

A Comparative Study of Machine Learning Techniques for Cell Annotation of scRNA-Seq Data



Abstract
:1. Introduction
2. Materials and Methods
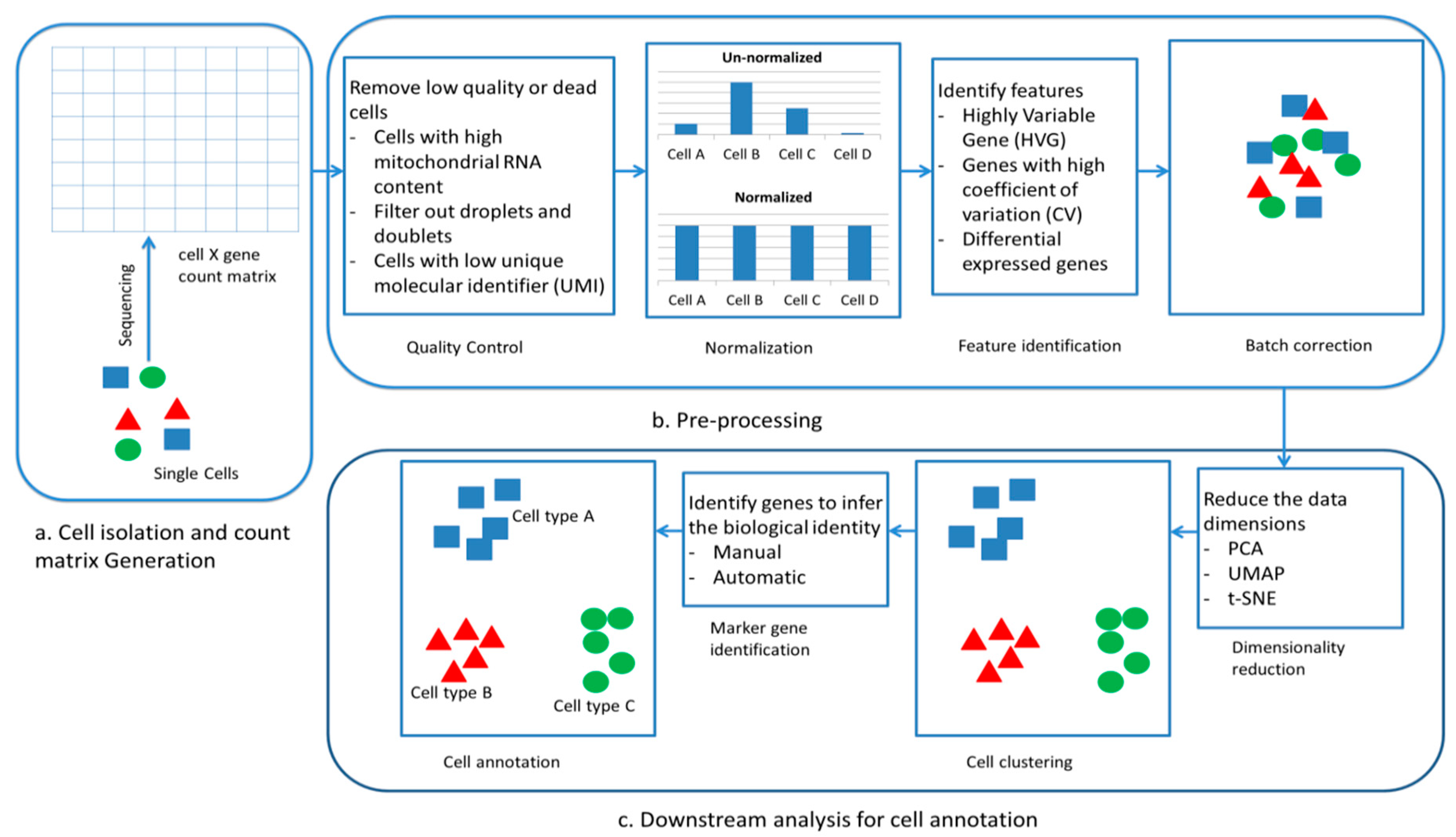
2.1. Single-Cell Annotation and Pre-Processing of scRNA-Seq
2.2. Machine Learning Techniques for Comparative Analysis of Single Cell Annotation
2.3. Hyper-Parameter Details
2.4. Dataset Description
3. Results and Discussion
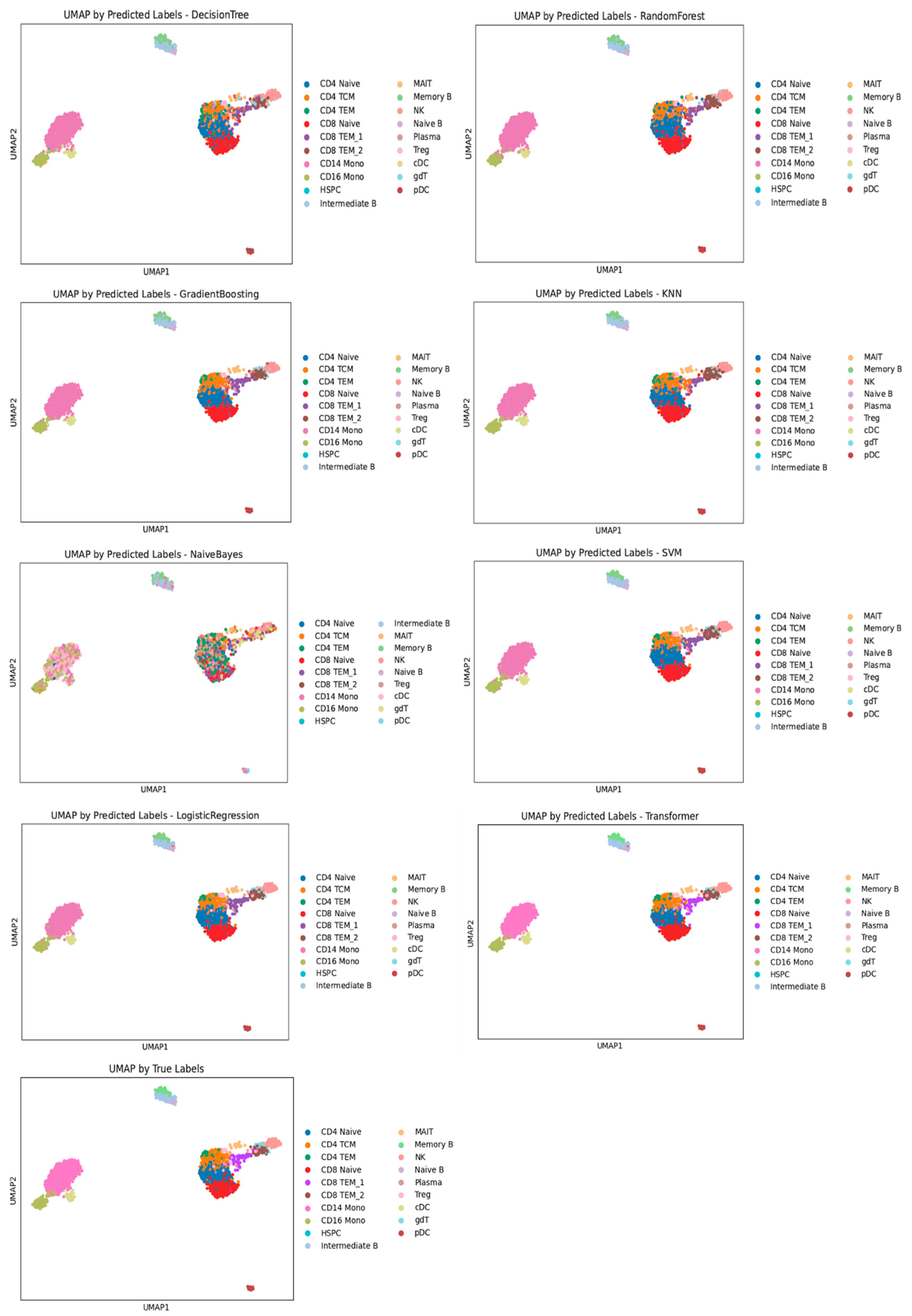
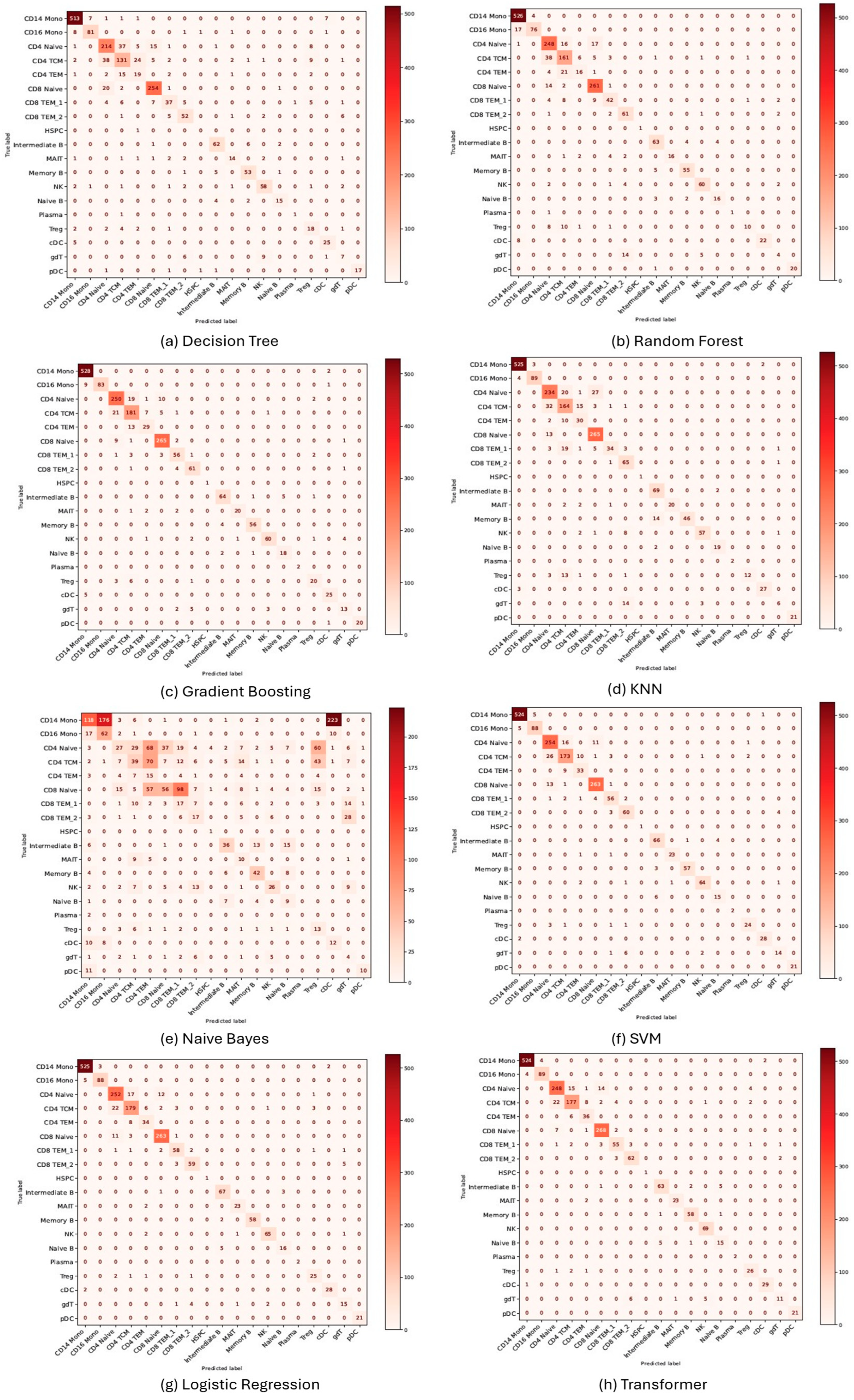
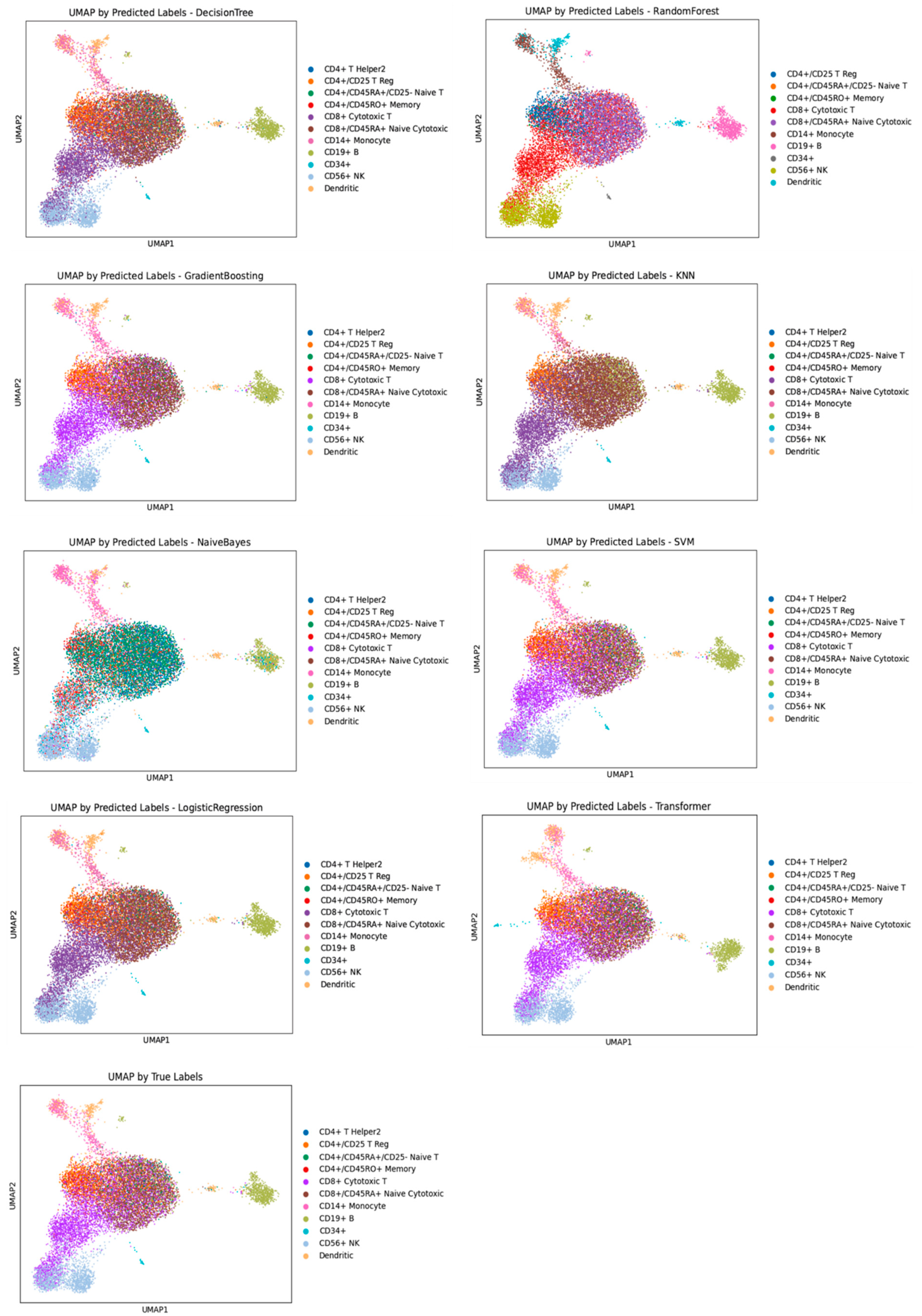
3.1. Performance Evaluation of Machine Learning Models for Cell Annotation of PBMC Datasets
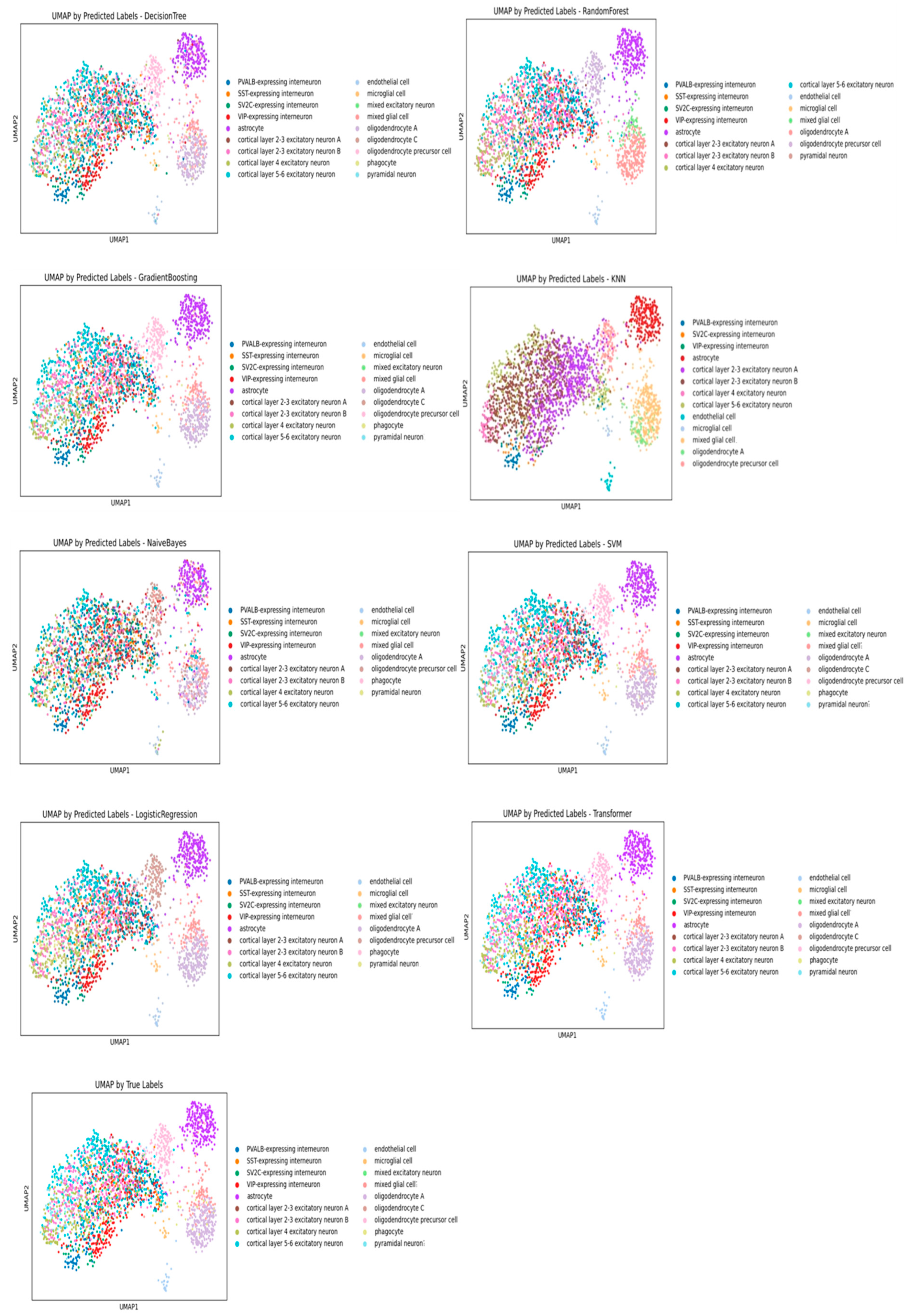
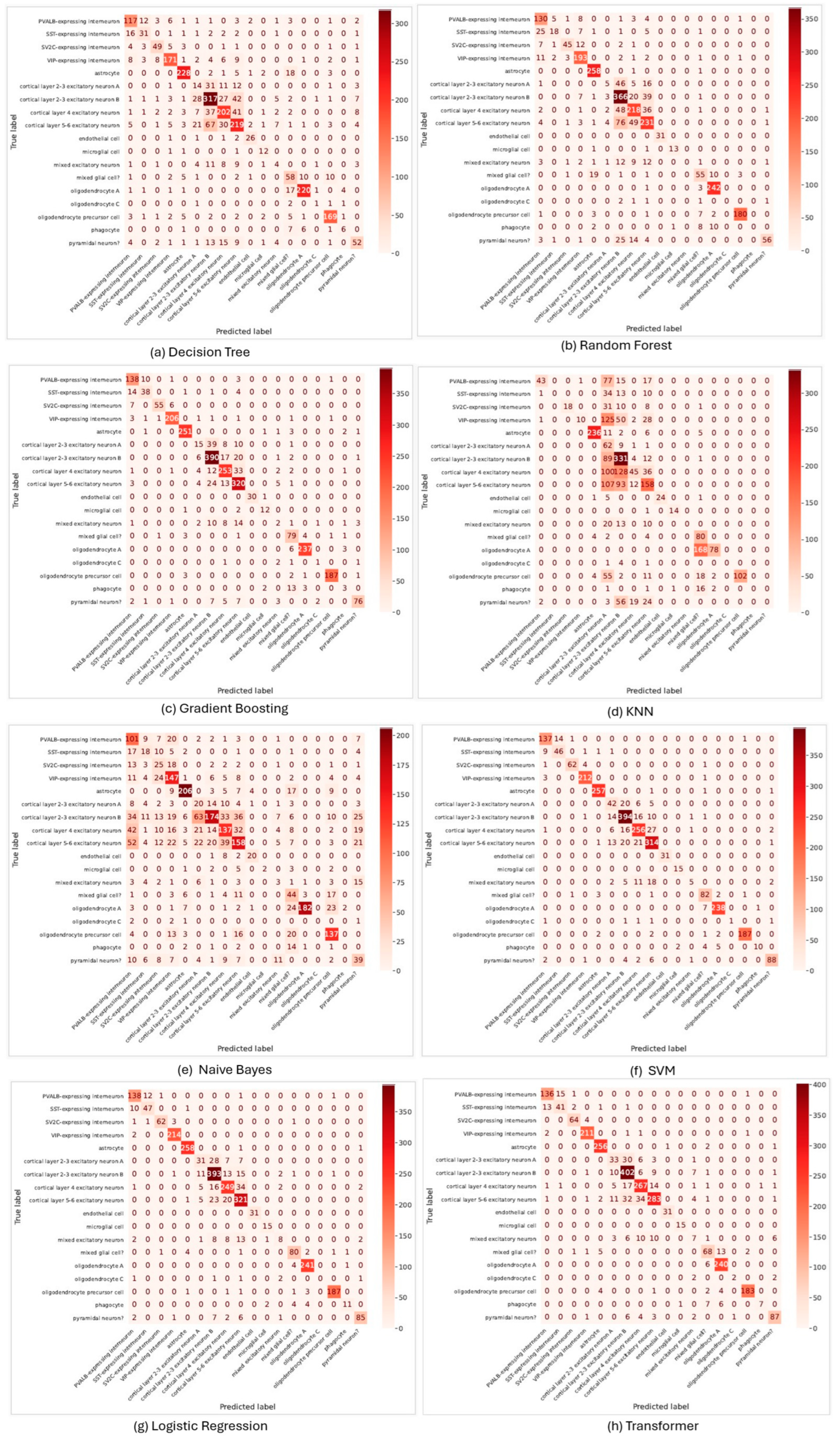
3.2. Performance Evaluation of Machine Learning Models for Cell Annotation on Multiple Sclerosis Dataset
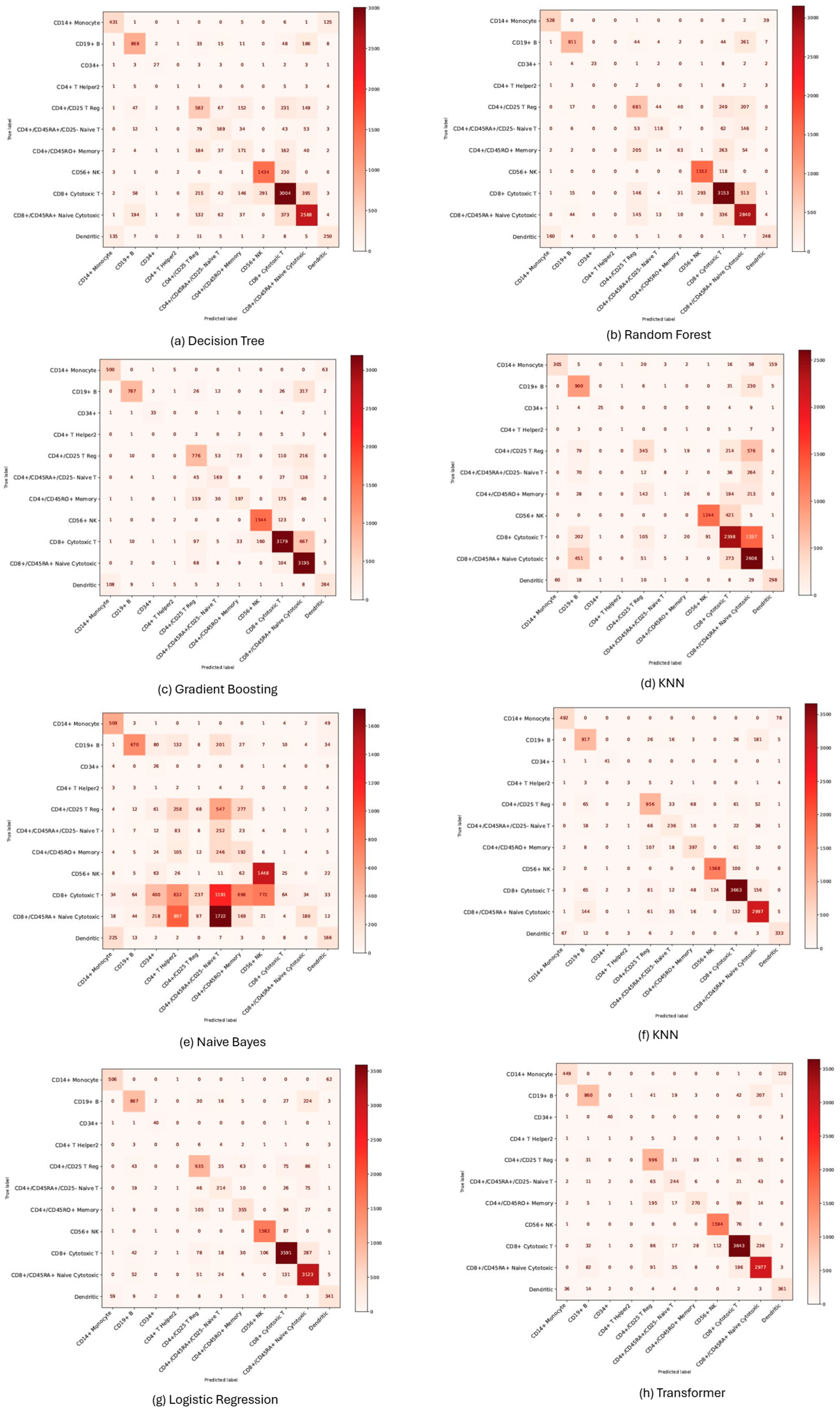
3.3. Performance Evaluation of Machine Learning Models for Cell Annotation of Zhengh68k Datasets
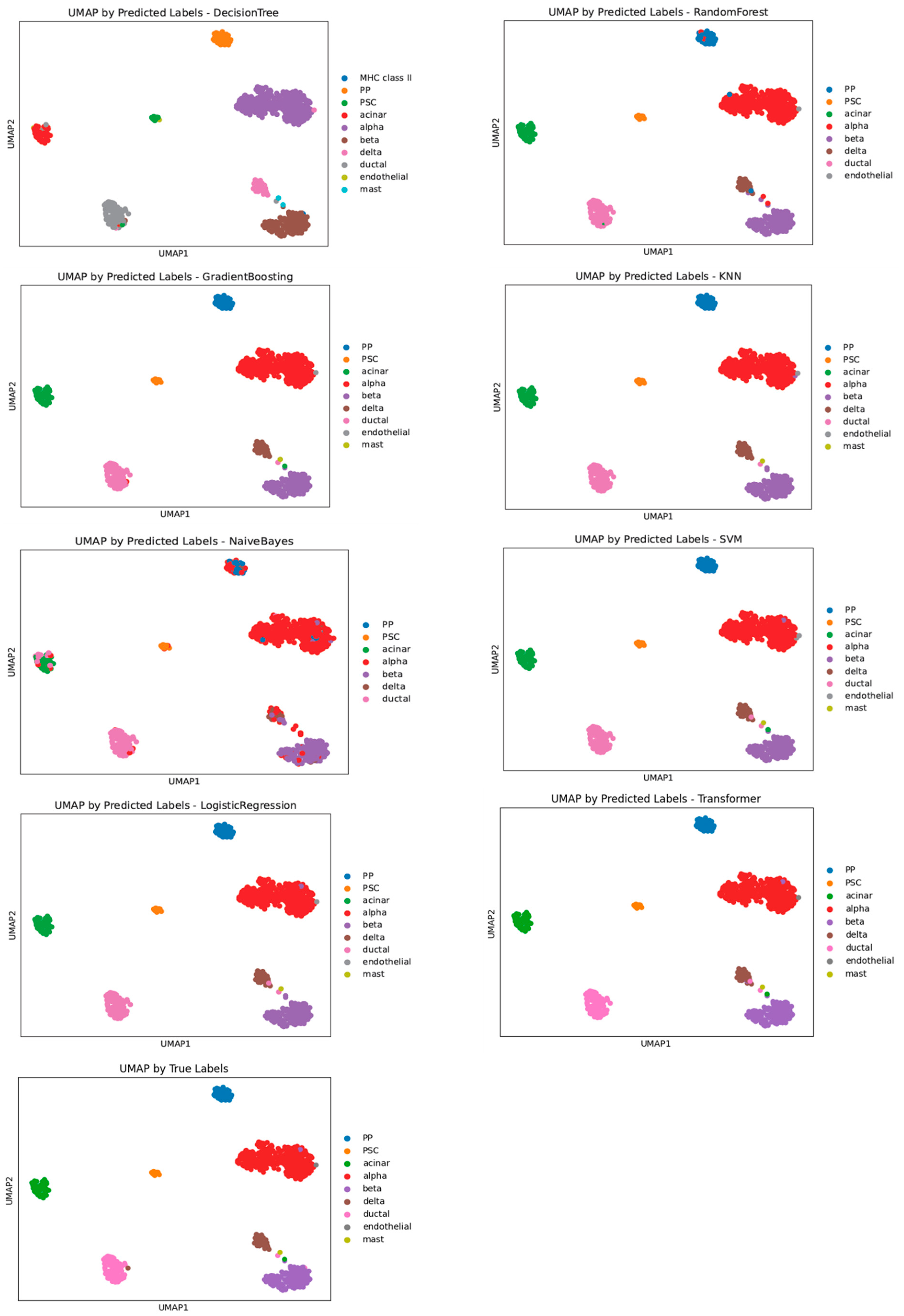
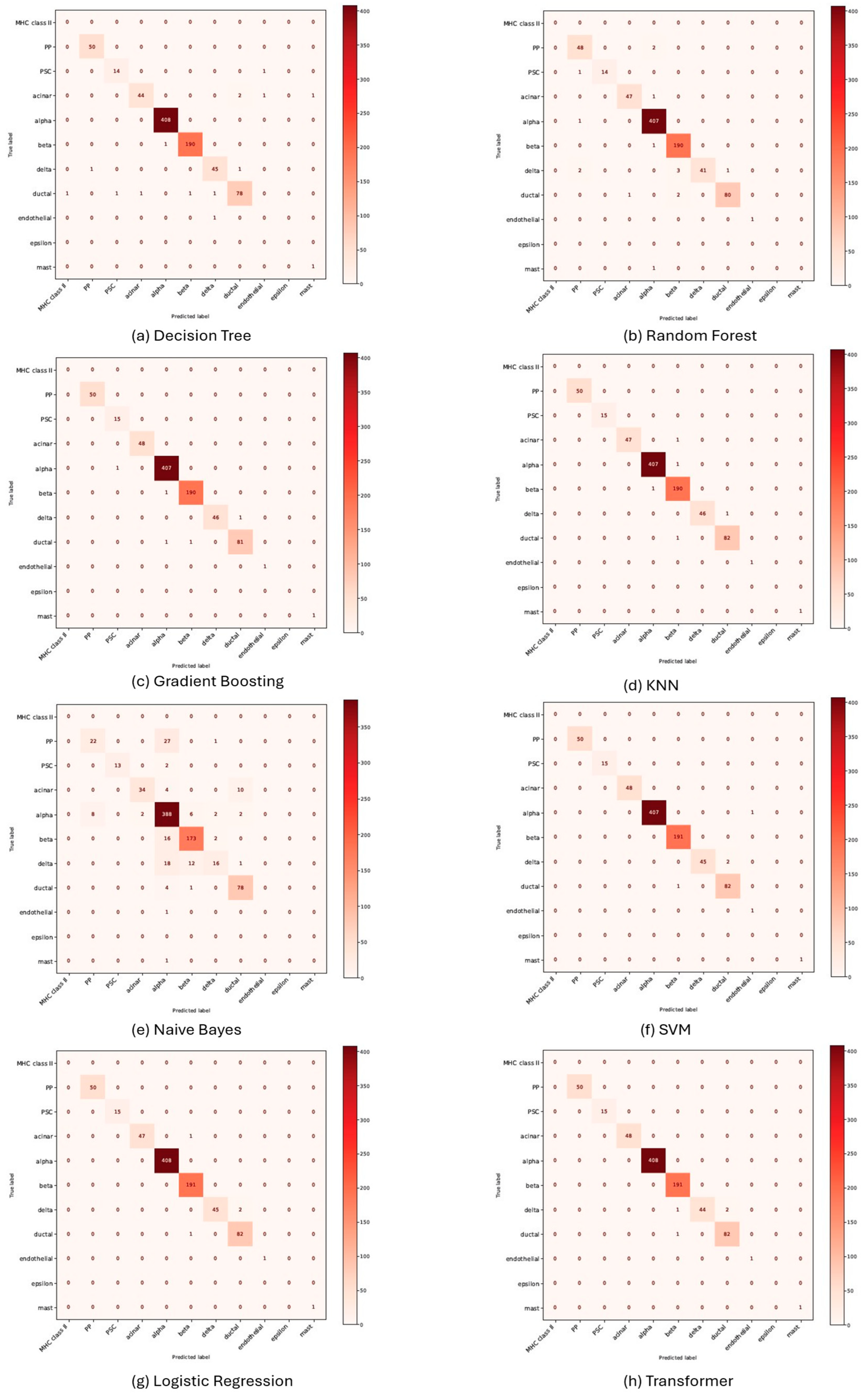
3.4. Performance Evaluation of Machine Learning Models for Cell Annotation of Pancreatic Dataset
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Zimmer, C. How Many Cells Are In Your Body? National Geographic Magazine, 23 October 2013. [Google Scholar]
- Lücken, M.D.; Burkhardt, D.B.; Cannoodt, R.; Lance, C.; Agrawal, A.; Aliee, H.; Chen, A.T.; Deconinck, L.; Detweiler, A.M.; Granados, A.A.; et al. A sandbox for prediction and integration of DNA, RNA, and protein data in single cells. In Proceedings of the NeurIPS 2021 Track Datasets and Benchmarks, Virtual, 6–14 December 2021. [Google Scholar]
- Tang, F.; Barbacioru, C.; Wang, Y.; Nordman, E.; Lee, C.; Xu, N.; Wang, X.; Bodeau, J.; Tuch, B.B.; Siddiqui, A.; et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nat. Methods 2009, 6, 377–382. [Google Scholar] [CrossRef] [PubMed]
- Stuart, T.; Butler, A.; Hoffman, P.; Hafemeister, C.; Papalexi, E.; Mauck, W.M., III; Hao, Y.; Stoeckius, M.; Smibert, P.; Satija, R. Comprehensive Integration of Single-Cell Data. Cell 2019, 177, 1888–1902.e21. [Google Scholar] [CrossRef] [PubMed]
