Update of Genetic Linkage Map and QTL Analysis for Growth Traits in Eucommia ulmoides Oliver


Abstract
1. Introduction
2. Materials and Methods
2.1. Mapping Population
2.2. DNA Extraction
2.3. Simple Sequence Repeat (SSR) Analysis
2.4. Segregation Analysis and Linkage Map Construction
2.5. Growth Traits Assessment
2.6. Quantitative Trait Loci (QTL) Analysis
3. Results
3.1. SSR Analysis
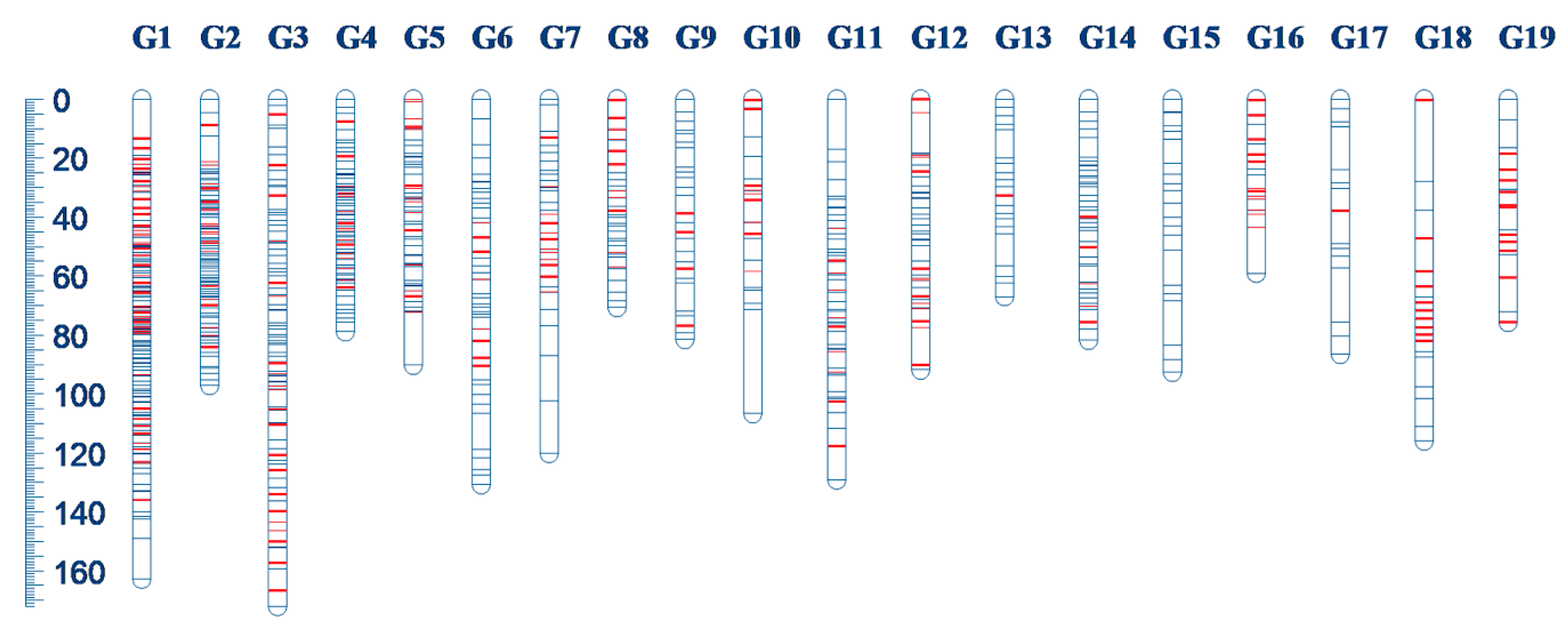
3.2. Genetics Linkage Map
3.3. Growth Traits
3.4. QTL Analysis
4. Discussion
4.1. SSR Marker and Segregation Distortion
4.2. Genetic Linkage Map
4.3. Growth Traits
4.4. QTL Analysis
4.5. Candidate Genes for QTLs
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Hu, S.Y. A contribution to our knowledge of Tu-chung—Eucommia ulmoides. Am. J. Chin. Med. 1979, 7, 5–37. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Xie, B.; Shao, S. Prospects and research progress of Gutta-percha. J. Central South For. Univ. 2003, 23, 95–98. [Google Scholar]
- Nakazawa, Y.; Bamba, T.; Takeda, T.; Uefuji, H.; Harada, Y.; Li, X.; Chen, R.; Inoue, S.; Tutumi, M.; Shimizu, T. Production of Eucommia-rubber from Eucommia ulmoides Oliv. (Hardy Rubber Tree). Plant Biotechnol. 2009, 26, 71–79. [Google Scholar] [CrossRef]
- Sih, C.J.; Ravikumar, P.R.; Huang, F.C.; Buckner, C.; Whitlock, H., Jr. Isolation and synthesis of pinoresinol diglucoside, a major antihypertensive principle of Tu-Chung (Eucommia ulmoides, Oliver). J. Am. Chem. Soc. 1976, 98, 5412–5413. [Google Scholar] [CrossRef]
- Kawasaki, T.; Uezono, K.; Nakazawa, Y. Antihypertensive mechanism of food for specified health use: “Eucommia leaf glycoside” and its clinical application. J. Health Sci. 2000, 22, 29–36. [Google Scholar]
- Hirata, T.; Kobayashi, T.; Wada, A.; Ueda, T.; Fujikawa, T.; Miyashita, H.; Ikeda, T.; Tsukamoto, S.; Nohara, T. Anti-obesity compounds in green leaves of Eucommia ulmoides. Bioorganic Med. Chem. Lett. 2011, 21, 1786–1791. [Google Scholar] [CrossRef]
- Jin, X.; Amitani, K.; Zamami, Y.; Takatori, S.; Hobara, N.; Kawamura, N.; Hirata, T.; Wada, A.; Kitamura, Y.; Kawasaki, H. Ameliorative effect of Eucommia ulmoides Oliv. leaves extract (ELE) on insulin resistance and abnormal perivascular innervation in fructose-drinking rats. J. Ethnopharmacol. 2010, 128, 672–678. [Google Scholar] [CrossRef]
- Chen, R.; Namimatsu, S.; Nakadozono, Y.; Bamba, T.; Nakazawa, Y.; Gyokusen, K. Efficient regeneration of Eucommia ulmoides from hypocotyl explant. Biol. Plant. 2008, 52, 713–717. [Google Scholar] [CrossRef]
- Du, H.; Du, L.; Li, F. Dynamic of Gutta-percha formation and accumulation in Samara of Eucommia ulmoides. For. Res. 2004, 17, 185–191. [Google Scholar]
- Du, H.Y.; Li, F.D.; Yang, S.B.; Fu, J.M.; Duan, J.H.; Du, L.Y.; Li, F.H. An elite variety for Samara use: Eucommia ulmoides “Huazhong NO. 9”. Scientia Silvae Sinicae 2008, 46, 182. [Google Scholar]
- Dong, J.; Ma, X.; Wei, Q.; Peng, S.; Zhang, S. Effects of growing location on the contents of secondary metabolites in the leaves of four selected superior clones of Eucommia ulmoides. Ind. Crop. Prod. 2011, 34, 1607–1614. [Google Scholar] [CrossRef]
- Wei, Y.C.; Li, Z.Q.; Li, Y.; Li, C. Genetic analysis of hybrid offspring morphological traits in Eucommia ulmoides Oliver. J. Northwest AF Univ. Nat. Sci. Ed. 2012, 40, 137–143. [Google Scholar]
- Priyadarshan, P.M. Breeding hevea rubber: Formal and molecular genetics. Adv. Genet. 2004, 52, 51–115. [Google Scholar] [PubMed]
- Grattapaglia, D.; Sederoff, R. Genetic linkage maps of Eucalyptus grandis and Eucalyptus urophylla using a pseudo-testcross: Mapping strategy and RAPD markers. Genetics 1994, 137, 1121–1137. [Google Scholar]
