Differential Regulation of Anthocyanins in Cerasus humilis Fruit Color Revealed by Combined Transcriptome and Metabolome Analysis


Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials and Treatments
2.2. Metabolite Extraction and Profiling
2.3. Qualitative and Quantitative Analysis of Metabolites
2.4. RNA Extraction and Transcriptome Sequencing
2.5. Expression Level Analysis
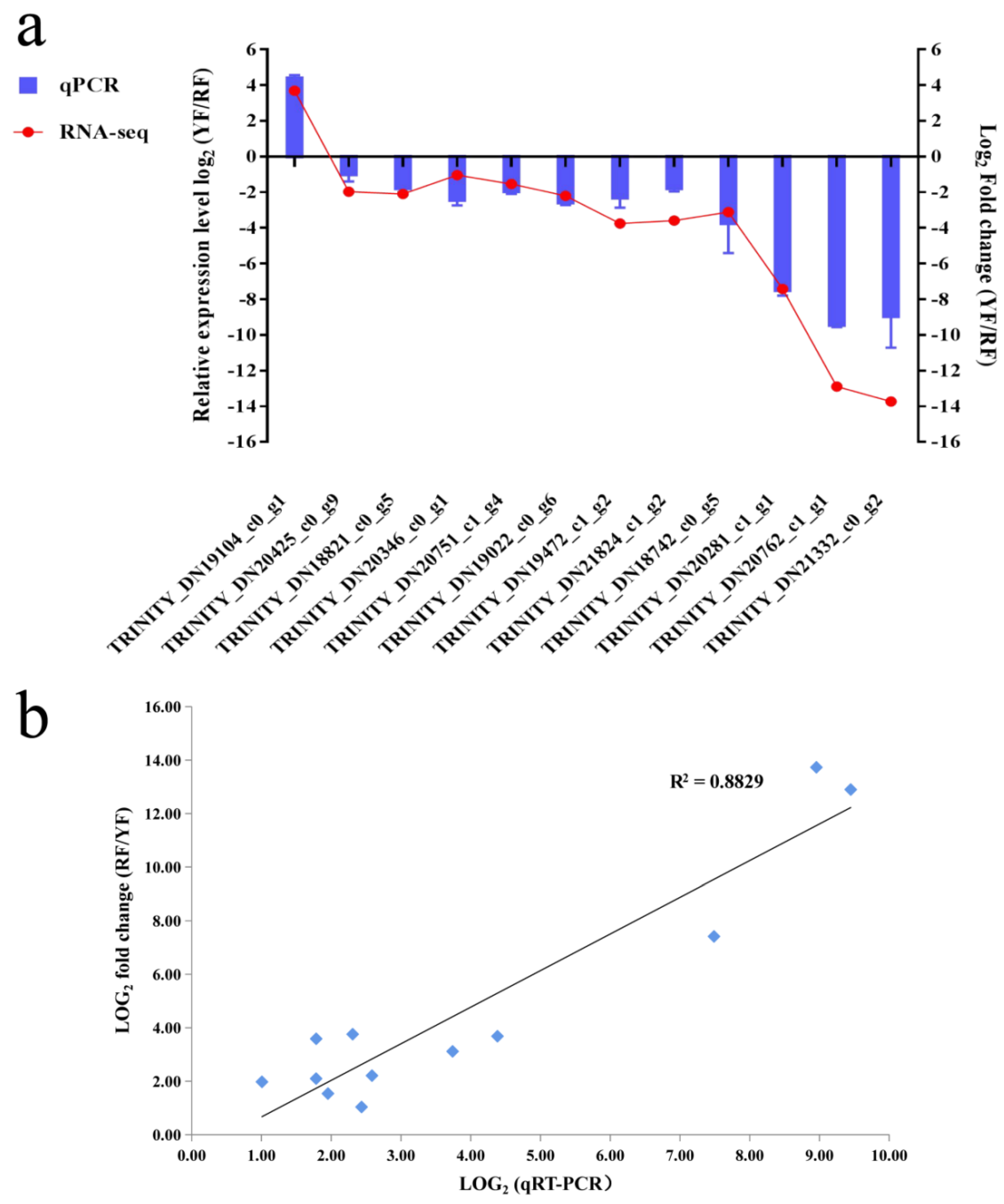
2.6. Quantitative Real-Time PCR and Expression Validation
2.7. Statistical Analysis
3. Results
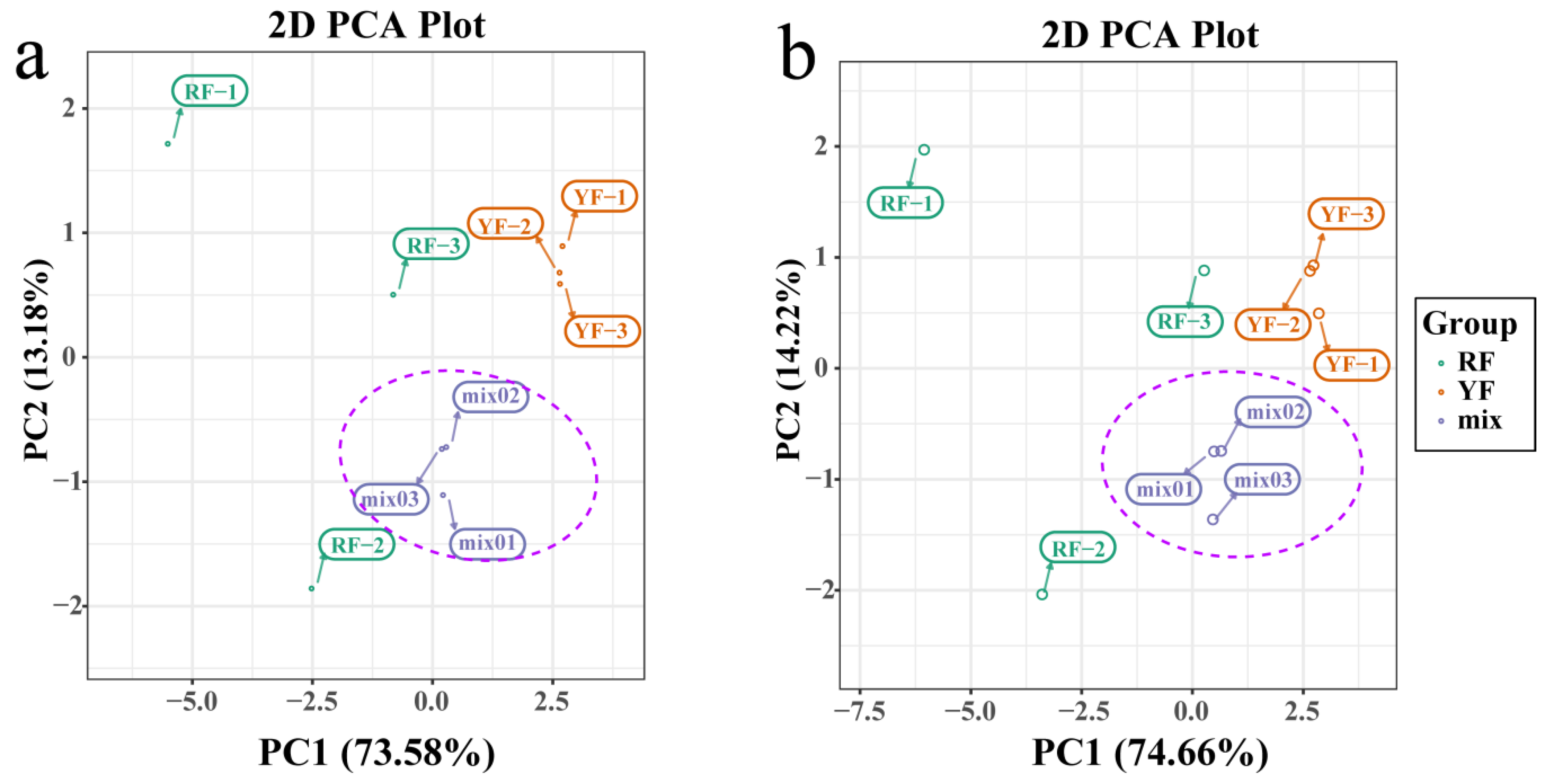
3.1. Metabolomics Analysis of C. humilis Fruit Peel
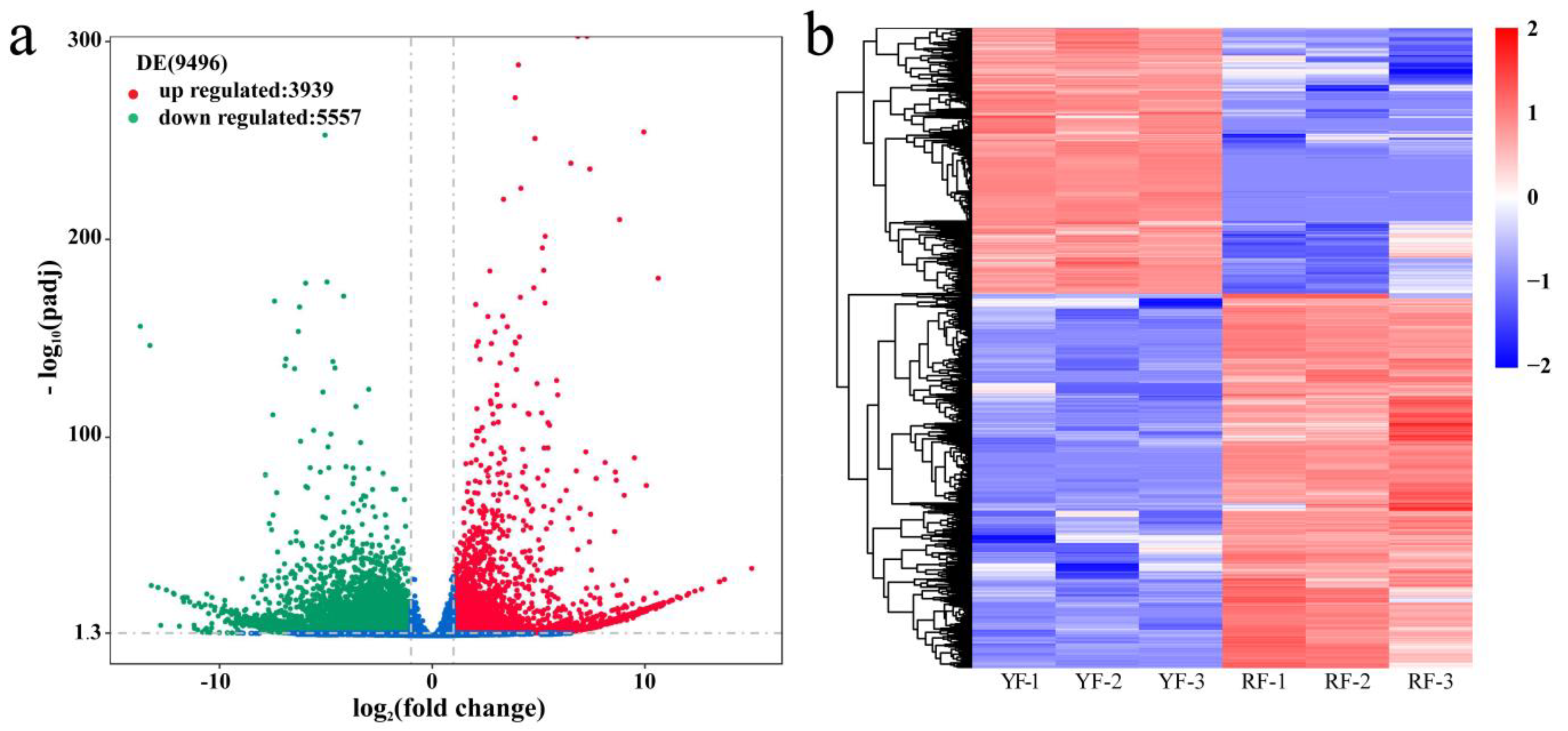
3.2. Assembly and Annotation of the C. humilis Fruit Peel Transcriptome
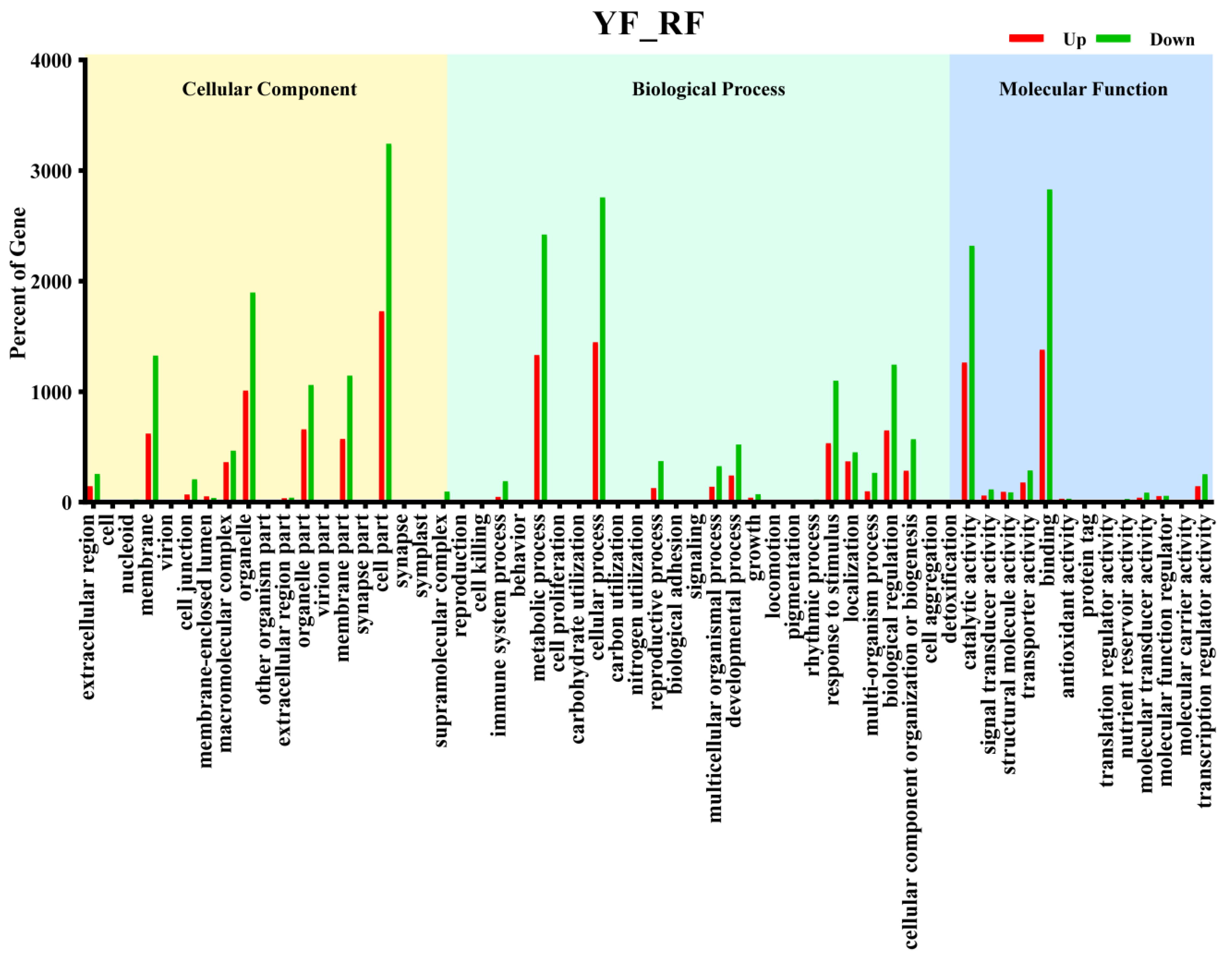
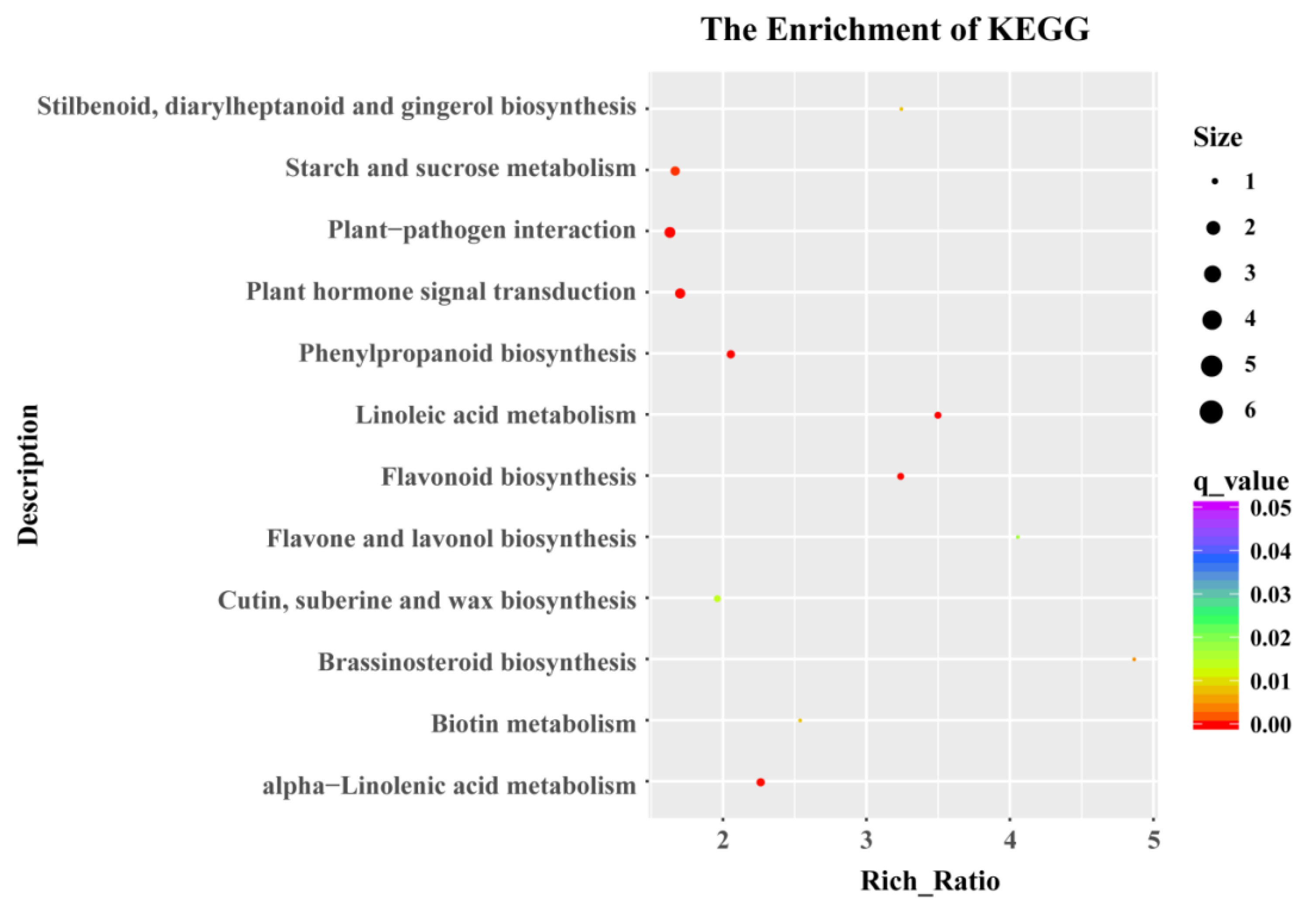
3.3. GO Enrichment and KEGG Pathway Analysis of DEGs
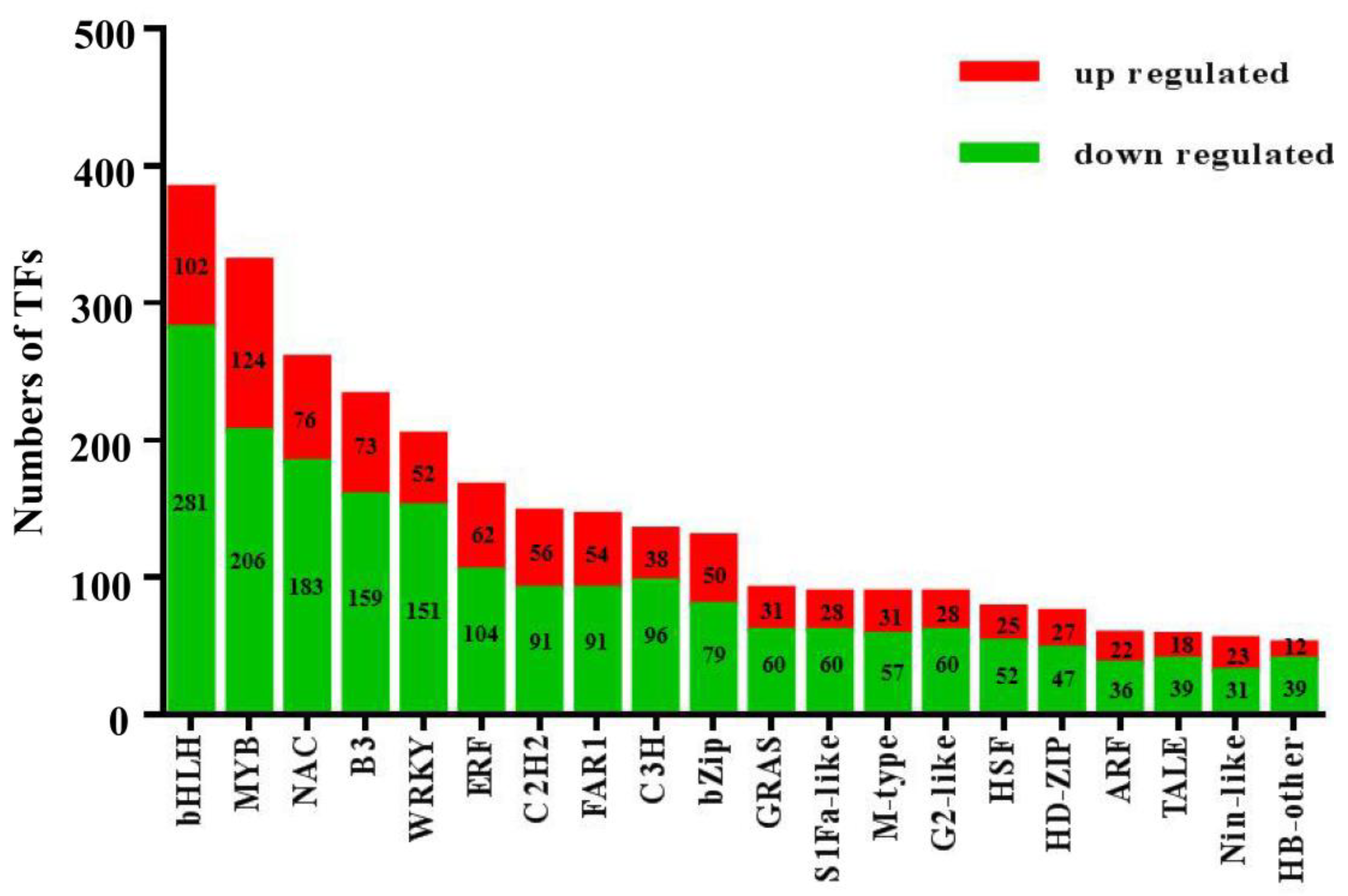
3.4. Analysis of Transcription Factors
3.5. Regulatory Network of Anthocyanin Biosynthetics
3.6. Integrated Analysis of the Transcriptome and Metabolome of Anthocyanins
4. Discussion
4.1. Effect of Content and Types of Anthocyanins on Coloration in Yellow and Red Fruit Peel
4.2. Key Genes in the Anthocyanin Pathway Affect Coloration in Yellow and Red Fruit Peel
4.3. Impact of Different Varieties on TFs
4.4. Integrated Analysis of the Transcriptome and Metabolome
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Yin, Z.; Ren, J.; Zhou, L.; Sun, L.; Wang, J.; Liu, Y.; Song, X. Water deficit mechanisms in perennial shrubs Cerasus humilis leaves revealed by physiological and proteomic analyses. Proteome Sci. 2016, 15, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, F.; Wang, J.W.; Sun, L.J.; Song, X.S. The molecular cloning and functional characterization of ChNAC1, a NAC transcription factor in Cerasus humilis. Plant Growth Regul. 2019, 89, 331–343. [Google Scholar] [CrossRef]
- Ren, J.; Sun, L.N.; Zhang, Q.Y.; Song, X.S. Drought Tolerance Is Correlated with the Activity of Antioxidant Enzymes in Cerasus humilis Seedlings. BioMed Res. Int. 2016, 2016, 9851095. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Mu, X.; Du, J.; Gao, Y.G.; Bai, D.; Jia, L.; Zhang, J.; Ren, H.; Xue, X. Flavonoid content and radical scavenging activity in fruits of Chinese dwarf cherry (Cerasus humilis) genotypes. J. For. Res. 2017, 29, 55–63. [Google Scholar] [CrossRef]