- Tirosh, I.; Izar, B.; Prakadan, S.M.; Wadsworth, M.H., II; Treacy, D.; Trombetta, J.J.; Rotem, A.; Rodman, C.; Lian, C.; Murphy, G.; et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 2016, 352, 189–196. [Google Scholar] [CrossRef]
- Trapnell, C. Defining cell types and states with single-cell genomics. Genome Res. 2015, 25, 1491–1498. [Google Scholar] [CrossRef]
- Grün, D.; Lyubimova, A.; Kester, L.; Wiebrands, K.; Basak, O.; Sasaki, N.; Clevers, H.; van Oudenaarden, A. Single-cell messenger RNA sequencing reveals rare intestinal cell types. Nature 2015, 525, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, A.H.; Camara, P.G.; Kandror, E.K.; Roberts, T.J.; Schieren, I.; Maniatis, T.; Rabadan, R. Single-cell topological RNA-seq analysis reveals insights into cellular differentiation and development. Nat. Biotechnol. 2017, 35, 551–560. [Google Scholar] [CrossRef]
- Kumar, P.; Tan, Y.; Cahan, P. Understanding development and stem cells using single cell-based analyses of gene expression. Development 2017, 144, 17–32. [Google Scholar] [CrossRef]
- Lyu, P.; Zhai, Y.; Li, T.; Qian, J. CellAnn: A comprehensive, super-fast, and user-friendly single-cell annotation web server. Bioinformatics 2023, 39, btad521. [Google Scholar] [CrossRef]
- Clarke, Z.A.; Andrews, T.S.; Atif, J.; Pouyabahar, D.; Innes, B.T.; MacParland, S.A.; Bader, G.D. Tutorial: Guidelines for annotating single-cell transcriptomic maps using automated and manual methods. Nat. Protoc. 2021, 16, 2749–2764. [Google Scholar] [CrossRef]
- Yang, F.; Wang, W.; Wang, F.; Fang, Y.; Tang, D.; Huang, J.; Lu, H.; Yao, J. scBERT as a large-scale pretrained deep language model for cell type annotation of single-cell RNA-seq data. Nat. Mach. Intell. 2022, 4, 852–866. [Google Scholar] [CrossRef]
- Cheng, C.; Chen, W.; Jin, H.; Chen, X. A Review of Single-Cell RNA-Seq Annotation, Integration, and Cell–Cell Communication. Cells 2023, 12, 1970. [Google Scholar] [CrossRef]
- Papalexi, E.; Satija, R. Single-cell RNA sequencing to explore immune cell heterogeneity. Nat. Rev. Immunol. 2018, 18, 35–45. [Google Scholar] [CrossRef]
