation For the TargetP, we used the mc-AuROC performance metric.
Toxin
The research work [
35] introduced a data set associating protein sequence to toxic or other physiological content. We used the hard setting, which provides a uniprot ID, sequence, and the label toxin content or non-toxin content, for 15,496 proteins. Finally, we kept only the proteins with unique domain composition, resulting in 2270 protein instances in total. This is a binary task, and the class distribution is shown in
Appendix E.
Evaluation As the Toxin data set is a binary task, we used AuROC as a performance metric.
NEW
The NEW data set [
36] contains the data for predicting the enzymatic function of proteins. For each of the 22,618 proteins, the data set provides the sequence and the EC number class. The primary enzyme class, the first digit of an EC number, is our label on this prediction task, resulting in a multi-class task. Finally, we removed all instances with duplicate domain composition, resulting in a total of 14,434 protein instances. The possible classes are six, and the class distribution is shown in
Appendix E.
Evaluation The NEW data set is a multi-class task; thus, we used mc-AuROC as a performance metric.
3.5.1. Data Partitioning
We divided each data set into 70/30% train and test splits. To perform model selection, we created inner three-fold cross-validation sets on the train split.
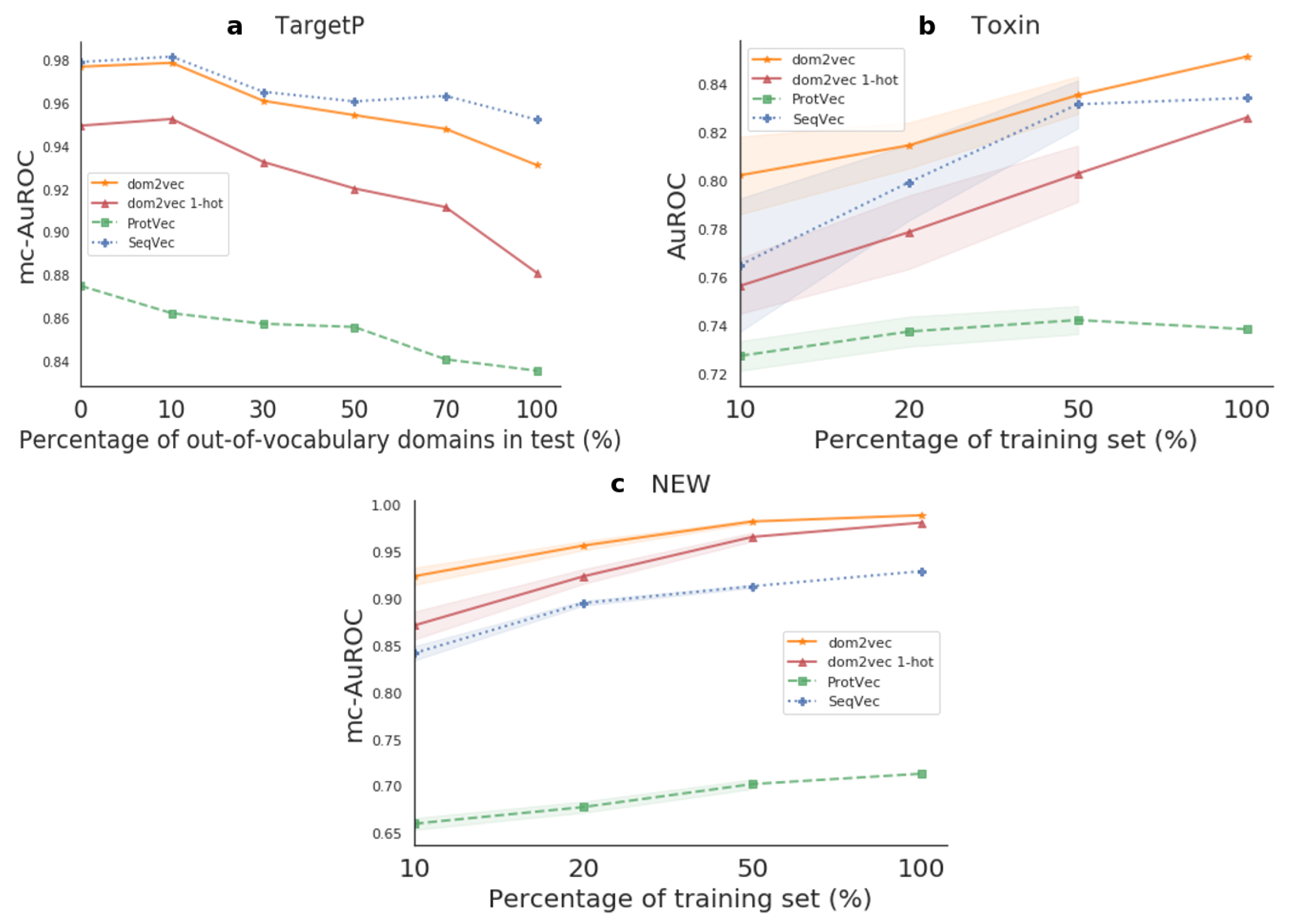
Out-of-vocabulary experiment We observed that the performance of classifiers depending on protein domains was highly dependent on the out-of-vocabulary (OOV) domains, as first discussed in [
37]. OOV domains are all the domains contained in the test set, but not in the train. For TargetP, Toxin, and NEW, we observed that approximately 60%, 20%, and 20% of test proteins contained
at least one OOV domain, respectively.