- Wang, R.F.; Huang, F.L.; Zhang, J.; Zhang, Q.Y.; Sun, L.N.; Song, X.S. Establishment of a high-frequency regeneration system in Cerasus humilis, an important economic shrub. J. For. Res. 2017, 21, 244–250. [Google Scholar] [CrossRef]
- Wang, P.; Mu, X.; Gao, Y.G.; Zhang, J.; Du, J. Successful induction and the systematic characterization of tetraploids in Cerasus humilis for subsequent breeding. Sci. Hortic. 2020, 265, 109216. [Google Scholar] [CrossRef]
- Fan, S.; Wu, P.; Xing, G. Nutritional Composition and Development of Chinese Dwarf Cherry (Cerasus Humilis (Bge.) Sok.). Int. J. Nutr. Food Sci. 2019, 8, 30. [Google Scholar] [CrossRef] [Green Version]
- Negi, P.; Chauhan, A.; Sadia, G.; Rohinishree, Y.; Ramteke, R. Antioxidant and antibacterial activities of various seabuckthorn ((Hippophae rhamnoides L.) seed extracts. Food Chem. 2005, 92, 119–124. [Google Scholar] [CrossRef]
- Bunea, A.; Rugina, D.; Sconta, Z.; Pop, R.M.; Pintea, A.; Socaciu, C.; Tabaran, F.; Grootaert, C.; Struijs, K.; VanCamp, J. Anthocyanin determination in blueberry extracts from various cultivars and their antiproliferative and apoptotic properties in B16-F10 metastatic murine melanoma cells. Phytochemistry 2013, 95, 436–444. [Google Scholar] [CrossRef]
- Borges, G.; Degeneve, A.; Mullen, W.; Crozier, A. Identification of flavonoid and phenolic antioxidants in black currants, blueberries, raspberries, red currants, and cranberries. J. Agric. Food Chem. 2010, 58, 3901–3909. [Google Scholar] [CrossRef]
- Yang, B.; He, S.; Liu, Y.; Liu, B.; Ju, Y.; Kang, D.; Sun, X.; Fang, Y. Transcriptomics integrated with metabolomics reveals the effect of regulated deficit irrigation on anthocyanin biosynthesis in Cabernet Sauvignon grape berries. Food Chem. 2020, 314, 126170. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Fang, J.; Qi, X.; Lin, M.; Zhong, Y.; Sun, L.; Cui, W. Combined Analysis of the Fruit Metabolome and Transcriptome Reveals Candidate Genes Involved in Flavonoid Biosynthesis in Actinidia arguta. Int. J. Mol. Sci. 2018, 19, 1471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Zhou, T.; Dai, Z.; Dai, X.; Li, W.; Cao, M.; Li, C.; Tsai, W.C.; Wu, X.; Zhai, J.; et al. Comparative Transcriptomics Provides Insight into Floral Color Polymorphism in a Pleione limprichtii Orchid Population. Int. J. Mol. Sci. 2019, 21, 247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, Y.; Sasaki, N.; Ohmiya, A. Biosynthesis of plant pigments: Anthocyanins, betalains and carotenoids. Plant J. 2008, 54, 733–749. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Giusti, M.M. Anthocyanins: Natural colorants with health-promoting properties. Annu. Rev. Food Sci. Technol. 2010, 1, 163–187. [Google Scholar] [CrossRef]
- Welch, C.R.; Wu, Q.; Simon, J.E. Recent Advances in Anthocyanin Analysis and Characterization. Curr. Anal. Chem. 2008, 4, 75–101. [Google Scholar] [CrossRef] [Green Version]
- Cheng, J.; Wei, G.; Zhou, H.; Gu, C.; Vimolmangkang, S.; Liao, L.; Han, Y. Unraveling the mechanism underlying the glycosylation and methylation of anthocyanins in peach. Plant Physiol. 2014, 166, 1044–1058. [Google Scholar] [CrossRef] [Green Version]
- Li, W.F.; Mao, J.; Yang, S.J.; Guo, Z.G.; Ma, Z.H.; Dawuda, M.M.; Zuo, C.W.; Chu, M.Y.; Chen, B.H. Anthocyanin accumulation correlates with hormones in the fruit skin of ’Red Delicious’ and its four generation bud sport mutants. BMC Plant Biol. 2018, 18, 363. [Google Scholar] [CrossRef] [Green Version]
- Xue, L.; Wang, Z.; Zhang, W.; Li, Y.; Wang, J.; Lei, J. Flower pigment inheritance and anthocyanin characterization of hybrids from pink-flowered and white-flowered strawberry. Sci. Hortic. 2016, 200, 143–150. [Google Scholar] [CrossRef]
- Yang, Y.; Yao, G.; Yue, W.; Zhang, S.; Wu, J. Transcriptome profiling reveals differential gene expression in proanthocyanidin biosynthesis associated with red/green skin color mutant of pear (Pyrus communis L.). Front. Plant Sci. 2015, 6, 795. [Google Scholar] [CrossRef] [Green Version]
- Kong, J.M.; Chia, L.S.; Goh, N.K.; Chia, T.F.; Brouillard, R. Analysis and biological activities of anthocyanins. Phytochemistry 2003, 64, 923–933. [Google Scholar] [CrossRef]
- Fang, H.; Dong, Y.; Yue, X.; Hu, J.; Jiang, S.; Xu, H.; Wang, Y.; Su, M.; Zhang, J.; Zhang, Z.; et al. The B-box zinc finger protein MdBBX20 integrates anthocyanin accumulation in response to ultraviolet radiation and low temperature. Plant Cell Environ. 2019, 42, 2090–2104. [Google Scholar] [CrossRef] [PubMed]
