
Characterization of a Novel Emaravirus Affecting Ash Species (Fraxinus spp.) in Europe

 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Material and RNA Extraction
2.2. HTS of Leaf Material from Diseased Fraxinus spp.
2.3. Determination of the Complete Genome of the Novel Virus
2.4. Detection of the Novel Virus and Association with Disease Symptoms in Fraxinus spp.
2.5. Variability Analysis of Virus Variants
2.6. Electron Microscopic Studies
3. Results
3.1. Identification and Assembly of the Complete Genome of a Novel Virus in Shoestring-Diseased Fraxinus spp.
3.2. Genome Organization of the Novel Virus Identified in Fraxinus spp. and Comparison with Members of the Genus Emaravirus
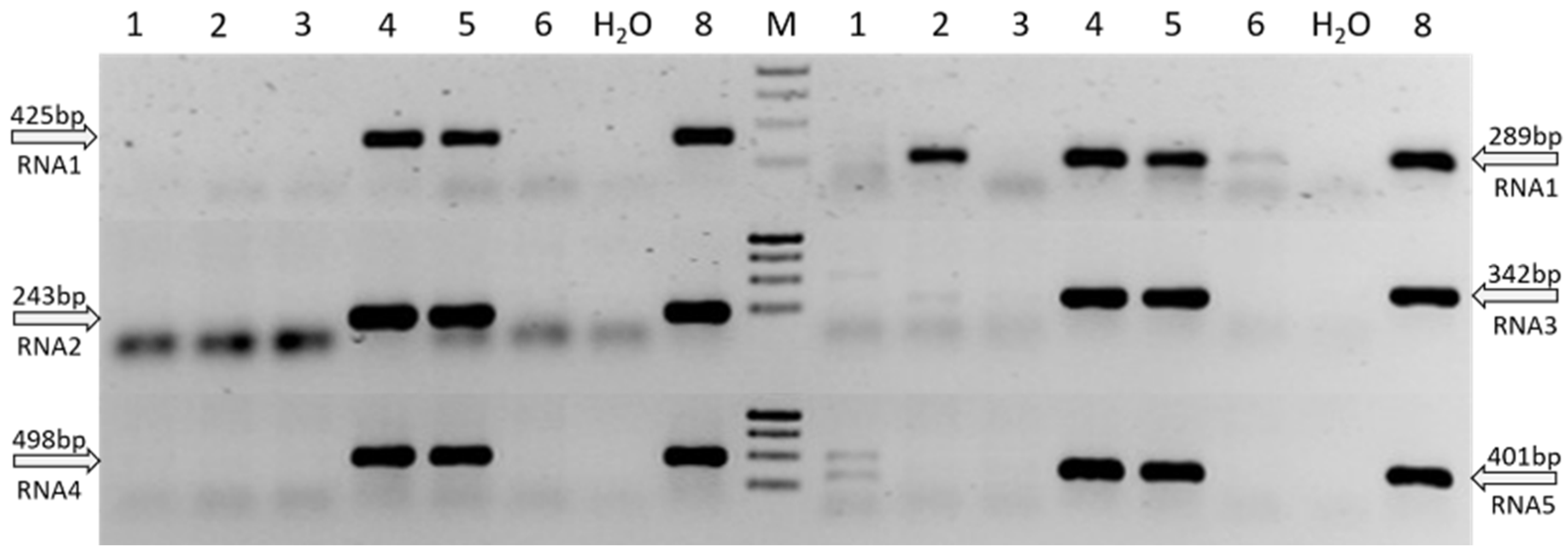
3.3. Establishment of a Virus Specific RT-PCR Detection
3.4. Geographical Distribution of ASaV and Association with Disease Symptoms
3.5. Genetic Variability of ASaV Based on Partial Sequences
3.6. Visualization of Double-Membrane Bodies in Virus-Infected Tissues
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schmelzer, K.; Schmelzer, E.S.; Schmidt, H. Viruskrankheiten und virusverdächtige Erscheinungen an Forstgehölzen. Arch. Forstwes. 1966, 5, 107–120. [Google Scholar]
- Nienhaus, F.; Hamacher, J. Virus diseases of European ash. Allg. Forstz. 1990, 385–386. [Google Scholar]
- Büttner, C.; von Bargen, S.; Bandte, M.; Mühlbach, H.P. Forest diseases caused by viruses. In Infectious Forest Diseases; Gonthier, P., Nicolotti, G., Eds.; CAB International: Wallingford, UK, 2013; pp. 50–75. [Google Scholar]
- Nienhaus, F.; Castello, J.D. Viruses in forest trees. Annu. Rev. Phytopathol. 1989, 27, 165–186. [Google Scholar] [CrossRef]
- Enderle, R.; Stenlid, J.; Vasaitis, R. An overview of ash (Fraxinus spp.) and the ash dieback disease in Europe. CAB Rev. 2019, 14, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Caudullo, W.; de Rigo, D. Fraxinus ornus in Europe: Distribution, habitat, usage and threats. In The European Atlas of Forest Tree Species: Modelling, Data and Information on Forest Tree Species; De Rigo, D., Caudullo, G., Houston-Durrant, T., San-Miguel-Ayanz, J., Eds.; Publishing Office of the European Union: Luxemburg, 2016; pp. 100–101. [Google Scholar]
- Beck, P.; Caudullo, W.; de Rigo, D. Fraxinus excelsior in Europe: Distribution, habitat, usage and threats. In The European Atlas of Forest Tree Species: Modelling, Data and Information on Forest Tree Species; De Rigo, D., Caudullo, G., Houston-Durrant, T., San-Miguel-Ayanz, J., Eds.; Publishing Office of the European Union: Luxemburg, 2016; pp. 98–99. [Google Scholar]
- Dobrowolska, D.; Hein, S.; Oosterbaan, A.; Wagner, S.; Clark, J.; Skovsgaard, J.P. A review of European ash (Fraxinus excelsior L.): Implication for silviculture. Forestry 2011, 84, 133–148. [Google Scholar] [CrossRef] [Green Version]
- “Klimabäume” Welche Arten Können in Zukunft Gepflanzt Werden? Available online: https://www.lwg.bayern.de/mam/cms06/landespflege/dateien/zukunft_klimabaeume.pdf (accessed on 12 July 2021).
