Single-Molecule Real-Time Sequencing of the Madhuca pasquieri (Dubard) Lam. Transcriptome Reveals the Diversity of Full-Length Transcripts


Abstract
:1. Introduction
2. Materials and Methods
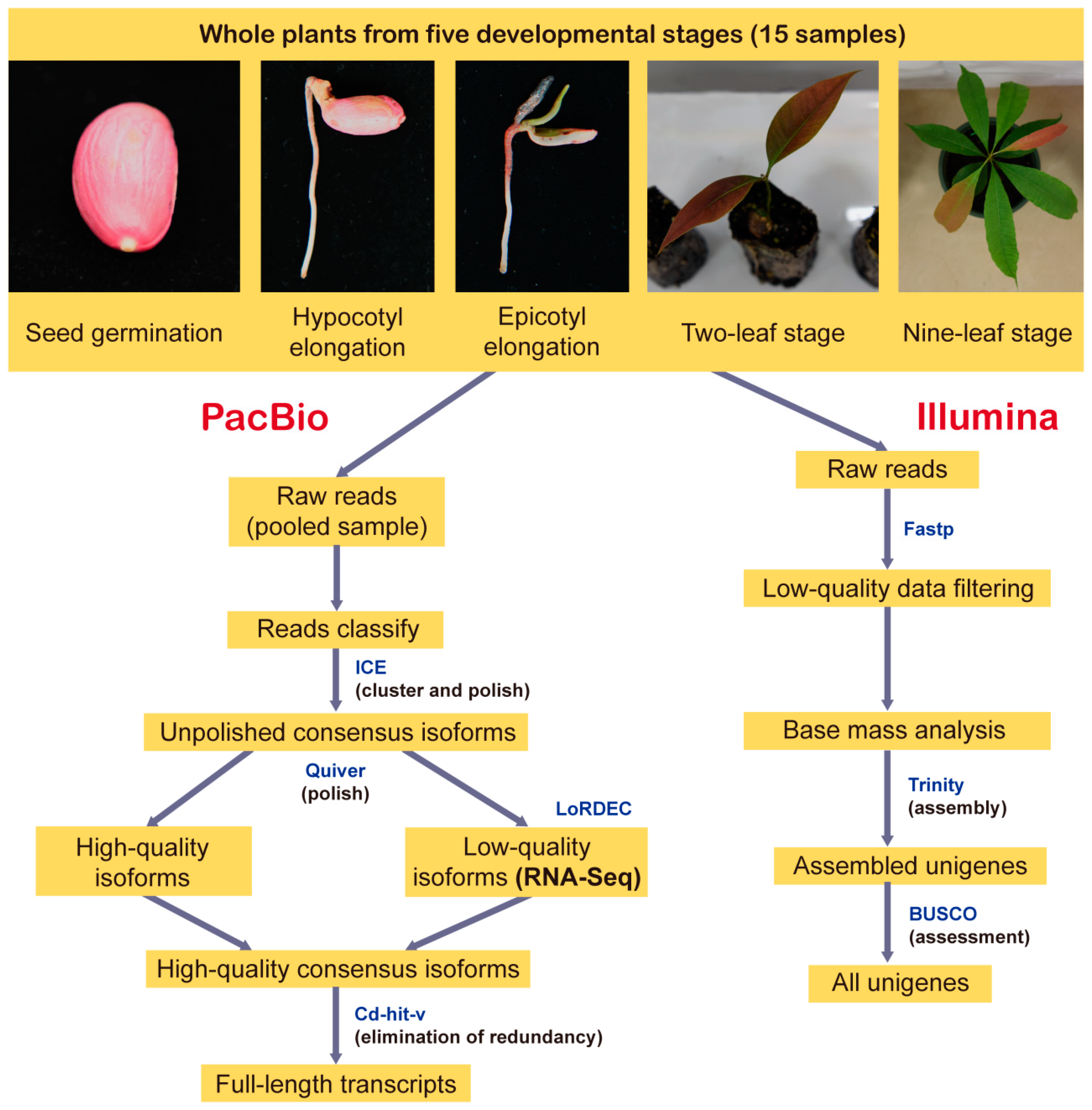
2.1. Plant Materials
2.2. Library Construction and SMRT Sequencing
2.3. Analysis of SMRT Sequencing Data
2.4. Illumina RNA Sequencing and De Novo Assembly of Short Reads
2.5. Evaluation of Sequencing Results
2.6. Prediction of Coding Sequences (CDSs), Simple Sequence Repeats (SSRs), and Transcription Factors (TFs)
2.7. Characterization of AS Events
2.8. LncRNA Identification from PacBio Sequences
2.9. Functional Annotation
3. Results
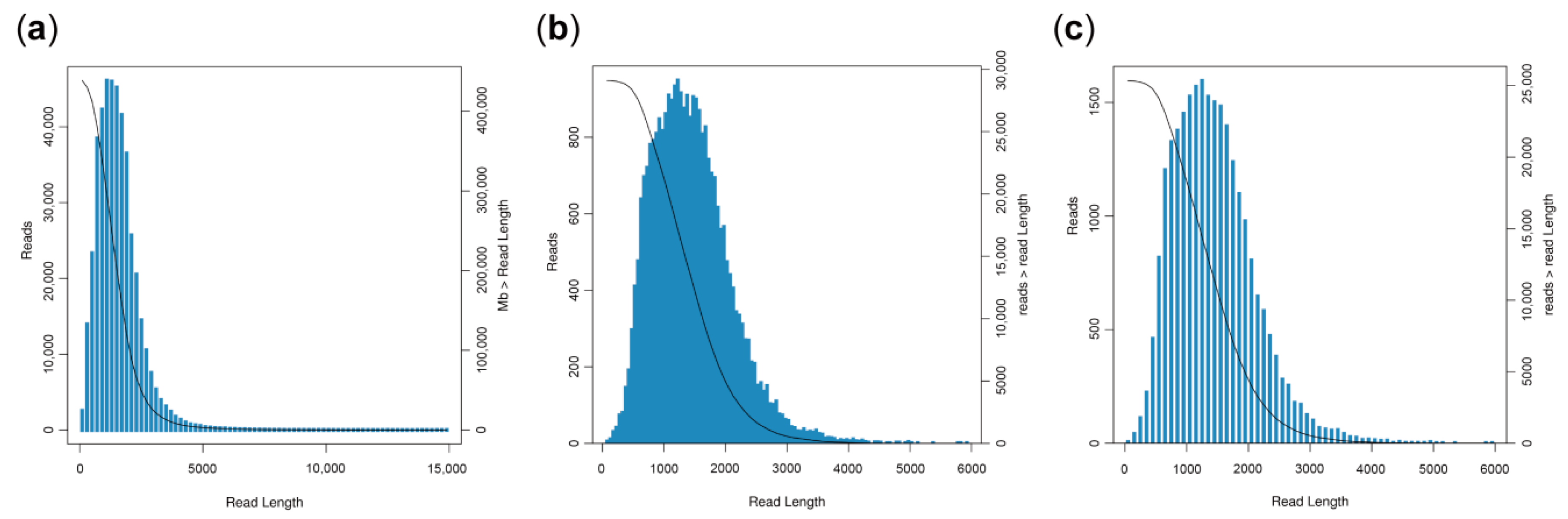
3.1. General Properties of Single-Molecule Long-Reads
3.2. Acquisition of High-Quality Sequences and Error Correction of Long Reads Using Illumina Data
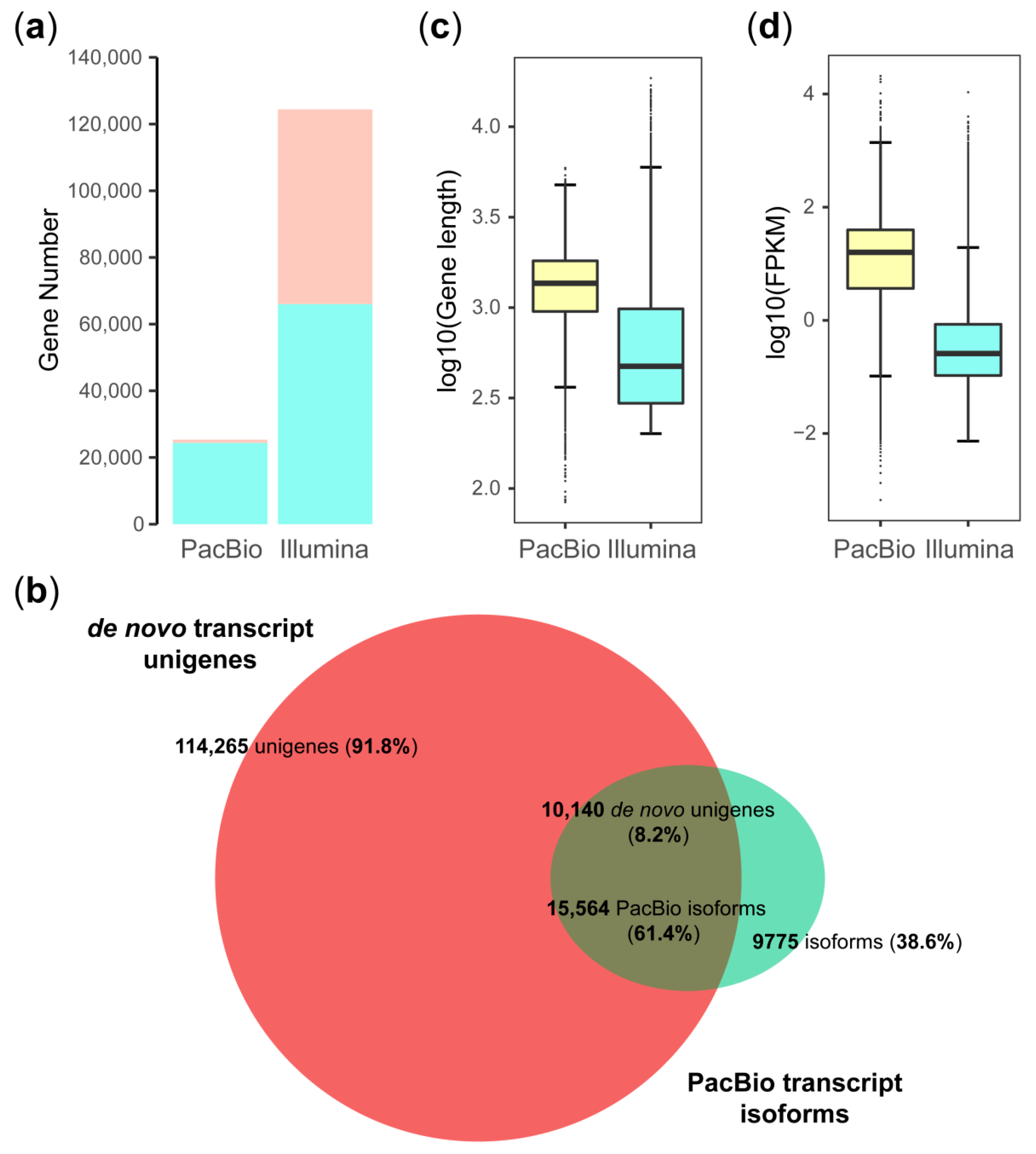
3.3. Comparison of PacBio and Illumina Transcripts and Sequencing Depth
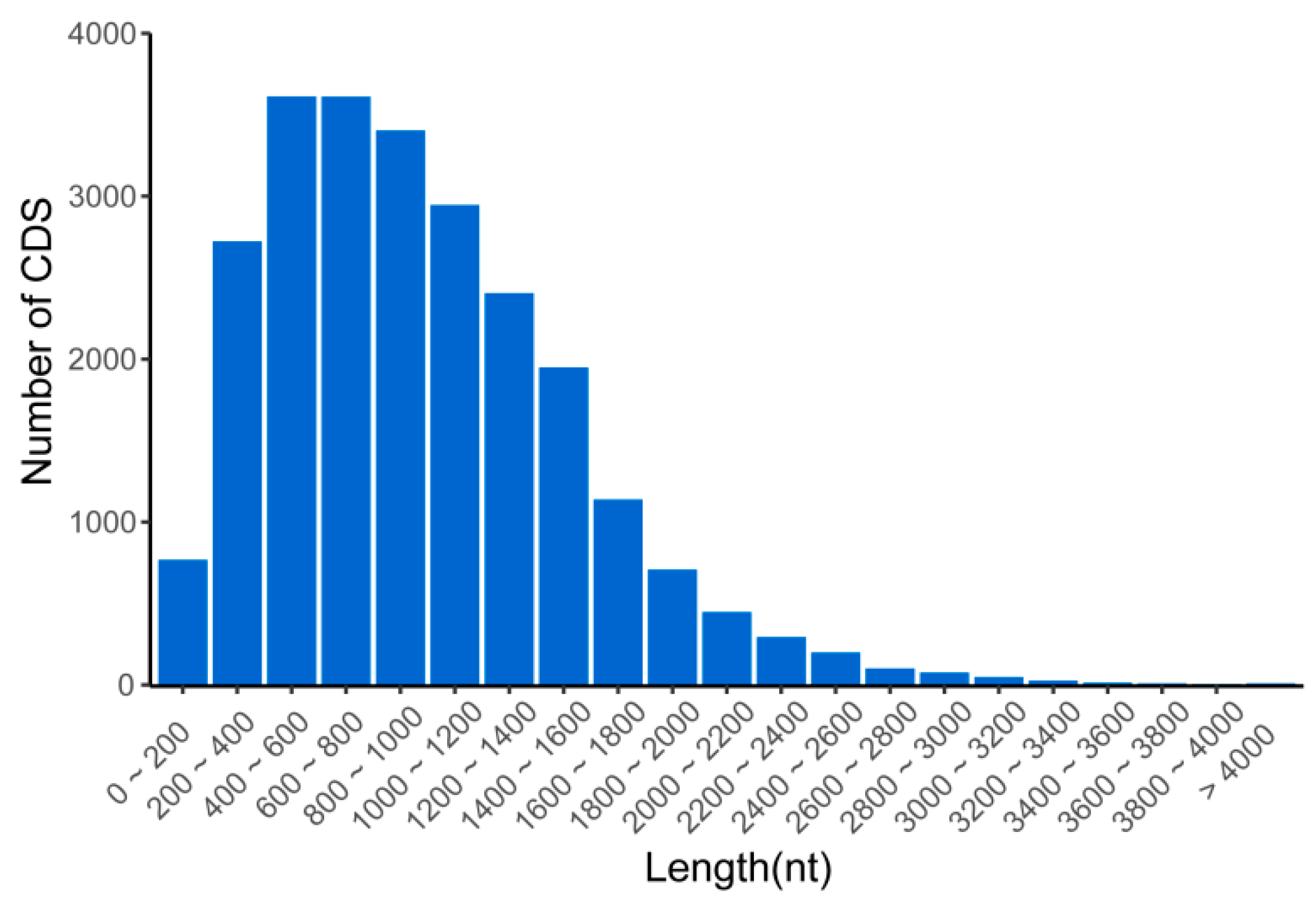
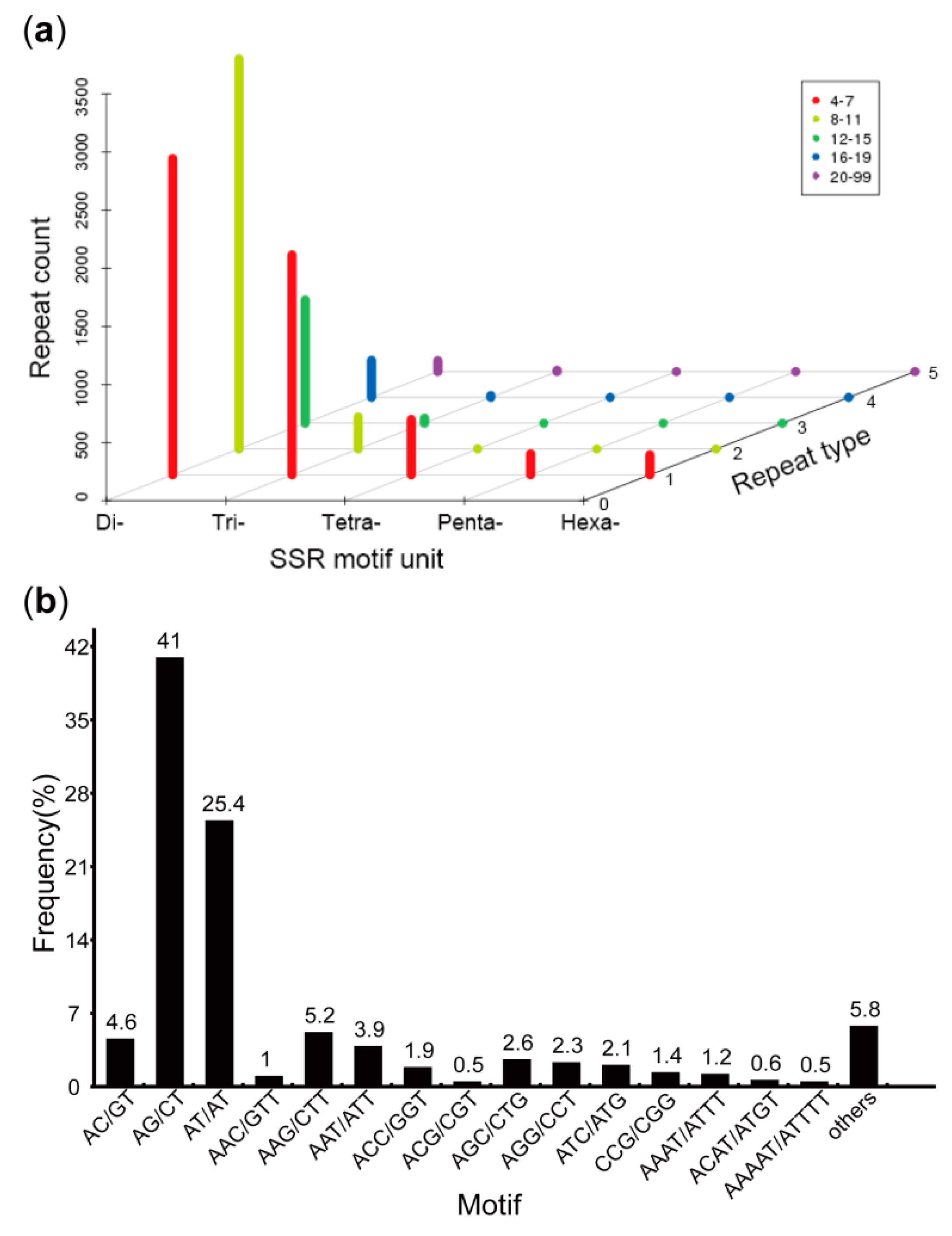
3.4. Prediction of CDSs, SSRs, and TFs
3.5. AS Events Detected from PacBio Sequel
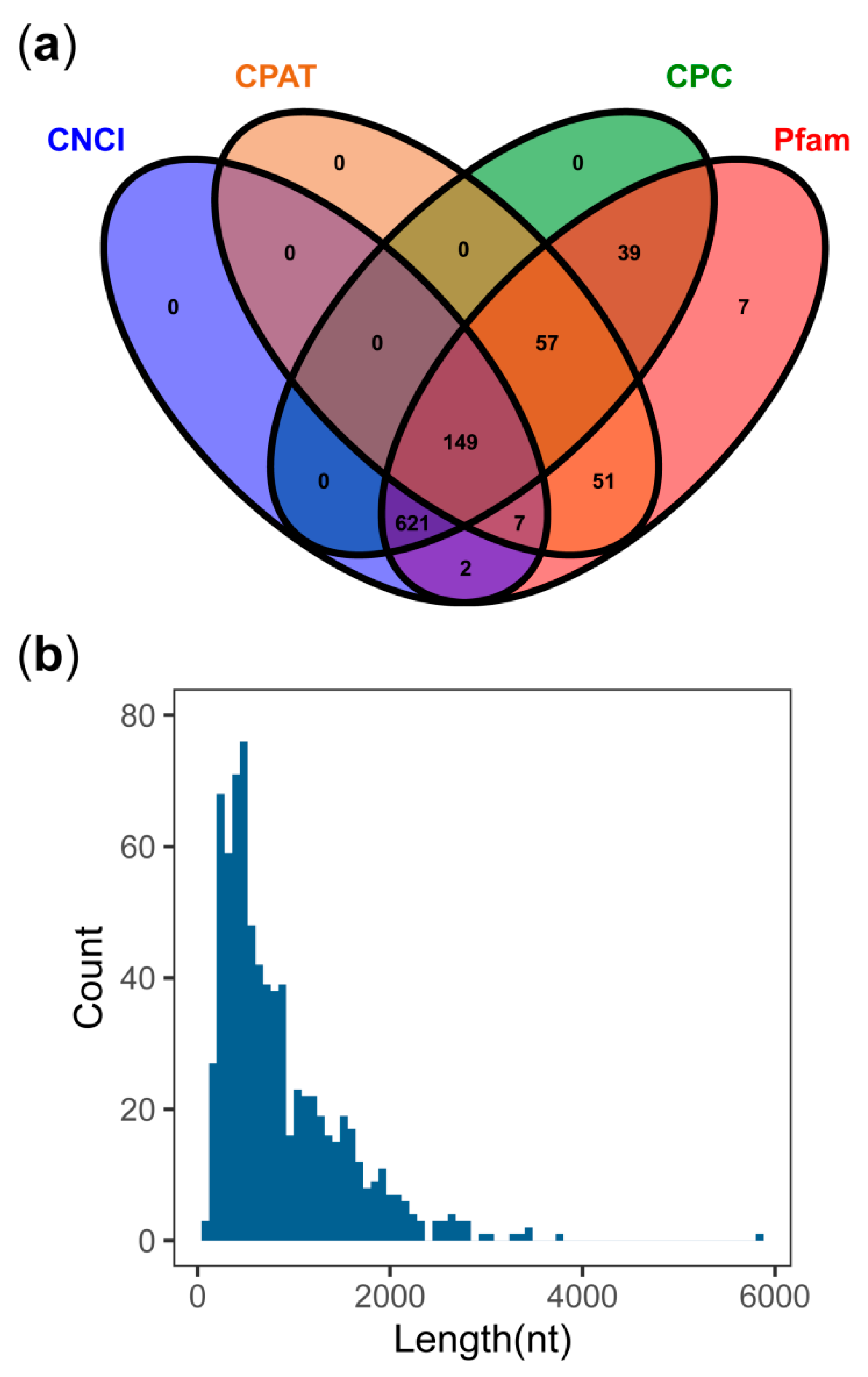
3.6. LncRNA Detected from PacBio Sequel
3.7. Functional Annotation of Transcripts
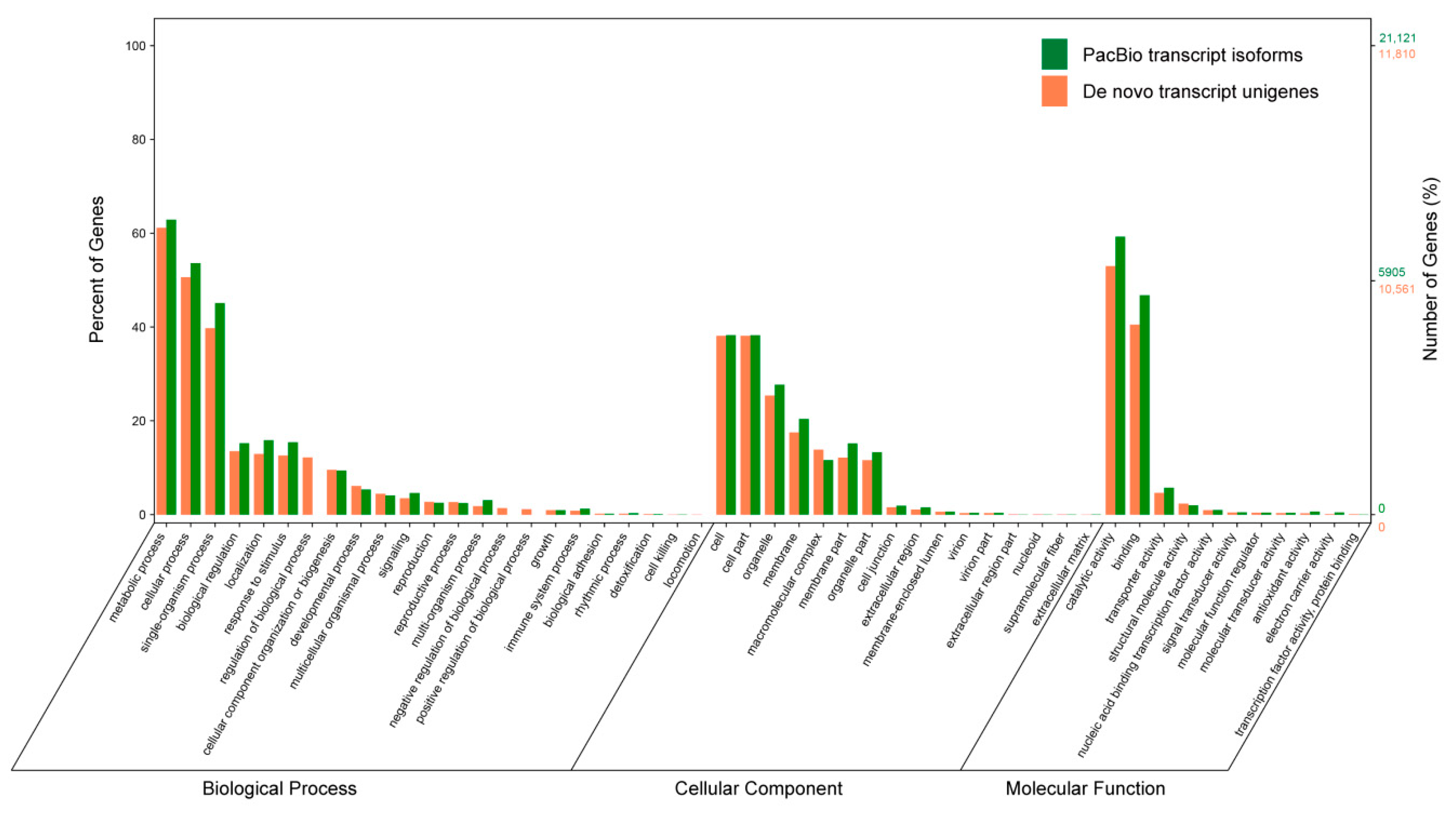
3.8. Gene Ontology (GO) Annotation
3.9. Analysis of KEGG Pathways and Gene Annotation Information
4. Discussion
4.1. Comparison of PacBio Transcripts and De Novo Unigenes
4.2. Analysis of Alternative Splicing in Transcriptomes
4.3. Analysis of lncRNAs Detected by PacBio Sequel
4.4. Analysis of Nr Annotation and Transcription Factors
4.5. Excavation of KEGG Annotation Pathways Gene Annotation Information in M. pasquieri
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Conesa, A.; Madrigal, P.; Tarazona, S.; Gomez-Cabrero, D.; Cervera, A.; McPherson, A.; Szczesniak, M.W.; Gaffney, D.J.; Elo, L.L.; Zhang, X.; et al. A survey of best practices for RNA-seq data analysis. Genome Biol. 2016, 17, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chao, Y.; Yuan, J.; Li, S.; Jia, S.; Han, L.; Xu, L. Analysis of transcripts and splice isoforms in red clover (Trifolium pratense L.) by single-molecule long-read sequencing. BMC Plant Biol. 2018, 18, 300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, N.; Cheng, F.; Zhong, Y.; Guo, X. Comparative transcriptome and coexpression network analysis of carpel quantitative variation in Paeonia rockii. BMC Genom. 2019, 20, 683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Jin, Y.; Huang, W.; Sun, Q.; Liu, F.; Huang, X. Full-length transcriptome sequences of ephemeral plant Arabidopsis pumila provides insight into gene expression dynamics during continuous salt stress. BMC Genom. 2018, 19, 717. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ju, D.; Yang, X.; Ma, D.; Wang, X. Comparative transcriptome analysis between resistant and susceptible rice cultivars responding to striped stem borer (SSB), Chilo suppressalis (Walker) infestation. Front. Physiol. 2018, 9, 1717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piriyapongsa, J.; Kaewprommal, P.; Vaiwsri, S.; Anuntakarun, S.; Wirojsirasak, W.; Punpee, P.; Klomsa-Ard, P.; Shaw, P.J.; Pootakham, W.; Yoocha, T.; et al. Uncovering full-length transcript isoforms of sugarcane cultivar Khon Kaen 3 using single-molecule long-read sequencing. PeerJ 2018, 6, e5818. [Google Scholar] [CrossRef] [Green Version]