- Tong, C.; Li, H.; Wang, Y.; Li, X.; Shi, J. Construction of high-density linkage maps of Populus deltoides × P. simonii using restriction-site associated DNA sequencing. PLoS ONE 2016, 11, e0150692. [Google Scholar] [CrossRef]
- Conson, A.R.O.; Taniguti, C.H.; Amadeu, R.R.; Andreotti, I.A.A.; de Souza, L.M.; dos Santos, L.H.B.; Rosa, J.R.B.F.; Mantello, C.C.; da Silva, C.C.; José Scaloppi, E., Jr.; et al. High-resolution genetic map and QTL analysis of growth-related traits of Hevea brasiliensis cultivated under suboptimal temperature and humidity conditions. Front. Plant Sci. 2018, 9. [Google Scholar] [CrossRef]
- Emeriewen, O.F.; Richter, K.; Piazza, S.; Micheletti, D.; Broggini, G.A.L.; Berner, T.; Keilwagen, J.; Hanke, M.-V.; Malnoy, M.; Peil, A. Towards map-based cloning of FB_Mfu10: Identification of a receptor-like kinase candidate gene underlying the Malus fusca fire blight resistance locus on linkage group. Mol. Breed. 2018, 38, 106. [Google Scholar] [CrossRef]
- Santos, C.; Nelson, C.D.; Zhebentyayeva, T.; Machado, H.; Costa, R.L. First interspecific genetic linkage map for Castanea sativa × Castanea crenata revealed QTLs for resistance to Phytophthora cinnamomi. PLoS ONE 2017, 12, e0184381. [Google Scholar] [CrossRef]
- Goonetilleke, S.N.; March, T.J.; Wirthensohn, M.G.; Arús, P.; Mather, D.E. Genotyping-by-sequencing in almond: SNP discovery, linkage mapping and marker design. G3 Genes Genomes Genet. 2017, 8, 161–172. [Google Scholar] [CrossRef]
- Xia, Z.; Zhang, S.; Wen, M.; Lu, C.; Sun, Y.; Zou, M.; Wang, W. Construction of an ultrahigh-density genetic linkage map for Jatropha curcas L. and identification of QTL for fruit yield. Biotechnol. Biofuels 2018, 11, 3. [Google Scholar] [CrossRef]
- Li, Y.; Wang, D.; Li, Z.; Wei, J.; Jin, C.; Liu, M. A molecular genetic linkage map of Eucommia ulmoides and quantitative trait loci (QTL) analysis for growth traits. Int. J. Mol. Sci. 2014, 15, 2053–2074. [Google Scholar] [CrossRef] [PubMed]
- Cangfu, J.; Zhouqi, L.; Yu, L.; Shuhui, W.; Long, L.; Minhao, L.; Jing, Y. Transcriptome analysis of terpenoid biosynthetic genes and simple sequence repeat marker screening in Eucommia ulmoides. Mol. Biol. Rep. 2020, 47, 1979–1990. [Google Scholar] [CrossRef]
- Wang, D.; Li, Y.; Li, L.; Wei, Y.; Li, Z. The first genetic linkage map of Eucommia ulmoides. J. Genet. 2014, 93, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Porebski, S.; Bailey, L.G.; Baum, B.R. Modification of a CTAB DNA extraction protocol for plants containing high polysaccharide and polyphenol components. Plant Mol. Biol. Rep. 1997, 15, 8–15. [Google Scholar] [CrossRef]
- Wu, M.; Du, H.; Wuyun, T.; Liu, P.; Jing, T. Characterization of Genomic MICROSATELLLITES and development of SSR markers of Eucommia ulmoides. For. Res. 2015, 28, 89–95. [Google Scholar]
- Ooijen, J.W.V. JoinMap 4. In Software for the Calculation of Genetic Linkage Maps in Experimental Populations; Kyazma B.V.: Wageningen, The Netherlands, 2006. [Google Scholar]
- Voorrips, R.E. MapChart: Software for the graphical presentation of linkage maps and QTLs. J. Hered. 2002, 93, 77–78. [Google Scholar] [CrossRef]
- Chakravarti, A.; Lasher, L.K.; Reefer, J.E. A maximum likelihood method for estimating genome length using genetic linkage data. Genetics 1991, 128, 175–182. [Google Scholar]
- Ooijen, J.W.; van Kessel, J.; van’t Verlaat, J.W.; Van, O.; van der Meer, J.; Ooijen, J.W.V. MapQTL 5. In Software for the Mapping of Quantitative Trait Loci in Experimental Populations; Kyazma B.V.: Wageningen, The Netherlands, 2004. [Google Scholar]