- Zhang, X.; Lan, Y.; Xu, J.; Quan, F.; Zhao, E.; Deng, C.; Luo, T.; Xu, L.; Liao, G.; Yan, M.; et al. CellMarker: A manually curated resource of cell markers in human and mouse. Nucleic Acids Res. 2019, 47, D721–D728. [Google Scholar] [CrossRef] [PubMed]
- Franzén, O.; Gan, L.M.; Björkegren, J.L.M. PanglaoDB: A web server for exploration of mouse and human single-cell RNA sequencing data. Database 2019, 2019, baz046. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Yan, M.; Zhang, G.; Liu, W.; Deng, C.; Liao, G.; Xu, L.; Luo, T.; Yan, H.; Long, Z.; et al. CancerSEA: A cancer single-cell state atlas. Nucleic Acids Res. 2019, 47, D900–D908. [Google Scholar] [CrossRef] [PubMed]
- Shao, X.; Liao, J.; Lu, X.; Xue, R.; Ai, N.; Fan, X. scCATCH: Automatic Annotation on Cell Types of Clusters from Single-Cell RNA Sequencing Data. iScience 2020, 23, 100882. [Google Scholar] [CrossRef]
- Cao, Y.; Wang, X.; Peng, G. SCSA: A cell type annotation tool for single-cell RNA-seq data. Front. Genet. 2020, 11, 490. [Google Scholar] [CrossRef]
- Zhang, A.W.; O’flanagan, C.; Chavez, E.A.; Lim, J.L.P.; Ceglia, N.; McPherson, A.; Wiens, M.; Walters, P.; Chan, T.; Hewitson, B.; et al. Probabilistic cell-type assignment of single-cell RNA-seq for tumor microenvironment profiling. Nat. Methods 2019, 16, 1007–1015. [Google Scholar] [CrossRef]
- Zhang, Z.; Luo, D.; Zhong, X.; Choi, J.H.; Ma, Y.; Wang, S.; Mahrt, E.; Guo, W.; Stawiski, E.W.; Modrusan, Z.; et al. SCINA: A Semi-Supervised Subtyping Algorithm of Single Cells and Bulk Samples. Genes 2019, 10, 531. [Google Scholar] [CrossRef]
- Ge, S.; Wang, H.; Alavi, A.; Xing, E.; Bar-Joseph, Z. Supervised Adversarial Alignment of Single-Cell RNA-seq Data. J. Comput. Biol. 2021, 28, 501–513. [Google Scholar] [CrossRef]
- Xie, P.; Gao, M.; Wang, C.; Zhang, J.; Noel, P.; Yang, C.; Von Hoff, D.; Han, H.; Zhang, M.Q.; Lin, W. SuperCT: A supervised-learning framework for enhanced characterization of single-cell transcriptomic profiles. Nucleic Acids Res. 2019, 47, e48. [Google Scholar] [CrossRef]
- Butler, A.; Hoffman, P.; Smibert, P.; Papalexi, E.; Satija, R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018, 36, 411–420. [Google Scholar] [CrossRef]