- Crifo, T.; Petrone, G.; Lo Cicero, L.; Lo Piero, A.R. Short cold storage enhances the anthocyanin contents and level of transcripts related to their biosynthesis in blood oranges. J. Agric. Food Chem. 2012, 60, 476–481. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Ling, W.; Guo, H.; Song, F.; Ye, Q.; Zou, T.; Li, D.; Zhang, Y.; Li, G.; Xiao, Y.; et al. Anti-inflammatory effect of purified dietary anthocyanin in adults with hypercholesterolemia: A randomized controlled trial. Nutr. Metab. Cardiovasc. Dis. 2013, 23, 843–849. [Google Scholar] [CrossRef]
- Inaguma, T.; Han, J.; Isoda, H. Improvement of insulin resistance by Cyanidin 3-glucoside, anthocyanin from black beans through the up-regulation of GLUT4 gene expression. BMC Proc. 2011, 5 (Suppl. 8), P21. [Google Scholar] [CrossRef] [Green Version]
- Hwang, J.W.; Kim, E.K.; Lee, S.J.; Kim, Y.S.; Choi, D.K.; Park, T.K.; Moon, S.H.; Jeon, B.T.; Park, P.J. Anthocyanin effectively scavenges free radicals and protects retinal cells from H2O2-triggered G2/M arrest. Eur. Food Res. Technol. 2011, 234, 431–439. [Google Scholar] [CrossRef]
- Kuhnen, S.; Menel Lemos, P.M.; Campestrini, L.H.; Ogliari, J.B.; Dias, P.F.; Maraschin, M. Carotenoid and anthocyanin contents of grains of Brazilian maize landraces. J. Sci. Food Agric. 2011, 91, 1548–1553. [Google Scholar] [CrossRef]
- Abdel-Aal, E.S.M.; Abou-Arab, A.A.; Gamel, T.H.; Hucl, P.; Young, J.C.; Rabalski, I. Fractionation of Blue Wheat Anthocyanin Compounds and Their Contribution to Antioxidant Properties. J. Agric. Food Chem. 2008, 56, 11171–11177. [Google Scholar] [CrossRef]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef]
- Reuter, J.A.; Spacek, D.V.; Snyder, M.P. High-throughput sequencing technologies. Mol. Cell 2015, 58, 586–597. [Google Scholar] [CrossRef] [Green Version]
- Xu, S.; Huang, Q.; Lin, C.; Lin, L.; Zhou, Q.; Lin, F.; He, E. Transcriptome comparison reveals candidate genes responsible for the betalain-/anthocyanidin-production in bougainvilleas. Funct. Plant Biol. 2016, 43, 278–286. [Google Scholar] [CrossRef] [PubMed]
- Yu, R.; Du, X.; Li, J.; Liu, L.; Hu, C.; Yan, X.; Xia, Y.; Xu, H. Identification and differential expression analysis of anthocyanin biosynthetic genes in root-skin color variants of radish (Raphanus sativus L.). Genes Genom. 2020, 42, 413–424. [Google Scholar] [CrossRef] [PubMed]
- Zou, S.; Wu, J.; Shahid, M.Q.; He, Y.; Lin, S.; Liu, Z.; Yang, X. Identification of key taste components in loquat using widely targeted metabolomics. Food Chem. 2020, 323, 126822. [Google Scholar] [CrossRef] [PubMed]