- Woolbright, S.A.; Rehill, B.J.; Lindroth, R.L.; Difazio, S.P.; Whitham, T.G. Large effect quantitative trait loci for salicinoid phenolic glycosides in Populus: Implications for gene discovery. Ecol. Evol. 2018, 8, 3726–3737. [Google Scholar] [CrossRef]
- Kefayati, S.; Ikhsan, A.S.; Sutyemez, M.; Paizila, A.; Kafkas, S. First simple sequence repeat-based genetic linkage map reveals a major QTL for leafing time in walnut (Juglans regia L.). Tree Genet. Genomes 2019, 15, 13. [Google Scholar] [CrossRef]
- Zhang, Z.; Gao, J.; Kong, D.; Wang, A.; Tang, S.; Li, Y.; Pang, X.M. Assessing genetic diversity in Ziziphus jujuba ‘Jinsixiaozao’ using morphological and microsatellite (SSR) markers. Biochem. Syst. Ecol. 2015, 61, 196–202. [Google Scholar] [CrossRef]
- Chang, Y.; Kim, H.B.; Oh, E.; Yi, K.; Song, K.J. Construction of genetic linkage maps of ‘Fina Sodea’ clementine (Citrus clementina) and Byungkyul (C. platymamma), a Korean landrace, based on RAPD and SSR markers. Hortic. Environ. Biotechnol. 2018, 59, 263–274. [Google Scholar] [CrossRef]
- Joy, K.; Yanfeng, D.; Xiaoyan, C.; Richard, M.; Zhongli, Z.; Xingxing, W.; Yuhong, W.; Zhenmei, Z.; Kunbo, W.; Fang, L. Simple sequence repeat (SSR) genetic linkage map of D genome diploid cotton derived from an interspecific cross between Gossypium davidsonii and Gossypium klotzschianum. Int. J. Mol. Sci. 2018, 19, 204. [Google Scholar]
- Jenczewski, E.; Gherardi, M.; Bonnin, I.; Prosperi, J.M.; Olivieri, I.; Huguet, T. Insight on segregation distortions in two intraspecific crosses between annual species of Medicago (Leguminosae). Theor. Appl. Genet. 1997, 94, 682–691. [Google Scholar] [CrossRef]
- Bradshaw, H.D.; Stettler, R.F. Molecular genetics of growth and development in Populus. II. Segregation distortion due to genetic load. Theor. Appl. Genet. 1994, 89, 551–558. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Lavalle, L.A.B.; Matheson, B.; Brubaker, C.L. A genetic map of an Australian wild Gossypium C genome and assignment of homoeologies with tetraploid cultivated cotton. Genome 2011, 54, 779–794. [Google Scholar] [CrossRef] [PubMed]
- Alheit, K.V.; Reif, J.C.; Maurer, H.P.; Hahn, V.; Weissmann, E.A.; Miedaner, T.; Würschum, T. Detection of segregation distortion loci in triticale (x Triticosecale Wittmack) based on a high-density DArT marker consensus genetic linkage map. BMC Genom. 2011, 12, 380. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wei, Y.C.; Li, Z.Q.; Wang, S.H.; Chang, L. Relationship between progeny growth performance and molecular marker-based genetic distances in Eucommia ulmoides parental genotypes. Genet. Mol. Res. 2014, 13, 4736–4746. [Google Scholar] [CrossRef]
- Motalebipour, E.Z.; Gozel, H.; Khodaeiaminjan, M.; Kafkas, S. SSR-based genetic linkage map construction in pistachio using an interspecific F1 population and QTL analysis for leaf and shoot traits. Mol. Breed. 2018, 38, 134. [Google Scholar] [CrossRef]
- Mortaza, K.; Salih, K.; Ziya, M.E.; Nergiz, C. In silico polymorphic novel SSR marker development and the first SSR-based genetic linkage map in pistachio. Tree Genet. Genomes 2018, 14, 45. [Google Scholar]
- Uttam, B.; Prasanta, K.S. Identification of drought responsive QTLs during vegetative growth stage of rice using a saturated GBS-based SNP linkage map. Euphytica 2018, 214, 38. [Google Scholar] [CrossRef]
- Yang, Z.; Zhenyong, C.; Zhengsong, P.; Yan, Y.; Mingli, L.; Shuhong, W. Development of a high-density linkage map and mapping of the three-pistil gene (Pis1) in wheat using GBS markers. BMC Genom. 2017, 18, 567. [Google Scholar] [CrossRef] [PubMed]