- Rather, G.A.; Sharma, A.; Pandith, S.A.; Kaul, V.; Nandi, U.; Misra, P.; Lattoo, S.K. De novo transcriptome analyses reveals putative pathway genes involved in biosynthesis and regulation of camptothecin in Nothapodytes nimmoniana (Graham) Mabb. Plant Mol. Biol. 2018, 96, 197–215. [Google Scholar] [CrossRef]
- Dharshini, S.; Chakravarthi, M.; Narayan, J.A.; Manoj, V.M.; Naveenarani, M.; Kumar, R.; Meena, M.; Ram, B.; Appunu, C. De novo sequencing and transcriptome analysis of a low temperature tolerant Saccharum spontaneum clone IND 00-1037. J. Biotechnol. 2016, 231, 280–294. [Google Scholar] [CrossRef]
- Xu, Q.; Zhu, J.; Zhao, S.; Hou, Y.; Li, F.; Tai, Y.; Wan, X.; Wei, C. Transcriptome profiling using single-molecule direct RNA sequencing approach for in-depth understanding of genes in secondary metabolism pathways of Camellia sinensis. Front. Plant Sci. 2017, 8, 1205. [Google Scholar] [CrossRef] [Green Version]
- Rhoads, A.; Au, K.F. PacBio sequencing and its applications. Genom. Proteom. Bioinf. 2015, 13, 278–289. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Jiang, X.; Wang, L.; Wang, W.; Fu, C.; Yan, X.; Geng, X. A survey of transcriptome complexity using PacBio single-molecule real-time analysis combined with Illumina RNA sequencing for a better understanding of ricinoleic acid biosynthesis in Ricinus communis. BMC Genom. 2019, 20, 456. [Google Scholar] [CrossRef] [PubMed]
- Chao, Y.; Yuan, J.; Guo, T.; Xu, L.; Mu, Z.; Han, L. Analysis of transcripts and splice isoforms in Medicago sativa L. by single-molecule long-read sequencing. Plant Mol. Biol. 2019, 99, 219–235. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.; Liu, H.; Zhang, J.; Yang, S.; Kong, G.; Chu, J.S.; Chen, N.; Wang, D. Single-molecule real-time transcript sequencing facilitates common wheat genome annotation and grain transcriptome research. BMC Genom. 2015, 16, 1039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.; Wang, H.; Cai, D.; Gao, Y.; Zhang, H.; Wang, Y.; Lin, C.; Ma, L.; Gu, L. Comprehensive profiling of rhizome-associated alternative splicing and alternative polyadenylation in moso bamboo (Phyllostachys edulis). Plant J. 2017, 91, 684–699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, J.; Xiang, Y.; Xiong, Y.; Lin, Z.; Xue, Y.; Mao, M.; Sun, L.; Zhou, Y.; Li, X.; Huang, Z. SMRT sequencing analysis reveals the full-length transcripts and alternative splicing patterns in Ananas comosus var. bracteatus. PeerJ 2019, 7, e7062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuo, C.; Blow, M.; Sreedasyam, A.; Kuo, R.C.; Ramamoorthy, G.K.; Torres-Jerez, I.; Li, G.; Wang, M.; Dilworth, D.; Barry, K.; et al. Revealing the transcriptomic complexity of switchgrass by PacBio long-read sequencing. Biotechnol. Biofuels 2018, 11, 170. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Zeng, Z.; Li, F.; Yang, X.; Gao, X.; Ma, Y.; Rao, J.; Wang, H.; Liu, T. A genomic resource derived from the integration of genome sequences, expressed transcripts and genetic markers in ramie. BMC Genom. 2019, 20, 476. [Google Scholar] [CrossRef]