- Yan, N.; Du, Y.; Liu, X.; Chu, M.; Shi, J.; Zhang, H.; Liu, Y.; Zhang, Z. A comparative UHPLC-QqQ-MS-based metabolomics approach for evaluating Chinese and North American wild rice. Food Chem. 2019, 275, 618–627. [Google Scholar] [CrossRef]
- Johanningsmeier, S.D.; Harris, G.K.; Klevorn, C.M. Metabolomic Technologies for Improving the Quality of Food: Practice and Promise. Annu. Rev. Food Sci. Technol. 2016, 7, 413–438. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Tian, C.; Zhang, Y.; Li, C.; Li, X.; Yu, Q.; Wang, S.; Wang, X.; Chen, X.; Feng, S. Transcriptomic and metabolomic analysis provides insights into anthocyanin and procyanidin accumulation in pear. BMC Plant Biol. 2020, 20, 129. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Yan, J.; Li, W.; Wang, Q.; Wang, C.; Guo, J.; Geng, D.; Guan, Q.; Ma, F. Integrative Analyses of Widely Targeted Metabolic Profiling and Transcriptome Data Reveals Molecular Insight into Metabolomic Variations during Apple (Malus domestica) Fruit Development and Ripening. Int. J. Mol. Sci. 2020, 21, 4797. [Google Scholar] [CrossRef]
- Wan, L.; Lei, Y.; Yan, L.; Liu, Y.; Pandey, M.K.; Wan, X.; Varshney, R.K.; Fang, J.; Liao, B. Transcriptome and metabolome reveal redirection of flavonoids in a white testa peanut mutant. BMC Plant Biol. 2020, 20, 161. [Google Scholar] [CrossRef] [Green Version]
- Holton, T.A.; Cornish, E.C. Genetics and Biochemistry of Anthocyanin Biosynthesis. Plant Cell. 1995, 7, 1071–1083. [Google Scholar] [CrossRef]
- Twyford, A.D.; Caola, A.M.; Choudhary, P.; Raina, R.; Friedman, J. Loss of Color Pigmentation Is Maintained at High Frequency in a Monkey Flower Population. Am. Nat. 2018, 191, 135–145. [Google Scholar] [CrossRef]
- Wishart, D.S.; Tzur, D.; Knox, C.; Eisner, R.; Guo, A.C.; Young, N.; Cheng, D.; Jewell, K.; Arndt, D.; Sawhney, S.; et al. HMDB: The Human Metabolome Database. Nucleic Acids Res. 2007, 35, D521–D526. [Google Scholar] [CrossRef] [PubMed]
- Fraga, C.G.; Clowers, B.H.; Moore, R.J.; Zink, E.M. Signature-Discovery Approach for Sample Matching of a Nerve-Agent Precursor Using Liquid Chromatography-Mass Spectrometry, XCMS, and Chemometrics. Anal Chem. 2010, 82, 4165–4173. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Gong, L.; Guo, Z.; Wang, W.; Zhang, H.; Liu, X.; Yu, S.; Xiong, L.; Luo, J. A novel integrated method for large-scale detection, identification, and quantification of widely targeted metabolites: Application in the study of rice metabolomics. Mol. Plant. 