- Zukunftsbäume für Die Stadt. Available online: https://galk.de/arbeitskreise/stadtbaeume/themenuebersicht/zukunftsbaeume-fuer-die-stadt (accessed on 12 July 2021).
- Kirisits, T. Eschenpathogen Chalara fraxinea nun auch in Kärnten nachgewiesen. Forstsch. Aktuell 2008, 45, 28–30. [Google Scholar]
- Wu, Q.; Ding, S.W.; Zhang, Y.; Zhu, S. Identification of viruses and viroids by next-generation sequencing and homology-dependent and homology-independent algorithms. Ann. Rev. Phytopathol. 2015, 53, 425–444. [Google Scholar] [CrossRef] [PubMed]
- Massart, S.; Olmos, A.; Jijakli, H.; Candresse, T. Current impact and future directions of high throughput sequencing in plant virus diagnostics. Virus Res. 2014, 188, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Gao, S.; Padmanabhan, C.; Li, R.; Galvez, M.; Gutierrez, D.; Fuentes, S.; Ling, K.S.; Kreuze, J.; Fei, Z. VirusDetect: An automated pipeline for efficient virus discovery using deep sequencing of small RNAs. Virology 2017, 500, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Massart, S.; Chiumenti, M.; De Jonghe, K.; Glover, R.; Haegeman, A.; Koloniuk, I.; Komínek, P.; Kreuze, J.; Kutnjak, D.; Lotos, L.; et al. Virus Detection by High-Throughput Sequencing of Small RNAs: Large-Scale Performance Testing of Sequence Analysis Strategies. Phytopathology 2019, 109, 488–497. [Google Scholar] [CrossRef] [Green Version]
- Villamor, D.E.V.; Ho, T.; Al Rwahnih, M.; Martin, R.R.; Tzanetakis, I.E. High throughput sequencing for plant virus detection and discovery. Phytopathology 2019, 109, 716–725. [Google Scholar] [CrossRef] [PubMed]
- Rumbou, A.; Candresse, T.; Marais, A.; Theil, S.; Langer, J.; Jalkanen, R.; Büttner, C. A novel badnavirus discovered from Betula sp. affected by birch leaf-roll disease. PLoS ONE 2018, 13, e0193888. [Google Scholar] [CrossRef]
- Rumbou, A.; Candresse, T.; Marais, A.; Svanella-Dumas, L.; Landgraf, M.; von Bargen, S.; Büttner, C. Unravelling the virome in birch: RNA-Seq reveals a complex of known and novel viruses. PLoS ONE 2020, 15, e0221834. [Google Scholar] [CrossRef] [PubMed]
- Rehanek, M.; von Bargen, S.; Bandte, M.; Karlin, D.G.; Büttner, C. A novel emaravirus comprising five RNA segments is associated with ringspot disease in oak. Arch. Virol. 2021, 166, 987–990. [Google Scholar] [CrossRef] [PubMed]
- Bandte, M.; Rehanek, M.; Leder, B.; von Bargen, S.; Büttner, C. Identification of an emaravirus in a common Oak (Quercus robur L.) Conservation seed orchard in germany: Implications for Oak health. Forests 2020, 11, 1174. [Google Scholar] [CrossRef]
- Rumbou, A.; Candresse, T.; von Bargen, S.; Büttner, C. Next-Generation Sequencing Reveals a Novel Emaravirus in Diseased Maple Trees from a German Urban Forest. Front. Microbiol. 2021, 11, 621179. [Google Scholar] [CrossRef] [PubMed]