- Edger, P.P.; VanBuren, R.; Colle, M.; Poorten, T.J.; Wai, C.M.; Niederhuth, C.E.; Alger, E.I.; Ou, S.; Acharya, C.B.; Wang, J.; et al. Single-molecule sequencing and optical mapping yields an improved genome of woodland strawberry (Fragaria vesca) with chromosome-scale contiguity. Gigascience 2018, 7, 1–7. [Google Scholar] [CrossRef] [Green Version]
- An, D.; Cao, H.X.; Li, C.; Humbeck, K.; Wang, W. Isoform sequencing and state-of-art applications for unravelling complexity of plant transcriptomes. Genes 2018, 9, 43. [Google Scholar] [CrossRef] [Green Version]
- Hoang, N.V.; Furtado, A.; Mason, P.J.; Marquardt, A.; Kasirajan, L.; Thirugnanasambandam, P.P.; Botha, F.C.; Henry, R.J. A survey of the complex transcriptome from the highly polyploid sugarcane genome using full-length isoform sequencing and de novo assembly from short read sequencing. BMC Genom. 2017, 18, 395. [Google Scholar] [CrossRef]
- Xu, Z.; Peters, R.J.; Weirather, J.; Luo, H.; Liao, B.; Zhang, X.; Zhu, Y.; Ji, A.; Zhang, B.; Hu, S.; et al. Full-length transcriptome sequences and splice variants obtained by a combination of sequencing platforms applied to different root tissues of Salvia miltiorrhiza and tanshinone biosynthesis. Plant J. 2015, 82, 951–961. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.Q.; Liu, H.; Zhou, S.S.; Zhang, D.X.; Zhao, W.; Wang, S.; Chen, F.; Sun, Y.Q.; Nie, S.; Jia, K.H.; et al. Genome sequence of Malania oleifera, a tree with great value for nervonic acid production. Gigascience 2019, 8, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, C.; Liu, H.; Ren, J.; Ye, X.; Feng, H.; Liu, Z. Single-molecule real-time sequencing facilitates the analysis of transcripts and splice isoforms of anthers in Chinese cabbage (Brassica rapa L. ssp. pekinensis). BMC Plant Biol. 2019, 19, 517. [Google Scholar] [CrossRef] [PubMed]
- Kuang, X.; Sun, S.; Wei, J.; Li, Y.; Sun, C. Iso-Seq analysis of the Taxus cuspidata transcriptome reveals the complexity of Taxol biosynthesis. BMC Plant Biol. 2019, 19, 210. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Lin, R.; Jiang, Y.; Jiang, S.; Xiong, Y.; Lian, H.; Zeng, Q.; Liu, X.; Liu, Z.J.; Chen, S. Transcriptome analysis and identification of genes associated with starch metabolism in Castanea henryi seed (Fagaceae). Int. J. Mol. Sci. 2020, 21, 1431. [Google Scholar] [CrossRef] [Green Version]
- Iralu, V.; Barbhuyan, H.S.A.; Upadhaya, K. Ecology of seed germination in threatened trees: A review. Energ. Ecol. Environ. 2019, 4, 189–210. [Google Scholar] [CrossRef]