- Zhigunov, A.V.; Ulianich, P.S.; Lebedeva, M.V.; Chang, P.L.; Nuzhdin, S.V.; Potokina, E.K. Development of F1 hybrid population and the high-density linkage map for European aspen (Populus tremula L.) using RADseq technology. BMC Plant Biol. 2017, 17, 180. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.Q.; Wang, L.Y.; Xu, L.Y.; Wu, L.Y.; Peng, M.; Zhang, C.C.; Wei, K.; Bai, P.X.; Li, H.L.; Cheng, H. SSR-based genetic mapping and QTL analysis for timing of spring bud flush, young shoot color, and mature leaf size in tea plant (Camellia sinensis). Tree Genet. Genomes 2016, 12, 52. [Google Scholar] [CrossRef]
- Beavis, W.D. The power and deceit of QTL experiments: Lessons from comparative QTL studies. In Proceedings of the Forty-Ninth Annual Corn and Sorghum Industry Research Conference; American Seed Trade Assocition: Washington, DC, USA, 1994; pp. 250–266. [Google Scholar]
- Bradshaw, H.D.; Stettler, R.F. Molecular genetics of growth and development in Populus. IV. mapping QTLs with large effects on growth, form, and phenology traits in a forest tree. Genetics 1997, 84, 143–153. [Google Scholar]
- Li, R.; Jiang, H.; Zhang, Z.; Zhao, Y.; Xie, J.; Wang, Q.; Zheng, H.; Hou, L.; Xiong, X.; Xin, D.; et al. Combined linkage mapping and BSA to identify QTL and candidate genes for plant height and the number of nodes on the main stem in Soybean. Int. J. Mol. Sci. 2019, 21, 42. [Google Scholar] [CrossRef]
- Yang, H.X.; Liu, T.; Liu, C.; Zhao, F. QTL detection for growth traits in Pinus elliottii var. elliottii and P. caribaea var. hondurensis. For. Ecosyst. 2013, 15, 196–205. [Google Scholar]
- Conner, P.J.; Brown, S.K.; Weeden, N.F. Molecular-marker analysis of quantitative traits for growth and development in juvenile apple trees. Theor. Appl. Genet. 1998, 96, 1027–1035. [Google Scholar] [CrossRef]
- Kenis, K.; Keulemans, J. Study of tree architecture of apple (Malus × domestica Borkh.) by QTL analysis of growth traits. Mol. Breed. 2007, 19, 193–208. [Google Scholar] [CrossRef]
- Thumma, B.R.; Baltunis, B.S.; Bell, J.C.; Emebiri, L.C.; Moran, G.F.; Southerton, S.G. Quantitative trait locus (QTL) analysis of growth and vegetative propagation traits in Eucalyptus nitens full-sib families. Tree Genet. Genomes 2010, 6, 877–889. [Google Scholar] [CrossRef]
- Freeman, J.S.; Whittock, S.P.; Potts, B.M.; Vaillancourt, R.E. QTL influencing growth and wood properties in Eucalyptus globulus. Tree Genet. Genomes 2009, 5, 713–722. [Google Scholar] [CrossRef]
- Fowler, S.; Lee, K.; Onouchi, H.; Samach, A.; Richardson, K.; Morris, B.; Coupland, G.; Putterill, J. GIGANTEA: A circadian clock-controlled gene that regulates photoperiodic flowering in Arabidopsis and encodes a protein with several possible membrane-spanning domains. EMBO J. 1999, 18, 4679–4688. [Google Scholar] [CrossRef] [PubMed]
- De Montaigu, A.; Coupland, G. The timing of GIGANTEA expression during day/night cycles varies with the geographical origin of Arabidopsis accessions. Plant Signal. Behav. 2017, 12, e1342026. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Böhlenius, H.; Rühl, M.; Chen, P.; Sane, S.; Zambrano, J.; Zheng, B.; Eriksson, M.; Nilsson, O. GIGANTEA-like genes control seasonal growth cessation in Populus. New Phytol. 2018, 218. [Google Scholar] [CrossRef] [PubMed]
- Basu, D.; Le, J.; El-Essal, E.-D.S.; Huang, S.; Zhang, C.; Mallery, E.L.; Koliantz, G.; Staiger, C.J.; Szymanski, D.B. DISTORTED3/SCAR2 is a putative Arabidopsis WAVE complex subunit that activates the Arp2/3 complex and is required for epidermal morphogenesis. Plant Cell 2005, 17, 502–524. [Google Scholar] [CrossRef] [PubMed]
- Grolig, F.; Moch, J.; Schneider, A.; Galland, P.; Palme, K. Actin cytoskeleton and organelle movement in the sporangiophore of the zygomycete Phycomyces blakesleeanus. Plant Biol. 2014, 16, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Li, L.-J.; Ren, F.; Gao, X.Q.; Wei, P.C.; Wang, X.C. The reorganization of actin filaments is required for vacuolar fusion of guard cells during stomatal opening in Arabidopsis. Plant Cell Environ. 2013, 36, 484–497. [Google Scholar] [CrossRef] [PubMed]