- Haghverdi, L.; Lun, A.T.L.; Morgan, M.D.; Marioni, J.C. Batch effects in single-cell RNA-sequencing data are corrected by matching mutual nearest neighbors. Nat. Biotechnol. 2018, 36, 421–427. [Google Scholar] [CrossRef]
- Ji, X.; Tsao, D.; Bai, K.; Tsao, M.; Xing, L.; Zhang, X. scAnnotate: An automated cell-type annotation tool for single-cell RNA-sequencing data. Bioinform. Adv. 2023, 3, vbad030. [Google Scholar] [CrossRef]
- Lin, Y.; Cao, Y.; Kim, H.J.; Salim, A.; Speed, T.P.; Lin, D.M.; Yang, P.; Yang, J.Y.H. scClassify: Sample size estimation and multiscale classification of cells using single and multiple reference. Mol. Syst. Biol. 2020, 16, e9389. [Google Scholar] [CrossRef]
- Tan, Y.; Cahan, P. SingleCellNet: A Computational Tool to Classify Single Cell RNA-Seq Data Across Platforms and Across Species. Cell Syst. 2019, 9, 207–213.e2. [Google Scholar] [CrossRef]
- Wagner, F.; Yanai, I. Moana: A robust and scalable cell type classification framework for single-cell RNA-Seq data. BioRxiv 2018. [Google Scholar] [CrossRef]
- Johnson, T.S.; Wang, T.; Huang, Z.; Yu, C.Y.; Wu, Y.; Han, Y.; Zhang, Y.; Huang, K.; Zhang, J. LAmbDA: Label ambiguous domain adaptation dataset integration reduces batch effects and improves subtype detection. Bioinformatics 2019, 35, 4696–4706. [Google Scholar] [CrossRef]
- LeCun, Y.; Bengio, Y. Convolutional networks for images, speech, and time series. In The Handbook of Brain Theory and Neural Networks; MIT Press: Cambridge, UK, 1995; Volume 3361. [Google Scholar]
- Khan, F.; Ayoub, S.; Gulzar, Y.; Majid, M.; Reegu, F.A.; Mir, M.S.; Soomro, A.B.; Elwasila, O. MRI-Based Effective Ensemble Frameworks for Predicting Human Brain Tumor. J. Imaging 2023, 9, 163. [Google Scholar] [CrossRef]
- Bao, S.; Li, K.; Yan, C.; Zhang, Z.; Qu, J.; Zhou, M. Deep learning-based advances and applications for single-cell RNA-sequencing data analysis. Brief. Bioinform. 2022, 23, bbab473. [Google Scholar] [CrossRef]