2013, 6, 1769–1780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, C.; Yang, H.; Xu, Q.; Wang, Y.; Sang, Z.; Yuan, H. Comparative metabolomics analysis of the response to cold stress of resistant and susceptible Tibetan hulless barley (Hordeum distichon). Phytochemistry 2020, 174, 112346. [Google Scholar] [CrossRef]
- Su, P.; Zhao, L.; Li, W.; Zhao, J.; Yan, J.; Ma, X.; Li, A.; Wang, H.; Kong, L. Integrated metabolo-transcriptomics and functional characterization reveals that the wheat auxin receptor TIR1 negatively regulates defense against Fusarium graminearum. J. Int. Plant Biol. 2020. [Google Scholar] [CrossRef]
- Simao, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [Green Version]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [Green Version]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Feng, Z.; Wang, X.; Wang, X.; Zhang, X. DEGseq: An R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 2010, 26, 136–138. [Google Scholar] [CrossRef]
- Zhou, C.; Mei, X.; Rothenberg, D.O.; Yang, Z.; Zhang, W.; Wan, S.; Yang, H.; Zhang, L. Metabolome and Transcriptome Analysis Reveals Putative Genes Involved in Anthocyanin Accumulation and Coloration in White and Pink Tea (Camellia sinensis) Flower. Molecules 2020, 25, 190. [Google Scholar] [CrossRef] [Green Version]
- He, Q.; Shen, Y.; Wang, M.; Huang, M.; Yang, R.; Zhu, S.; Wang, L.; Xu, Y.; Wu, R. Natural variation in petal color in Lycoris longituba revealed by anthocyanin components. PLoS ONE 2011, 6, e22098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Wang, L.; Liu, Z.; Zhao, Z.; Zhao, J.; Wang, Z.; Zhou, G.; Liu, P.; Liu, M. Transcriptome and metabolome profiling unveil the mechanisms of Ziziphus jujuba Mill. peel coloration. Food Chem. 2020, 312, 125903. [Google Scholar] [CrossRef] [PubMed]
- Ono, E.; Fukuchi-Mizutani, M.; Nakamura, N.; Fukui, Y.; Yonekura-Sakakibara, K.; Yamaguchi, M.; Nakayama, T.; Tanaka, T.; Kusumi, T.; Tanaka, Y. Yellow flowers generated by expression of the aurone biosynthetic pathway. Proc. Natl. Acad. Sci. USA 2006, 103, 11075–11080. [Google Scholar] [CrossRef] [Green Version]