- Hao, X.; Li, H.; Hua, Y.; Yu, K.; Walter, M.; Qi, T.; Zhang, B.; Ma, Q. Actin dynamic polymerization is required for the expression of nonhost resistance in pepper against Blumeria graminis f. sp. tritici. Physiol. Mol. Plant Pathol. 2014, 86, 64–72. [Google Scholar] [CrossRef]
- Lanza, M.; Garcia-Ponce, B.; Castrillo, G.; Catarecha, P.; Sauer, M.; Rodriguez-Serrano, M.; Páez-García, A.; Sánchez-Bermejo, E.; TC, M.; Leo del Puerto, Y. Role of actin cytoskeleton in brassinosteroid signaling and in its integration with the auxin response in plants. Dev. Cell 2012, 22, 1275–1285. [Google Scholar] [CrossRef]
- Wagner, R.; Sydow, L.V.; Aigner, H.; Netotea, S.; Funk, C. Deletion of FtsH11 protease has impact on chloroplast structure and function in Arabidopsis thaliana when grown under continuous light. Plant Cell Environ. 2016, 39, 2530–2544. [Google Scholar] [CrossRef]
- Karata, K.; Inagawa, T.; Wilkinson, A.J.; Tatsuta, T.; Ogura, T. Dissecting the role of a conserved motif (the second region of homology) in the AAA family of ATPases: Site-directed mutagenesis of the ATP-dependent protease FtsH. J. Biol. Chem. 1999, 274, 26225–26232. [Google Scholar] [CrossRef]
- Kolodziejczak, M.; Skibior, B.R.; Janska, H. m-AAA complexes are not crucial for the survival of arabidopsis under optimal growth conditions despite their importance for mitochondrial translation. Plant Cell Physiol. 2018, 59, 1006. [Google Scholar] [CrossRef] [PubMed]
- Gan, W.; Rhoads, R.E. Internal initiation of translation directed by the 5′-untranslated region of the mRNA for eIF4G, a factor involved in the picornavirus-induced switch from cap-dependent to internal initiation. J. Biol. Chem. 1996, 271, 623–626. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Jolley, B.; Caldwell, C.; Gallie, D.R. Eukaryotic translation initiation factor eIFiso4G is required to regulate violaxanthin de-epoxidase expression in Arabidopsis. J. Biol. Chem. 2014, 289, 13926–13936. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Linkage Group | Number of Markers | Length | Mean Distance | Number of Added SSR |
|---|---|---|---|---|
| G1 | 151 | 162.93 | 1.08 | 44 |
| G2 | 94 | 97.34 | 1.04 | 19 |
| G3 | 93 | 172.21 | 1.85 | 21 |
| G4 | 72 | 79.09 | 1.10 | 15 |
| G5 | 53 | 90.51 | 1.71 | 16 |
| G6 | 42 | 130.89 | 3.12 | 8 |
| G7 | 33 | 120.22 | 3.64 | 13 |
| G8 | 31 | 70.71 | 2.28 | 11 |
| G9 | 30 | 81.71 | 2.72 | 4 |
| G10 | 21 | 106.84 | 5.09 | 9 |
| G11 | 55 | 129.42 | 2.35 | 10 |
| G12 | 42 | 91.90 | 2.19 | 14 |
| G13 | 20 | 67.09 | 3.35 | 1 |
| G14 | 42 | 81.82 | 1.95 | 5 |
| G15 | 21 | 92.88 | 4.42 | 0 |
| G16 | 17 | 59.13 | 3.48 | 12 |
| G17 | 15 | 86.63 | 5.78 | 1 |
| G18 | 19 | 116.12 | 6.11 | 10 |
| G19 | 18 | 75.85 | 4.21 | 11 |
| Total | 869 | 1913.29 | 2.20 | 224 |
| Mean | 46 | 100.70 | 2.20 | 11.79 |
| Traits 1 | Min. | Max. | Mean | Median | SD 2 | CV 3/% | Max/Min |
|---|---|---|---|---|---|---|---|
| H2010/cm | 9.00 | 85.00 | 39.25 | 37.00 | 16.80 | 42.81 | 9.44 |
| H2011/cm | 50.00 | 216.00 | 138.71 | 139.00 | 36.62 | 26.40 | 4.32 |
| H2012/cm | 120.00 | 310.00 | 224.84 | 230.00 | 48.38 | 21.52 | 2.58 |
| H2013/cm | 170.00 | 480.00 | 332.02 | 340.00 | 74.44 | 22.42 | 2.82 |
| H2014/cm | 178.00 | 500.00 | 380.84 | 400.00 | 79.23 | 20.80 | 2.81 |
| H2015/cm | 120.00 | 620.00 | 390.16 | 380.00 | 110.48 | 28.32 | 5.17 |
| H2016/cm | 260.00 | 640.00 | 457.04 | 450.00 | 80.05 | 17.51 | 2.46 |
| H2017/cm | 320.00 | 770.00 | 589.44 | 595.00 | 66.40 | 11.27 | 2.41 |