- von Bargen, S.; Al Kubrusli, R.; Gaskin, T.; Fürl, S.; Hüttner, F.; Blystad, D.-R.; Karlin, D.G.; Jalkanen, R.; Büttner, C. Characterisation of a novel Emaravirus identified in mosaic-diseased Eurasian aspen (Populus tremula). Ann. App. Biol. 2020, 176, 210–222. [Google Scholar] [CrossRef]
- Elbeaino, T.; Digiaro, M.; Mielke-Ehret, N.; Muehlbach, H.P.; Martelli, G.P. ICTV Virus Taxonomy Profile: Fimoviridae. J. Gen. Virol. 2018, 99, 1478–1479. [Google Scholar] [CrossRef] [PubMed]
- Mielke-Ehret, N.; Mühlbach, H.P. Emaravirus: A novel genus of multipartite, negative strand RNA plant viruses. Viruses 2012, 4, 1515–1536. [Google Scholar] [CrossRef] [Green Version]
- Kormelink, R.; Garcia, M.L.; Goodin, M.; Sasaya, T.; Haenni, A.L. Negative-strand RNA viruses: The plant-infecting counterparts. Virus Res. 2011, 162, 184–202. [Google Scholar] [CrossRef] [PubMed]
- Mielke, N.; Muehlbach, H.P. A novel, multipartite, negative-strand RNA virus is associated with the ringspot disease of European mountain ash (Sorbus aucuparia L.). J. Gen. Virol. 2007, 88, 1337–1346. [Google Scholar] [CrossRef] [PubMed]
- ICTV Taxonomy Release 2020. Available online: https://talk.ictvonline.org/taxonomy (accessed on 6 July 2021).
- von Bargen, S.; Dieckmann, H.L.; Candresse, T.; Mühlbach, H.-P.; Roßbach, J.; Büttner, C. Determination of the complete genome sequence of European mountain ash ringspot-associated emaravirus from Sorbus intermedia reveals two additional genome segments. Arch. Virol. 2019, 164, 1937–1941. [Google Scholar] [CrossRef] [PubMed]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef]
- Boetzer, M.; Henkel, C.V.; Jansen, H.J.; Butler, D.; Pirovano, W. Scaffolding pre-assembled contigs using SSPACE. Bioinformatics 2011, 27, 578–579. [Google Scholar] [CrossRef] [Green Version]
- Boetzer, M.; Pirovano, W. Toward almost closed genomes with GapFiller. Genome Biol. 2012, 13, R56. [Google Scholar] [CrossRef] [Green Version]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Di Bello, P.L.; Ho, T.; Tzanetakis, I.E. The evolution of emaraviruses is becoming more complex: Seven segments identified in the causal agent of Rose rosette disease. Virus Res. 2015, 210, 241–244. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elbeaino, T.; Digiaro, M.; Alabdullah, A.; De Stradis, A.; Minafra, A.; Mielke, N.; Castellano, M.A.; Martelli, G.P. A multipartite single-stranded negative-sense RNA virus is the putative agent of fig mosaic disease. J. Gen. Virol. 2009, 90, 1281–1288. [Google Scholar] [CrossRef]
- Menzel, W.; Jelkmann, W.; Maiss, E. Detection of four apple viruses by multiplex RT-PCR assays with coamplification of plant mRNA as internal control. J. Virol. Methods 2002, 99, 81–92. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, E.S. Use of lead citrate at high pH as an electron-opaque stain in electron microscopy. J. Cell Biol. 1963, 17, 208–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaafar, Y.Z.; Herz, K.; Hartrick, J.; Fletcher, J.; Blouin, A.G.; MacDiarmid, R.; Ziebell, H. Investigating the Pea Virome in Germany—Old Friends and New Players in the Field. Front. Microbiol. 2020, 11, 583242. [Google Scholar] [CrossRef] [PubMed]