- Zhou, T.; Li, Z.H.; Bai, G.Q.; Feng, L.; Chen, C.; Wei, Y.; Chang, Y.X.; Zhao, G.F. Transcriptome sequencing and development of genic SSR markers of an endangered Chinese endemic Genus Dipteronia Oliver (Aceraceae). Molecules 2016, 21, 166. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Zhang, X.; Wang, Y.H.; Shen, S.K. De Novo assembly of transcriptome and development of novel EST-SSR markers in Rhododendron rex Levl. through Illumina Sequencing. Front. Plant Sci. 2017, 8, 1664. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Li, M.; Hou, L.; Zhang, Z.; Pang, X.; Li, Y. De novo transcriptome assembly and population genetic analyses for an endangered Chinese endemic Acer miaotaiense (Aceraceae). Genes 2018, 9, 378. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Dong, M.; Zhang, Y.; Qi, S.; Liu, X.; Zhang, J.; Zhao, J. Development and characterization of simple sequence repeat markers for, and genetic diversity analysis of Liquidambar formosana. Forests 2020, 11, 203. [Google Scholar] [CrossRef] [Green Version]
- Chen, F.; Hu, Y.; Vannozzi, A.; Wu, K.; Cai, H.; Qin, Y.; Mullis, A.; Lin, Z.; Zhang, L. The WRKY transcription factor family in model plants and crops. Crit. Rev. Plant Sci. 2018, 36, 311–335. [Google Scholar] [CrossRef]
- Wang, B.; Tseng, E.; Regulski, M.; Clark, T.A.; Hon, T.; Jiao, Y.; Lu, Z.; Olson, A.; Stein, J.C.; Ware, D. Unveiling the complexity of the maize transcriptome by single-molecule long-read sequencing. Nat. Commun. 2016, 7, 11708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Mei, W.; Soltis, P.S.; Soltis, D.E.; Barbazuk, W.B. Detecting alternatively spliced transcript isoforms from single-molecule long-read sequences without a reference genome. Mol. Ecol. Resour. 2017, 17, 1243–1256. [Google Scholar] [CrossRef] [PubMed]
- Ge, Y.; Cheng, Z.; Si, X.; Ma, W.; Tan, L.; Zang, X.; Wu, B.; Xu, Z.; Wang, N.; Zhou, Z.; et al. Transcriptome profiling provides insight into the genes in carotenoid biosynthesis during the mesocarp and seed developmental stages of avocado (Persea americana). Int. J. Mol. Sci. 2019, 20, 4117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, H.; Yang, W.; Zheng, Z.; Niu, Z.; Yang, Y.; Wan, D.; Liu, J.; Ma, T. Analysis of alternative splicing and alternative polyadenylation in Populus alba var. pyramidalis by single-molecular long-read sequencing. Front. Genet. 2020, 11, 48. [Google Scholar] [CrossRef]
- Luo, D.; Zhou, Q.; Wu, Y.; Chai, X.; Liu, W.; Wang, Y.; Yang, Q.; Wang, Z.; Liu, Z. Full-length transcript sequencing and comparative transcriptomic analysis to evaluate the contribution of osmotic and ionic stress components towards salinity tolerance in the roots of cultivated alfalfa (Medicago sativa L.). BMC Plant Biol. 2019, 19, 32. [Google Scholar] [CrossRef] [Green Version]
- Deng, N.; Hou, C.; Ma, F.; Liu, C.; Tian, Y. Single-molecule long-read sequencing reveals the diversity of full-length transcripts in leaves of Gnetum (Gnetales). Int. J. Mol. Sci. 2019, 20, 6350. [Google Scholar] [CrossRef] [Green Version]
- Kang, S.H.; Lee, J.Y.; Lee, T.H.; Park, S.Y.; Kim, C.K. De novo transcriptome assembly of the Chinese pearl barley, adlay, by full-length isoform and short-read RNA sequencing. PLoS ONE 2018, 13, e0208344. [Google Scholar] [CrossRef]
- Hallegger, M.; Llorian, M.; Smith, C.W. Alternative splicing: Global insights. FEBS J. 2010, 277, 856–866. [Google Scholar] [CrossRef] [Green Version]