| H2018/cm | 400.00 | 770.00 | 690.20 | 700.00 | 43.47 | 6.30 | 1.93 |
| H2019/cm | 440.00 | 880.00 | 740.46 | 740.00 | 63.29 | 8.55 | 2.00 |
| GD2010/mm | 1.27 | 9.37 | 4.85 | 4.81 | 1.65 | 34.12 | 7.38 |
| GD2011/mm | 3.93 | 19.61 | 11.87 | 12.02 | 3.06 | 25.74 | 4.99 |
| GD2012/mm | 10.02 | 30.41 | 19.20 | 18.83 | 4.56 | 23.73 | 3.03 |
| GD2013/mm | 10.42 | 38.27 | 22.37 | 22.02 | 6.08 | 27.16 | 3.67 |
| GD2014/mm | 13.55 | 47.60 | 25.66 | 24.58 | 7.05 | 27.49 | 3.51 |
| GD2015/mm | 21.33 | 82.69 | 46.41 | 42.80 | 14.35 | 30.91 | 3.88 |
| GD2016/mm | 16.04 | 96.29 | 56.70 | 54.00 | 15.00 | 26.45 | 6.00 |
| GD2017/mm | 39.67 | 135.99 | 69.95 | 67.32 | 14.60 | 20.88 | 3.43 |
| GD2018/mm | 36.46 | 126.27 | 79.56 | 80.65 | 15.10 | 18.98 | 3.46 |
| GD2019/mm | 39.09 | 123.59 | 84.18 | 85.18 | 15.23 | 18.09 | 3.16 |
| C2012/cm | 17.50 | 93.00 | 56.60 | 56.00 | 14.99 | 26.49 | 5.31 |
| C2013/cm | 33.50 | 117.50 | 77.49 | 78.50 | 15.52 | 20.02 | 3.51 |
| C2014/cm | 45.00 | 145.00 | 88.75 | 87.50 | 17.18 | 19.36 | 3.22 |
| C2015/cm | 32.50 | 190.00 | 100.04 | 90.00 | 33.79 | 33.77 | 5.85 |
| C2016/cm | 52.00 | 232.50 | 147.72 | 145.00 | 28.92 | 19.58 | 4.47 |
| C2017/cm | 127.50 | 275.00 | 203.20 | 200.00 | 27.16 | 13.37 | 2.16 |
| C2018/cm | 125.00 | 315.00 | 211.89 | 204.50 | 43.85 | 20.70 | 2.52 |
| C2019/cm | 110.00 | 345.00 | 196.88 | 190.00 | 40.99 | 20.82 | 3.14 |
| Trait 1 | QTL 2 | GW 3 | LG 4 | Peak position/cM | LOD 5 | Marker 6 | Var./% 7 | KW 8 |
|---|---|---|---|---|---|---|---|---|
| H2010 | qh1-1 | 5.1 * | G18 | 6.000 | 5.56 | EQ457–200a | 50.4 | * |
| qh1-2 | 5.1 * | G1 | 49.833 | 6.40 | em2me13–70 | 19.6 | ******* | |
| qh1-3 | 5.1 * | G1 | 75.405 | 6.77 | EQ580–180b | 19.8 | ******* | |
| H2011 | qh2-1 | 5.0 * | G1 | 49.488 | 5.90 | em13me4–620 | 17.3 | ******* |
| H2012 | qh3-1 | 5.2 | G13 | 14.309 | 4.65 | em13me4–360 | 31.4 | ***** |
| qh3-2 | 5.2 | G18 | 42.652 | 3.67 | EQ1107–110b | 61.8 | - | |
| H2013 | qh4-1 | 5.7 | G13 | 10.309 | 3.85 | em13me4–360 | 40.7 | ** |
| qh4-2 | 5.7 | G13 | 49.603 | 3.35 | em3me9–250 | 50.0 | **** | |
| H2014 | qh5-1 | 7.6 | G13 | 14.309 | 5.57 | em13me4–360 | 54.4 | - |
| qh5-2 | 7.6 | G13 | 53.603 | 6.77 | em3me9–250 | 68.7 | - | |
| qh5-3 | 7.6 | G18 | 41.652 | 5.50 | em53me13–220 | 60.8 | - | |
| H2015 | qh6-1 | 5.2 * | G8 | 70.462 | 5.48 | em1me6–170 | 38.5 | ** |
| qh6-2 | 5.2 * | G19 | 3.000 | 6.62 | em32me7–125 | 43.7 | ** | |
| qh6-3 | 5.2 * | G1 | 47.811 | 14.19 | em1me26–2400 | 53.0 | - | |
| qh6-4 | 5.2 * | G1 | 74.980 | 17.69 | em49me3–150 | 42.9 | ******* | |
| qh6-5 | 5.2 * | G1 | 121.509 | 9.45 | DZ159–161c | 39.4 | *** | |
| H2016 | qh7-1 | 5.0 * | G1 | 47.811 | 11.40 | em1me26–2400 | 42.1 | - |
| qh7-2 | 5.0 * | G1 | 74.980 | 13.49 | em49me3–150 | 33.9 | ******* | |
| qh7-3 | 5.0 * | G1 | 121.509 | 7.60 | DZ159–161c | 38.3 | ** | |
| H2017 | qh8-1 | 4.6 * | G1 | 45.720 | 5.39 | DZ165–301c | 15.1 | ******* |
| qh8-2 | 4.6 * | G1 | 89.173 | 5.83 | E8M8–115 | 23.2 | - | |
| H2018 | qh9-1 | 12.5 | G1 | 82.364 | 5.14 | E8M8–125 | 19.7 | **** |
| qh9-2 | 12.5 | G7 | 27.381 | 8.62 | em3me2–510 | 41.0 | - | |
| H2019 | qh10-1 | 6.7 | G5 | 46.693 | 4.70 | em7me12–365 | 29.7 | - |
| qh10-2 | 6.7 | G1 | 82.364 | 3.38 | E8M8–125 | 13.8 | * | |
| GR-H | qgr-h-1 | 5.7 * | G1 | 49.833 | 6.69 | em2me13–70 | 19.0 | ******* |
| qgr-h-2 | 5.7 * | G1 | 97.243 | 6.77 | em13me4–480c | 18.5 | ******* | |
| qgr-h-3 | 5.7 | G5 | 46.182 | 4.95 | em7me12–365 | 39.0 | - | |
| qgr-h-4 | 5.7 | G10 | 20.233 | 3.04 | em21me6–600 | 16.9 | - | |
| qgr-h-5 | 5.7 | G18 | 9.000 | 4.99 | EQ457–200a | 52.7 | - | |
| GD2010 | qgd1-1 | 5.0 * | G1 | 49.833 | 6.62 | em2me13–70 | 27.7 | ******* |