- Wani, S.A.; Khan, S.A.; Quadri, S.M.K. scJVAE: A novel method for integrative analysis of multimodal single-cell data. Comput. Biol. Med. 2023, 158, 106865. [Google Scholar] [CrossRef]
- Wani, S.A.; Quadri, S.M.K. Evaluation of Computational Methods for Single Cell Multi-Omics Integration. Procedia Comput. Sci. 2022, 218, 2744–2754. [Google Scholar] [CrossRef]
- Majid, M.; Gulzar, Y.; Ayoub, S.; Khan, F.; Reegu, F.A.; Mir, M.S.; Jaziri, W.; Soomro, A.B. Enhanced Transfer Learning Strategies for Effective Kidney Tumor Classification with CT Imaging. Int. J. Adv. Comput. Sci. Appl. 2023, 14, 421–432. [Google Scholar] [CrossRef]
- Alquicira-Hernandez, J.; Sathe, A.; Ji, H.P.; Nguyen, Q.; Powell, J.E. ScPred: Accurate supervised method for cell-type classification from single-cell RNA-seq data. Genome Biol. 2019, 20, 264. [Google Scholar] [CrossRef]
- Shao, X.; Yang, H.; Zhuang, X.; Liao, J.; Yang, P.; Cheng, J.; Lu, X.; Chen, H.; Fan, X. ScDeepSort: A pre-trained cell-type annotation method for single-cell transcriptomics using deep learning with a weighted graph neural network. Nucleic Acids Res. 2021, 49, e122. [Google Scholar] [CrossRef]
- Hou, W.; Ji, Z. Assessing GPT-4 for cell type annotation in single-cell RNA-seq analysis. Nat. Methods 2024, 21, 1462–1465. [Google Scholar] [CrossRef]
- Cui, H.; Wang, C.; Maan, H.; Pang, K.; Luo, F.; Duan, N.; Wang, B. scGPT: Toward building a foundation model for single-cell multi-omics using generative AI. Nat. Methods 2024, 21, 1470–1480. [Google Scholar] [CrossRef]
- Devlin, J.; Chang, M.W.; Lee, K.; Toutanova, K. BERT: Pre-training of deep bidirectional transformers for language understanding. In Proceedings of the NAACL HLT 2019—2019 Conference of the North American Chapter of the Association for Computational Linguistics: Human Language Technologies—Proceedings of the Conference, Minneapolis, MN, USA, 2–7 June 2019. [Google Scholar]
- de Kanter, J.K.; Lijnzaad, P.; Candelli, T.; Margaritis, T.; Holstege, F.C.P. CHETAH: A selective, hierarchical cell type identification method for single-cell RNA sequencing. Nucleic Acids Res. 2019, 47, e95. [Google Scholar] [CrossRef]