For the TargetP containing the highest OOV, we experimented to compensate for the high degree of OOV. We split the test set into shorter sets by an increasing degree of OOV, namely 0%, 10%, 30%, 50%, 70%, and 100%. Then, we trained models for the whole train set and benchmarked the performance on each of these test subsets.
Generalization experiment For the Toxin and NEW data sets, experiencing low OOV, we sought to investigate the generalization of the produced classifier. We increased the number of training examples that the model was allowed to learn from and we benchmarked always in the entire test set. To do so, we created training splits of size 10%, 20%, and 50% of the whole train set. To perform significance testing, we trained on 10 random subsamples for each training split percentage, and then tested on the separate step set. We used the paired sample t-test, the Benjamini–Hochberg multiple-test, to compare the performance between a pair of classifiers on the test set.
3.5.2. Simple Neural Models for Prediction
We consider a set of simple, well-established neural models to combine the InterPro annotation embeddings for each protein to perform downstream tasks, that is, for
extrinsic evaluation tasks. In particular, we use FastText [
38], convolutional neural networks (CNNs) [
39], and recurrent neural networks (RNNs) with long- and short-term memory (LSTM) cells [
40] and bi-directional LSTMs.
5. Conclusions
In this work, we presented dom2vec, an approach for learning quantitatively assessable protein domain embeddings using the word2vec method on domain architectures from InterPro annotations.
We have shown that dom2vec adequately captured the domain SCOPe structural information, EC enzymatic function, and the GO molecular function of each domain with such available metadata information. However, dom2vec produced moderate results in the domain hierarchy evaluation task. After investigating the properties of domain families that dom2vec produces these moderate results, we concluded that dom2vec cannot capture the domain hierarchy, mostly for domain families of low cardinality. We argue that by using more complex classifiers compared to , we could gain in hierarchy performance, but this was not the scope of our evaluation.
Importantly, we did discover that
dom2vec embeddings captured the most distinctive biological characteristics of domains, secondary structure, and enzymatic and molecular function for an individual domain. That is,
word2vec produced domain embeddings which clustered
sufficiently well by their structure and function class. Therefore, our finding supported the accepted modular evolution of proteins [
1], in a data-driven way. It also made possible a striking analogy between words in natural language that clustered together in
word2vec space [
14], and protein domains in domain architectures that clustered together in
dom2vec space. Therefore, we parallel the semantic and lexical similarity of words to the functional and structural resemblance of domains. This analogy may augment the research on understanding the nature of rules underlying the domain architecture grammar [
7]. We are confident that this interpretability aspect of
dom2vec will allow researchers to apply it reliably, so as to predict biological features of novel domain architectures and proteins with identifiable InterPro annotations.
In downstream task evaluation, dom2vec significantly outperformed domain 1-hot vectors and state-of-the-art sequence-based embeddings for the Toxin and NEW data sets. For the TargetP, dom2vec was comparable to the best-performing sequence-based embedding, Seqvec, for OOV up to 30%. Therefore, we recommend using dom2vec in combination with sequence embeddings to boost prediction performance.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