- McManus, C.J.; Graveley, B.R. RNA structure and the mechanisms of alternative splicing. Curr. Opin. Genet. Dev. 2011, 21, 373–379. [Google Scholar] [CrossRef] [Green Version]
- Reddy, A.S.; Marquez, Y.; Kalyna, M.; Barta, A. Complexity of the alternative splicing landscape in plants. Plant Cell 2013, 25, 3657–3683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Dai, C.; Hu, C.; Liu, Z.; Kang, C. Global identification of alternative splicing via comparative analysis of SMRT- and Illumina-based RNA-seq in strawberry. Plant J. 2017, 90, 164–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaudhary, S.; Jabre, I.; Reddy, A.S.N.; Staiger, D.; Syed, N.H. Perspective on alternative splicing and proteome complexity in plants. Trends Plant Sci. 2019, 24, 496–506. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Zhang, X.; Wang, S.; Tan, C.; Zhou, G.; Li, C. Involvement of alternative splicing in barley seed germination. PLoS ONE 2016, 11, e0152824. [Google Scholar] [CrossRef] [Green Version]
- Chao, Q.; Gao, Z.; Zhang, D.; Zhao, B.; Dong, F.; Fu, C.; Liu, L.; Wang, B. The developmental dynamics of the Populus stem transcriptome. Plant Biotechnol. J. 2019, 17, 206–219. [Google Scholar] [CrossRef] [Green Version]
- Qian, X.; Sun, Y.; Zhou, G.; Yuan, Y.; Li, J.; Huang, H.; Xu, L.; Li, L. Single-molecule real-time transcript sequencing identified flowering regulatory genes in Crocus sativus. BMC Genom. 2019, 20, 857. [Google Scholar] [CrossRef]
- Li, Y.; Mi, X.; Zhao, S.; Zhu, J.; Guo, R.; Xia, X.; Liu, L.; Liu, S.; Wei, C. Comprehensive profiling of alternative splicing landscape during cold acclimation in tea plant. BMC Genom. 2020, 21, 65. [Google Scholar] [CrossRef] [Green Version]
- Steijger, T.; Abril, J.F.; Engstrom, P.G.; Kokocinski, F.; Consortium, R.; Hubbard, T.J.; Guigo, R.; Harrow, J.; Bertone, P. Assessment of transcript reconstruction methods for RNA-seq. Nat. Methods 2013, 10, 1177–1184. [Google Scholar] [CrossRef] [Green Version]
- Shang, X.; Cao, Y.; Ma, L. Alternative splicing in plant genes: A means of regulating the environmental fitness of plants. Int. J. Mol. Sci. 2017, 18, 432. [Google Scholar] [CrossRef]
- Zhao, L.; Zhang, H.; Kohnen, M.V.; Prasad, K.; Gu, L.; Reddy, A.S.N. Analysis of transcriptome and epitranscriptome in plants using PacBio Iso-Seq and Nanopore-Based direct RNA sequencing. Front. Genet. 2019, 10, 253. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Wang, P.; Liang, F.; Ye, Z.; Li, J.; Shen, C.; Pei, L.; Wang, F.; Hu, J.; Tu, L.; et al. A global survey of alternative splicing in allopolyploid cotton: Landscape, complexity and regulation. New Phytol. 2018, 217, 163–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdel-Ghany, S.E.; Hamilton, M.; Jacobi, J.L.; Ngam, P.; Devitt, N.; Schilkey, F.; Ben-Hur, A.; Reddy, A.S. A survey of the sorghum transcriptome using single-molecule long reads. Nat. Commun. 