- Cao, X.; Xing, L.; Majd, E.; He, H.; Gu, J.; Zhang, X. A Systematic Evaluation of Supervised Machine Learning Algorithms for Cell Phenotype Classification Using Single-Cell RNA Sequencing Data. Front. Genet. 2022, 13, 836798. [Google Scholar] [CrossRef]
- Rokach, L.; Maimon, O. Decision Trees. In Data Mining and Knowledge Discovery Handbook; Springer: Boston, MA, USA, 2006. [Google Scholar] [CrossRef]
- Rokach, L. Decision forest: Twenty years of research. Inf. Fusion 2016, 27, 111–125. [Google Scholar] [CrossRef]
- Pouyan, M.B.; Kostka, D. Random forest based similarity learning for single cell RNA sequencing data. Bioinformatics 2018, 34, i79–i88. [Google Scholar] [CrossRef]
- Majid, M.; Gulzar, Y.; Ayoub, S.; Khan, F.; Reegu, F.A.; Mir, M.S.; Jaziri, W.; Soomro, A.B. Using Ensemble Learning and Advanced Data Mining Techniques to Improve the Diagnosis of Chronic Kidney Disease. Int. J. Adv. Comput. Sci. Appl. 2023, 14, 470–480. [Google Scholar] [CrossRef]
- Natekin, A.; Knoll, A. Gradient boosting machines, a tutorial. Front. Neurorobotics 2013, 7, 2. [Google Scholar] [CrossRef]
- Ding, S.; Wang, D.; Zhou, X.; Chen, L.; Feng, K.; Xu, X.; Huang, T.; Li, Z.; Cai, Y. Predicting Heart Cell Types by Using Transcriptome Profiles and a Machine Learning Method. Life 2022, 12, 228. [Google Scholar] [CrossRef]
- Dann, E.; Henderson, N.C.; Teichmann, S.A.; Morgan, M.D.; Marioni, J.C. Differential abundance testing on single-cell data using k-nearest neighbor graphs. Nat. Biotechnol. 2021, 40, 245–253. [Google Scholar] [CrossRef]
- Sun, Z.; Chen, L.; Xin, H.; Jiang, Y.; Huang, Q.; Cillo, A.R.; Tabib, T.; Kolls, J.K.; Bruno, T.C.; Lafyatis, R.; et al. A Bayesian mixture model for clustering droplet-based single-cell transcriptomic data from population studies. Nat. Commun. 2019, 10, 1649. [Google Scholar] [CrossRef]
- Khan, F.; Gulzar, Y.; Ayoub, S.; Majid, M.; Mir, M.S.; Soomro, A.B. Least square-support vector machine based brain tumor classification system with multi model texture features. Front. Appl. Math. Stat. 2023, 9, 1324054. [Google Scholar] [CrossRef]
- Saygili, G.; OzgodeYigin, B. Continual learning approaches for single cell RNA sequencing data. Sci. Rep. 2023, 13, 15286. [Google Scholar] [CrossRef]