- Shi, S.G.; Li, S.J.; Kang, Y.X.; Liu, J.J. Molecular characterization and expression analyses of an anthocyanin synthase gene from Magnolia sprengeri Pamp. Appl. Biochem. Biotech. 2015, 175, 477–488. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Gong, Q.; Ni, X.; Zhou, Y.; Gao, Z. UFGT: The Key Enzyme Associated with the Petals Variegation in Japanese Apricot. Front. Plant Sci. 2017, 8, 108. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Pan, W.; Jin, S.; Lin, S. Combined metabolomic and transcriptomic analysis reveals key candidate genes involved in the regulation of flavonoid accumulation in Anoectochilus roxburghii. Process Biochem. 2020, 91, 339–351. [Google Scholar] [CrossRef]
- Meng, X.; Yin, B.; Feng, H.L.; Zhang, S.; Liang, X.Q.; Meng, Q.W. Overexpression of R2R3-MYB gene leads to accumulation of anthocyanin and enhanced resistance to chilling and oxidative stress. Biol. Plant. 2014, 58, 121–130. [Google Scholar] [CrossRef]
- Yao, G.; Ming, M.; Allan, A.C.; Gu, C.; Li, L.; Wu, X.; Wang, R.; Chang, Y.; Qi, K.; Zhang, S.; et al. Map-based cloning of the pear gene MYB114 identifies an interaction with other transcription factors to coordinately regulate fruit anthocyanin biosynthesis. Plant J. 2017, 92, 437–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Z.; Li, G.; Liu, L.; Zhang, Q.; Han, Z.; Chen, X.; Li, B. A R2R3-MYB Transcription Factor, VvMYBC2L2, Functions as a Transcriptional Repressor of Anthocyanin Biosynthesis in Grapevine (Vitis vinifera L.). Molecules 2018, 24, 92. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Index | Q1 (Da) | Q3 (Da) | Molecular Weight (Da) | Compounds | RF | SE | YF | SE |
|---|---|---|---|---|---|---|---|---|
| pma1590 | 463.12 | 301.1 | 463.12 | Peonidin O-hexoside | 4.88 × 106 | 8.01 × 105 | 2.84 × 106 | 2.04 × 106 |
| pmb0542 | 535.10 | 287.2 | 535.10 | Cyanidin 3-O-malonylhexoside | 6.49 × 104 | 3.55 × 104 | 5.02 × 104 | 1.47 × 104 |
| pmb0545 | 477.10 | 315.1 | 477.10 | Rosinidin O-hexoside | 8.57 × 104 | 1.42 × 104 | 5.00 × 104 | 3.57 × 104 |
| pmb0837 | 577.10 | 289.1 | 576.10 | Procyanidin A3 | 9.04 × 105 | 2.31 × 105 | 5.68 × 105 | 3.37 × 105 |
| pmb2957 | 465.10 | 285.2 | 466.10 | Cyanidin O-syringic acid | 7.77 × 107 | 2.76 × 107 | 5.27 × 107 | 2.50 × 107 |
| pmb2962 | 473.10 | 269.1 | 474.10 | Pelargonidin O-acetylhexoside | 4.27 × 105 | 3.03 × 105 | 3.65 × 105 | 6.22 × 104 |