2016, 7, 11706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, J.; Cao, H.; Liu, J.D.; Zuo, J.H.; Fang, Y.; Lin, C.T.; Sun, R.Z.; Li, W.L.; Liu, Y.X. Insights into transcriptional characteristics and homoeolog expression bias of embryo and de-embryonated kernels in developing grain through RNA-Seq and Iso-Seq. Funct. Integr. Genom. 2019, 19, 919–932. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Yu, X.; Cheng, Z.; Zeng, C.; Li, W.; Zhang, L.; Peng, M. Large-scale analysis of the cassava transcriptome reveals the impact of cold stress on alternative splicing. J. Exp. Bot. 2020, 71, 422–434. [Google Scholar] [CrossRef]
- Wang, B.; Regulski, M.; Tseng, E.; Olson, A.; Goodwin, S.; McCombie, W.R.; Ware, D. A comparative transcriptional landscape of maize and sorghum obtained by single-molecule sequencing. Genome Res. 2018, 28, 921–932. [Google Scholar] [CrossRef] [Green Version]
- Long, Y.; Wang, X.; Youmans, D.T.; Cech, T.R. How do lncRNAs regulate transcription? Sci. Adv. 2017, 3, eaao2110. [Google Scholar] [CrossRef] [Green Version]
- Cui, J.; Luan, Y.; Jiang, N.; Bao, H.; Meng, J. Comparative transcriptome analysis between resistant and susceptible tomato allows the identification of lncRNA16397 conferring resistance to Phytophthora infestans by co-expressing glutaredoxin. Plant J. 2017, 89, 577–589. [Google Scholar] [CrossRef] [Green Version]
- Rai, M.I.; Alam, M.; Lightfoot, D.A.; Gurha, P.; Afzal, A.J. Classification and experimental identification of plant long non-coding RNAs. Genomics 2019, 111, 997–1005. [Google Scholar] [CrossRef]
- Tian, J.; Feng, S.; Liu, Y.; Zhao, L.; Tian, L.; Hu, Y.; Yang, T.; Wei, A. Single-molecule long-read sequencing of Zanthoxylum bungeanum Maxim. transcriptome: Identification of aroma-related genes. Forests 2018, 9, 765. [Google Scholar] [CrossRef] [Green Version]
- Deng, F.; Zhang, X.; Wang, W.; Yuan, R.; Shen, F. Identification of Gossypium hirsutum long non-coding RNAs (lncRNAs) under salt stress. BMC Plant Biol. 2018, 18, 23. [Google Scholar] [CrossRef]
- Chen, R.; Li, M.; Zhang, H.; Duan, L.; Sun, X.; Jiang, Q.; Zhang, H.; Hu, Z. Continuous salt stress-induced long non-coding RNAs and DNA methylation patterns in soybean roots. BMC Genom. 2019, 20, 730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Harata-Lee, Y.; Denton, M.D.; Feng, Q.; Rathjen, J.R.; Qu, Z.; Adelson, D.L. Long read reference genome-free reconstruction of a full-length transcriptome from Astragalus membranaceus reveals transcript variants involved in bioactive compound biosynthesis. Cell Discov. 2017, 3, 17031. [Google Scholar] [CrossRef] [PubMed]
- Cheng, B.; Furtado, A.; Henry, R.J. Long-read sequencing of the coffee bean transcriptome reveals the diversity of full-length transcripts. Gigascience 2017, 6, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Bahloul, Y.; Dauchot, N.; Machtoun, I.; Gaboun, F.; Van Cutsem, P. Development and characterization of microsatellite loci for the Moroccan endemic endangered species Argania spinosa (Sapotaceae). Appl. Plant Sci. 2014, 2, 1300071. [Google Scholar] [CrossRef]