- Hosmer, D.W.; Lemeshow, S.; Sturdivant, R.X.; Regression, A.L. Applied Logistic Regression; John Wiley & Sons: Hoboken, NJ, USA, 2013. [Google Scholar] [CrossRef]
- Vaswani, A.; Shazeer, N.; Parmar, N.; Uszkoreit, J.; Jones, L.; Gomez, A.N.; Kaiser, Ł.; Polosukhin, I. Attention is all you need. In Proceedings of the 31st Conference on Neural Information Processing Systems (NIPS 2017), Long Beach, CA, USA, 4–9 December 2017. [Google Scholar]
- 10X Genomics. PBMC-Multiome. [Online]. Available online: https://www.10xgenomics.com/resources/datasets/pbmc-from-a-healthy-donor-granulocytes-removed-through-cell-sorting-10-k-1-standard-1-0-0 (accessed on 12 December 2022).
- Schirmer, L.; Velmeshev, D.; Holmqvist, S.; Kaufmann, M.; Werneburg, S.; Jung, D.; Vistnes, S.; Stockley, J.H.; Young, A.; Steindel, M.; et al. Neuronal vulnerability and multilineage diversity in multiple sclerosis. Nature 2019, 573, 75–82. [Google Scholar] [CrossRef]
- Zheng, G.X.Y.; Terry, J.M.; Belgrader, P.; Ryvkin, P.; Bent, Z.W.; Wilson, R.; Ziraldo, S.B.; Wheeler, T.D.; McDermott, G.P.; Zhu, J.; et al. Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 2017, 8, 14049. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | Key Hyper-Parameters | Training Configuration |
|---|---|---|
| Decision Tree | Criterion: gini, Max Depth: None, Min Samples Split: 2, | 80% train data, pruning via max depth |
| Random Forest | Estimators: 100, Max Features: sqrt, Max Depth: Tuned, Bootstrap: True | 80% train data, majority voting |
| Gradient Boosting (XGBoost) | Estimators: 300, Learning Rate: 0.1, Max Depth: 3, Early Stopping | Gradient descent to minimize log loss, early stopping |
| Support Vector Machine (SVM) | Kernel: RBF, Regularization (C): 1, Tolerance: 1 × 10−3 | Linear kernel, normalized input |
| Logistic Regression | Regularization: L2, Max Iterations: 500, Multi-Class: Multinomial | Normalized input, convergence with cross-validation |
| K-Nearest Neighbours (KNN) | Neighbours (k): 5, Distance Metric: Euclidean, Weighting: distance-based | Distance calculations for all data points |