| qgd1-4 | 5.0 * | G1 | 75.405 | 10.18 | EQ580–180b | 27.0 | ******* | |
| qgd1-5 | 5.0 * | G18 | 6.000 | 5.76 | EQ457–200a | 54.1 | * | |
| GD2011 | qgd2-1 | 4.8 * | G1 | 49.488 | 5.03 | em13me4–620 | 17.5 | ******* |
| qgd2-2 | 4.8 * | G1 | 107.738 | 4.98 | em1me4–140c | 14.0 | ****** | |
| GD2012 | qgd3-1 | 5.0 | G3 | 106.842 | 3.03 | E7M4–350 | 16.4 | - |
| qgd3-2 | 5.0 | G4 | 42.836 | 3.30 | em4me7–250 | 14.2 | *** | |
| qgd3-3 | 5.0 | G10 | 54.253 | 3.17 | EQ962–250 | 15.2 | ** | |
| qgd3-4 | 5.0 | G13 | 12.306 | 3.61 | em13me4–360 | 34.4 | **** | |
| qgd3-5 | 5.0 | G19 | 3.000 | 3.68 | em32me7–125 | 50.5 | ** | |
| GD2013 | qgd4-1 | 4.9 | G2 | 79.346 | 3.29 | UBC886–2200 | 23.6 | ** |
| qgd4-2 | 4.9 | G4 | 35.118 | 4.33 | em1me1–750 | 26.3 | - | |
| qgd4-3 | 4.9 | G10 | 56.583 | 3.38 | EQ962–250 | 18.3 | - | |
| qgd4-4 | 4.9 | G13 | 12.309 | 3.34 | em13me4–360 | 30.2 | **** | |
| GD2014 | qgd5-1 | 5.6 | G1 | 47.811 | 9.11 | em1me26–2400 | 39.2 | - |
| qgd5-2 | 5.6 | G1 | 89.939 | 6.70 | DZ159–260 | 23.9 | ** | |
| GD2015 | qgd6-1 | 10.3 * | G8 | 70.462 | 11.04 | em1me6–170 | 59.2 | *** |
| qgd6-2 | 10.3 * | G18 | 114.366 | 10.74 | UBC842–300 | 72.7 | - | |
| qgd6-3 | 10.3 * | G1 | 74.980 | 26.21 | em49me3–150 | 56.8 | ******* | |
| qgd6-4 | 10.3 * | G1 | 121.509 | 12.04 | DZ159–161c | 49.5 | *** | |
| qgd6-5 | 10.3 * | G1 | 47.811 | 15.94 | em1me26–2400 | 56.5 | - | |
| GD2016 | qgd7-1 | 5.4 * | G8 | 70.709 | 5.80 | em1me6–170 | 34.0 | ** |
| qgd7-2 | 5.4 * | G1 | 45.720 | 11.14 | DZ165–301c | 28.6 | ******* | |
| qgd7-3 | 5.4 * | G1 | 74.980 | 16.50 | em49me3–150 | 39.7 | ******* | |
| qgd7-4 | 5.4 * | G1 | 122.509 | 8.53 | em25me28–110c | 32.0 | ******* | |
| GD2017 | qgd8-1 | 5.5 * | G1 | 45.720 | 10.81 | DZ165–301c | 27.9 | ******* |
| qgd8-2 | 5.5 * | G1 | 81.112 | 14.48 | em1me14–900 | 48.8 | ******* | |
| GD2018 | qgd9-1 | 4.9 * | G1 | 58.103 | 11.40 | DZ4–141c | 29.2 | ******* |
| qgd9-2 | 4.9 * | G1 | 74.980 | 13.49 | em49me3–150 | 37.7 | ******* | |
| qgd9-3 | 4.9 * | G1 | 121.509 | 7.50 | DZ159–161c | 37.3 | * | |
| GD2019 | qgd10-1 | 5.9 * | G5 | 47.445 | 31.36 | em7me12–360 | 74.9 | ** |
| qgd10-2 | 5.9 * | G16 | 27.163 | 37.52 | em39-me11–2300 | 74.8 | - | |
| GR-GD | qgr-gd-1 | 6.2 * | G1 | 75.405 | 10.87 | EQ580–180b | 29.4 | ******* |
| qgr-gd-2 | 6.2 | G5 | 54.954 | 4.75 | em5me7–260 | 36.6 | - | |
| qgr-gd-3 | 6.2 | G6 | 88.880 | 3.06 | EQ2190–150 | 11.7 | **** | |
| qgr-gd-4 | 6.2 | G12 | 1.000 | 4.45 | EQ1652–190b | 51.2 | - | |
| qgr-gd-5 | 6.2 | G13 | 15.309 | 3.28 | em1me3–90 | 45.6 | - | |
| qgr-gd-6 | 6.2 * | G18 | 8.000 | 7.06 | EQ457–200a | 59.6 | - | |
| C2012 | qc3-1 | 4.8 | G1 | 54.562 | 4.56 | em1me26–110 | 13.0 | ******* |
| C2013 | qc4-1 | 4.9 | G1 | 86.551 | 3.28 | em9me5–310 | 17.3 | - |
| qc4-2 | 4.9 | G5 | 45.182 | 3.05 | em56me4–150 | 13.6 | - | |
| C2014 | qc5-1 | 4.8 | G1 | 29.078 | 3.89 | em6me8–330c | 11.1 | - |
| qc5-2 | 4.8 | G1 | 74.980 | 4.00 | em49me3–150 | 18.7 | - | |
| C2015 | qc6-1 | 5.5 * | G5 | 45.182 | 5.77 | em56me4–150 | 36.1 | ** |
| qc6-2 | 5.5 * | G16 | 3.000 | 5.88 | EQ2084-230 | 34.8 | - | |
| qc6-3 | 5.5 * | G1 | 28.901 | 8.40 | em6me8–330c | 31.2 | ******* | |
| qc6-4 | 5.5 * | G1 | 47.811 | 11.83 | em1me26–2400 | 56.6 | - | |
| qc6-5 | 5.5 * | G1 | 81.121 | 15.57 | em1me14–900 | 52.0 | ******* | |
| C2016 | qc7-1 | 4.8 * | G1 | 68.393 | 5.06 | UBC808–420 | 19.8 | ** |
| C2017 | qc8-1 | 4.7 * | G1 | 75.405 | 6.35 | EQ580–180b | 18.3 | ******* |
| C2018 | qc9-1 | 5.2 * | G1 | 48.658 | 5.53 | E1M8–245 | 23.9 | ****** |
| qc9-2 | 5.2 * | G1 | 105.117 | 7.56 | em13me4–190c | 22.3 | ******* | |