- Jo, S.; Kim, H.-W.; Kim, Y.-K.; Cheon, S.-H.; Kim, K.-J. The first complete plastome sequence from the family Sapotaceae, Pouteria campechiana(Kunth) Baehni. Mitochondrial DNA B 2016, 1, 734–736. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Zhang, X.; Wang, H.; Zhu, Z.; Wang, H. Complete plastome sequence of Manilkara zapota (L.) P.Royen (Sapotaceae). Mitochondrial DNA B 2019, 4, 3114–3115. [Google Scholar] [CrossRef] [Green Version]
- Niu, Y.; Ni, S.; Liu, Z.; Zheng, C.; Mao, C.; Shi, C.; Liu, J. The complete chloroplast genome of tropical and sub-tropical fruit tree Lucuma nervosa (Sapotaceae). Mitochondrial DNA B 2018, 3, 440–441. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Yuan, X.; Chen, Z.; Luo, T. The complete chloroplast genome sequence of Vitellaria paradoxa. Mitochondrial DNA B 2019, 4, 2403–2404. [Google Scholar] [CrossRef] [Green Version]
- Han, Z.; Wang, B.; Tian, L.; Wang, S.; Zhang, J.; Guo, S.; Zhang, H.; Xu, L.; Chen, Y. Comprehensive dynamic transcriptome analysis at two seed germination stages in maize (Zea mays L.). Physiol. Plant. 2020, 168, 205–217. [Google Scholar] [CrossRef]
- Kushwaha, S.K.; Grimberg, A.; Carlsson, A.S.; Hofvander, P. Charting oat (Avena sativa) embryo and endosperm transcription factor expression reveals differential expression of potential importance for seed development. Mol. Genet. Genom. 2019, 294, 1183–1197. [Google Scholar] [CrossRef] [Green Version]
- Song, Q.; Cheng, S.; Chen, Z.; Nie, G.; Xu, F.; Zhang, J.; Zhou, M.; Zhang, W.; Liao, Y.; Ye, J. Comparative transcriptome analysis revealing the potential mechanism of seed germination stimulated by exogenous gibberellin in Fraxinus hupehensis. BMC Plant Biol. 2019, 19, 199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liew, L.C.; Narsai, R.; Wang, Y.; Berkowitz, O.; Whelan, J.; Lewsey, M.G. Temporal tissue-specific regulation of transcriptomes during barley (Hordeum vulgare) seed germination. Plant J. 2020, 101, 700–715. [Google Scholar] [CrossRef] [PubMed]
- Groszmann, M.; Bylstra, Y.; Lampugnani, E.R.; Smyth, D.R. Regulation of tissue-specific expression of SPATULA, a bHLH gene involved in carpel development, seedling germination, and lateral organ growth in Arabidopsis. J. Exp. Bot. 2010, 61, 1495–1508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Q.; Bai, X.; Wu, X.; Xiang, D.; Wan, Y.; Luo, Y.; Shi, X.; Li, Q.; Zhao, J.; Qin, P.; et al. Transcriptome profiling identifies transcription factors and key homologs involved in seed dormancy and germination regulation of Chenopodium quinoa. Plant Physiol. Biochem. 2020, 151, 443–456. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Htwe, Y.M.; Li, J.; Shi, P.; Zhang, D.; Zhao, Z.; Ihase, L.O. Integrative omics analysis on phytohormones involved in oil palm seed germination. BMC Plant Biol. 2019, 19, 363. [Google Scholar] [CrossRef] [PubMed]
- Kurita, M.; Mishima, K.; Tsubomura, M.; Takashima, Y.; Nose, M.; Hirao, T.; Takahashi, M. Transcriptome analysis in male strobilus induction by gibberellin treatment in Cryptomeria japonica D. Don. Forests 2020, 11, 633. [Google Scholar] [CrossRef]