| pme0094 | 447.30 | 285.1 | 448.30 | Cyanidin 3-O-glucoside (Kuromanin) | 8.81 × 106 | 2.57 × 106 | 5.69 × 106 | 3.12 × 106 |
| pme0431 | 577.00 | 289.1 | 576.13 | Procyanidin A1 | 5.34 × 106 | 3.00 × 106 | 4.17 × 106 | 1.17 × 106 |
| pme0432 | 575.00 | 287.1 | 576.13 | Procyanidin A2 | 4.84 × 104 | 2.84 × 104 | 3.84 × 104 | 1.00 × 104 |
| pme0434 | 577.00 | 125.2 | 578.14 | Procyanidin B2 | 5.29 × 106 | 5.74 × 105 | 2.93 × 106 | 2.36 × 106 |
| pme0436 | 577.10 | 125.2 | 578.14 | Procyanidin B3 | 5.70 × 106 | 1.99 × 106 | 3.85 × 106 | 1.86 × 106 |
| pme1773 | 595.00 | 287.2 | 595.00 | Cyanidin 3-O-rutinoside (Keracyanin) | 7.50 × 107 | 4.54 × 107 | 6.02 × 107 | 1.48 × 107 |
| pme1777 | 611.00 | 287.1 | 611.00 | Cyanidin 3,5-O-diglucoside (Cyanin) | 6.50 × 105 | 5.03 × 105 | 5.76 × 105 | 7.36 × 104 |
| pme3392 | 433.10 | 271.1 | 433.10 | Pelargonidin 3-O-beta-D-glucoside (Callistephin chloride) | 1.1.3 × 108 | 5.29 × 107 | 8.29 × 107 | 3.01 × 107 |
| pmf0027 | 449.10 | 287.2 | 448.10 | Cyanidin 3-O-galactoside | 3.10 × 107 | 1.08 × 107 | 2.09 × 107 | 1.01 × 107 |
| pmf0203 | 463.12 | 301.1 | 498.09 | Peonidin 3-O-glucoside chloride | 4.26 × 106 | 4.38 × 105 | 2.35 × 106 | 1.91 × 106 |
| Sample | Raw_Reads | Clean_Reads | Q30 | GC |
|---|---|---|---|---|
| RF-1(S4A) | 50709208 | 46869074 | 93.42% | 46.1% |
| RF-2(S4B) | 49852844 | 46851150 | 93.56% | 46.23% |
| RF-3(S4C) | 48348076 | 45129428 | 93.45% | 46.02% |
| YF-1(YA) | 44470648 | 40894112 | 93.91% | 46.37% |
| YF-2(YB) | 45243738 | 41408178 | 93.92% | 46.57% |
| YF-3(YC) | 46114526 | 41893004 | 93.99% | 46.49% |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ji, X.; Ren, J.; Lang, S.; Wang, D.; Zhu, L.; Song, X. Differential Regulation of Anthocyanins in Cerasus humilis Fruit Color Revealed by Combined Transcriptome and Metabolome Analysis. Forests 2020, 11, 1065. https://doi.org/10.3390/f11101065
Ji X, Ren J, Lang S, Wang D, Zhu L, Song X. Differential Regulation of Anthocyanins in Cerasus humilis Fruit Color Revealed by Combined Transcriptome and Metabolome Analysis. Forests. 2020; 11(10):1065. https://doi.org/10.3390/f11101065
Chicago/Turabian StyleJi, Xiaolong, Jing Ren, Shaoyu Lang, Di Wang, Liwei Zhu, and Xingshun Song. 2020. "Differential Regulation of Anthocyanins in Cerasus humilis Fruit Color Revealed by Combined Transcriptome and Metabolome Analysis" Forests 11, no. 10: 1065. https://doi.org/10.3390/f11101065