| C2019 | qc10-1 | 4.9 * | G1 | 88.173 | 7.68 | em8me10–230c | 20.8 | ******* |
| GR-C | qgr-c-1 | 5.4 * | G1 | 49.488 | 8.25 | em13me4–620 | 38.3 | - |
| qgr-c-2 | 5.4 | G1 | 110.483 | 7.74 | em4me3–2300 | 27.8 | ** | |
| qgr-c-3 | 5.4 | G5 | 46.182 | 3.60 | em7me12–365 | 23.2 | *** | |
| qgr-c-4 | 5.4 | G7 | 102.562 | 3.09 | em4me7–190 | 26.5 | *** | |
| qgr-c-5 | 5.4 | G10 | 64.006 | 3.92 | em14me8–270c | 11.2 | **** | |
| qgr-c-6 | 5.4 | G15 | 30.956 | 5.29 | em6me3–550 | 34.4 | - |
| NO. | LG1 | Interval/cM | QTL | ||
|---|---|---|---|---|---|
| H 2 | GD 3 | C 4 | |||
| Locus 1 | G1 | 27.901~29.168 | qc5-1, qc6-3 | ||
| Locus 2 | G1 | 45.526~54.202 | qh1-2, qh2-1, qh6-3, qh7-1, qh8-1, qgr-h-1, | qgd1-1, qgd2-1, qgd5-1, qgd6-5, qgd7-2, qgd8-1, qgd9-1 | qc3-1, qc6-4, qc9-1, qgr-c-1 |
| Locus 3 | G1 | 71.761~75.489 | qh1-3, qh6-4, qh7-2, qh8-2, qh9-1, qh10-2 | qgd1-4, qgd5-2, qgd6-3, qgd7-3, qgd8-2, qgd9-2, qgr-gd-1 | qc5-2, qc7-1, qc8-1 |
| Locus 4 | G1 | 86.881~90.899 | qc4-1, qc6-5, qc10-1 | ||
| Locus 5 | G1 | 95.989~99.254 | qgr-h-2 | ||
| Locus 6 | G1 | 103.685~109.72 | qgd2-2 | qc9-2, qgr-c-2 | |
| Locus 7 | G1 | 120.509~123.215 | qh6-5, qh7-3 | qgd6-4, qgd9-3, qgd7-4 | |
| Locus 8 | G2 | 78.034~80.215 | qgd4-1 | ||
| Locus 9 | G3 | 104.69~108.842 | qgd3-1 | ||
| Locus 10 | G4 | 31.947~47.632 | qgd3-2, qgd4-2 | ||
| Locus 11 | G5 | 43.445~52.63 | qh10-1, qgr-h-3 | qgr-gd-2, qgd10-1 | qc4-2, qc6-1, qgr-c-3 |
| Locus 12 | G6 | 84.245~94.599 | qgr-gd-3 | ||
| Locus 13 | G7 | 26.381~28.441 | qh9-2 | ||
| Locus 14 | G7 | 95.113~113.562 | qgr-c-4 | ||
| Locus 15 | G8 | 57.394~70.709 | qh6-1 | qgd6-1, qgd7-1 | |
| Locus 16 | G10 | 13.624~25.233 | qgr-h-4 | ||
| Locus 17 | G10 | 45.839~60.583 | qgd3-3, qgd4-3 | qgr-c-5 | |
| Locus 18 | G12 | 0.000~3.000 | qgr-gd-4 | ||
| Locus 19 | G13 | 10.309~17.309 | qh3-1, qh4-1, qh5-1 | qgd3-4, qgd4-4, qgr-gd-5 | |
| Locus 20 | G13 | 45.603~52.603 | qh4-2, qh5-2 | ||
| Locus 21 | G15 | 30.583~33.586 | qgr-c-6 | ||
| Locus 22 | G16 | 0.000~8.214 | qc6-2 | ||
| Locus 23 | G16 | 25.613~28.613 | qgd10-2 | ||
| Locus 24 | G18 | 0.000~25.000 | qh1-1, qgr-h-5 | qgd1-5, qgr-gd-6 | |
| Locus 25 | G18 | 38.652~45.652 | qh3-2, qh5-3 | ||
| Locus 26 | G18 | 112.366~115.366 | qgd6-2 | ||
| Locus 27 | G19 | 0.000~18.476 | qh6-2 | qgd3-5 | |
| Trait | Marker | Gene ID | Annotation | KO/GO ID |
|---|---|---|---|---|
| H2010, GD2010, GR-H, GR-GD | EQ457 | Unigene0019357 | gigantea protein | |
| H2010, GD2010, C2017, GR-GD | EQ580 | Unigene0000958 | protein ABIL1-like | GO:0016485 |
| GD2010, GR-GD | EQ767 | Unigene0003585 | cell division protease, ftsH isoform 1 | |
| H2012 | EQ1107 | Unigene0007449 | translation initiation factor 4G-like | K03260 |
| GD2012, GD2013 | EQ962 | Unigene0005909 | ||
| C2015 | EQ2084 | Unigene0016652 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jin, C.; Li, Z.; Li, Y.; Wang, S.; Li, L.; Liu, M. Update of Genetic Linkage Map and QTL Analysis for Growth Traits in Eucommia ulmoides Oliver. Forests 2020, 11, 311. https://doi.org/10.3390/f11030311
Jin C, Li Z, Li Y, Wang S, Li L, Liu M. Update of Genetic Linkage Map and QTL Analysis for Growth Traits in Eucommia ulmoides Oliver. Forests. 2020; 11(3):311. https://doi.org/10.3390/f11030311
Chicago/Turabian StyleJin, Cangfu, Zhouqi Li, Yu Li, Shuhui Wang, Long Li, and Minhao Liu. 2020. "Update of Genetic Linkage Map and QTL Analysis for Growth Traits in Eucommia ulmoides Oliver" Forests 11, no. 3: 311. https://doi.org/10.3390/f11030311
APA StyleJin, C., Li, Z., Li, Y., Wang, S., Li, L., & Liu, M. (2020). Update of Genetic Linkage Map and QTL Analysis for Growth Traits in Eucommia ulmoides Oliver. Forests, 11(3), 311. https://doi.org/10.3390/f11030311