- Gazara, R.K.; de Oliveira, E.A.G.; Rodrigues, B.C.; Nunes da Fonseca, R.; Oliveira, A.E.A.; Venancio, T.M. Transcriptional landscape of soybean (Glycine max) embryonic axes during germination in the presence of paclobutrazol, a gibberellin biosynthesis inhibitor. Sci. Rep. 2019, 9, 9601. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Liu, S.; Yuan, G.; Zhao, P.; Yang, W.; Jia, J.; Cheng, L.; Qi, D.; Chen, S.; Liu, G. Comparative transcriptome analysis provides insights into the distinct germination in sheepgrass (Leymus chinensis) during seed development. Plant Physiol. Biochem. 2019, 139, 446–458. [Google Scholar] [CrossRef]
- Shen, Q.; Zhang, S.; Liu, S.; Chen, J.; Ma, H.; Cui, Z.; Zhang, X.; Ge, C.; Liu, R.; Li, Y.; et al. Comparative transcriptome analysis provides insights into the seed germination in cotton in response to chilling stress. Int. J. Mol. Sci. 2020, 21, 2067. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.; Shi, X.; Fang, S.; Zhang, Y.; You, C.; Ma, H.; Lin, F. Comparative transcriptome analysis combining SMRT and NGS sequencing provides novel insights into sex differentiation and development in mud crab (Scylla paramamosain). Aquaculture 2019, 513, 734447. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Family | PacBio | Illumina | Family | PacBio | Illumina | Family | PacBio | Illumina |
|---|---|---|---|---|---|---|---|---|
| ERF | 121 | 154 | TCP | 13 | 28 | ARR-B | 2 | 8 |
| WRKY | 96 | 83 | BBR-BPC | 12 | 9 | CPP | 2 | 11 |
| GRAS | 87 | 88 | ARF | 11 | 24 | LSD | 2 | 8 |
| NAC | 71 | 99 | B3 | 11 | 37 | M-type | 2 | 22 |
| bHLH | 70 | 172 | BES1 | 11 | 10 | S1Fa-like | 2 | 6 |
| C3H | 68 | 72 | DBB | 10 | 13 | YABBY | 2 | 9 |
| bZIP | 49 | 141 | Dof | 9 | 42 | CAMTA | 1 | 7 |
| C2H2 | 46 | 241 | GeBP | 9 | 6 | E2F/DP | 1 | 7 |
| MYB_related | 45 | 82 | CO-like | 6 | 12 | GRF | 1 | 11 |
| TALE | 38 | 29 | ZF-HD | 6 | 22 | HB-other | 1 | 11 |
| MYB | 28 | 124 | FAR1 | 5 | 38 | Whirly | 1 | 3 |
| EIL | 27 | 6 | LBD | 5 | 32 | HB-PHD | 0 | 2 |
| HD-ZIP | 25 | 55 | NF-YA | 5 | 11 | HRT-like | 0 | 1 |
| Trihelix | 25 | 47 | NF-YB | 5 | 30 | LFY | 0 | 1 |
| GATA | 23 | 48 | SBP | 5 | 22 | SAP | 0 | 2 |
| Nin-like | 19 | 13 | AP2 | 4 | 20 | SRS | 0 | 8 |
| G2-like | 18 | 40 | RAV | 4 | 2 | STAT | 0 | 1 |
| HSF | 17 | 32 | WOX | 4 | 5 | |||
| NF-YC | 14 | 18 | NF-X1 | 3 | 2 | |||
| MIKC | 13 | 20 | VOZ | 3 | 1 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kan, L.; Liao, Q.; Su, Z.; Tan, Y.; Wang, S.; Zhang, L. Single-Molecule Real-Time Sequencing of the Madhuca pasquieri (Dubard) Lam. Transcriptome Reveals the Diversity of Full-Length Transcripts. Forests 2020, 11, 866. https://doi.org/10.3390/f11080866
Kan L, Liao Q, Su Z, Tan Y, Wang S, Zhang L. Single-Molecule Real-Time Sequencing of the Madhuca pasquieri (Dubard) Lam. Transcriptome Reveals the Diversity of Full-Length Transcripts. Forests. 2020; 11(8):866. https://doi.org/10.3390/f11080866
Chicago/Turabian StyleKan, Lei, Qicong Liao, Zhiyao Su, Yushan Tan, Shuyu Wang, and Lu Zhang. 2020. "Single-Molecule Real-Time Sequencing of the Madhuca pasquieri (Dubard) Lam. Transcriptome Reveals the Diversity of Full-Length Transcripts" Forests 11, no. 8: 866. https://doi.org/10.3390/f11080866