| Naive Bayes | Distribution: Gaussian, Variance Smoothing: 1 × 10−9 | Log-transformed input for comparability |
| Transformer | Embedding Dimension: 512, Encoder Layers: 6, Attention Heads: 8, Dropout: 0.1, Optimizer: Adam, Learning Rate: 0.0001 | Cross-entropy loss, trained for 10 epochs |
| Model | PBMC Dataset | MS Dataset | Zhengh68K | Pancreatic Dataset | ||||
|---|---|---|---|---|---|---|---|---|
| Accuracy | F1-Score | Accuracy | F1-Score | Accuracy | F1-Score | Accuracy | F1-Score | |
| Decision Tree | 0.8712 | 0.8730 | 0.7038 | 0.5339 | 0.6958 | 0.6946 | 0.9834 | 0.9846 |
| Random Forest | 0.8788 | 0.8686 | 0.7576 | 0.7373 | 0.7317 | 0.7177 | 0.9810 | 0.9803 |
| Gradient Boosting | 0.9299 | 0.9289 | 0.8512 | 0.8419 | 0.7790 | 0.7729 | 0.9941 | 0.9969 |
| KNN | 0.5152 | 0.4020 | 0.4458 | 0.4397 | 0.5959 | 0.5764 | 0.9921 | 0.9958 |
| Naive Bayes | 0.5019 | 0.5117 | 0.5245 | 0.5339 | 0.2613 | 0.2441 | 0.8578 | 0.8456 |
| SVM | 0.9583 | 0.9573 | 0.8823 | 0.8774 | 0.8476 | 0.8472 | 0.9953 | 0.9976 |
| Logistic Regression | 0.9527 | 0.9515 | 0.8801 | 0.8744 | 0.8440 | 0.8415 | 0.9921 | 0.9934 |
| Transformer | 0.9337 | 0.9299 | 0.8660 | 0.8610 | 0.8354 | 0.8330 | 0.9943 | 0.9958 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wani, S.A.; Quadri, S.; Mir, M.S.; Gulzar, Y. A Comparative Study of Machine Learning Techniques for Cell Annotation of scRNA-Seq Data. Algorithms 2025, 18, 232. https://doi.org/10.3390/a18040232
Wani SA, Quadri S, Mir MS, Gulzar Y. A Comparative Study of Machine Learning Techniques for Cell Annotation of scRNA-Seq Data. Algorithms. 2025; 18(4):232. https://doi.org/10.3390/a18040232
Chicago/Turabian StyleWani, Shahid Ahmad, SMK Quadri, Mohammad Shuaib Mir, and Yonis Gulzar. 2025. "A Comparative Study of Machine Learning Techniques for Cell Annotation of scRNA-Seq Data" Algorithms 18, no. 4: 232. https://doi.org/10.3390/a18040232
APA StyleWani, S. A., Quadri, S., Mir, M. S., & Gulzar, Y. (2025). A Comparative Study of Machine Learning Techniques for Cell Annotation of scRNA-Seq Data. Algorithms, 18(4), 232. https://doi.org/10.3390/a18040232