- Kubota, K.; Usugi, T.; Tomitaka, Y.; Shimomoto, Y.; Takeuchi, S.; Kadono, F.; Yanagisawa, H.; Chiaki, Y.; Tsuda, S. Perilla Mosaic Virus Is a Highly Divergent Emaravirus Transmitted by Shevtchenkella sp. (Acari: Eriophyidae). Phytopathology 2020, 110, 1352–1361. [Google Scholar] [CrossRef]
- Hassan, M.; Di Bello, P.L.; Keller, K.E.; Martin, R.R.; Sabanadzovic, S.; Tzanetakis, I.E. A new, widespread emaravirus discovered in blackberry. Virus Res. 2017, 235, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Csorba, T.; Burgyan, J. Antiviral Silencing and Suppression of Gene Silencing in Plants. In Current Research Topics in Plant Virology; Wang, A., Zhou, X., Eds.; Springer: Berlin/Heidelberg, Germany, 2016; pp. 1–33. [Google Scholar]
- Pumplin, N.; Voinnet, O. RNA silencing suppression by plant pathogens: Defense, counter defense and counter-counter-defense. Nat. Rev. Microbiol. 2013, 11, 745–760. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.K.; Hein, G.L.; Graybosch, R.A.; Tatineni, S. Octapartite negative-sense RNA genome of High Plains wheat mosaic virus encodes two suppressors of RNA silencing. Virology 2018, 518, 152–162. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.K.; Hein, G.L.; Tatineni, S. P7 and P8 proteins of High Plains wheat mosaic virus, a negative-strand RNA virus, employ distinct mechanisms of RNA silencing suppression. Virology 2019, 535, 20–31. [Google Scholar] [CrossRef] [Green Version]
- Olmedo-Velarde, A.; Park, A.C.; Sugano, J.; Uchida, J.Y.; Kawate, M.; Borth, W.B.; Hu, J.S.; Melzer, M.J. Characterization of Ti ringspot-associated virus, a novel emaravirus associated with an emerging ringspot disease of Cordyline fruticosa. Plant Dis. 2019, 103, 2345–2352. [Google Scholar] [CrossRef] [PubMed]
- McGavin, W.J.; Mitchell, C.; Cock, P.J.A.; Wright, K.M.; MacFarlane, S.A. Raspberry leaf blotch virus, a putative new member of the genus Emaravirus, encodes a novel genomic RNA. J. Gen. Virol. 2012, 93, 430–437. [Google Scholar] [CrossRef]
- Zheng, Y.; Navarro, B.; Wang, G.; Wang, Y.; Yang, Z.; Xu, W.; Zhu, C.; Wang, L.; Di Serio, F.; Hong, N. Actinidia chlorotic ringspot-associated virus: A novel emaravirus infecting kiwifruit plants. Mol. Plant Pathol. 2017, 18, 569–581. [Google Scholar] [CrossRef] [PubMed]
- Tatineni, S.; McMechan, A.J.; Wosula, E.N.; Wegulo, S.N.; Graybosch, R.A.; French, R.; Hein, G.L. An eriophyid mite-transmitted plant virus contains eight genomic RNA segments with unusual heterogeneity in the nucleocapsid protein. J. Virol. 2014, 88, 11834–11845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scotto–Lavino, E.; Du, G.; Frohman, M.A. 3′ end cDNA amplification using classic RACE. Nat. Protoc. 2006, 1, 2742–2745. [Google Scholar] [CrossRef]
- Scotto–Lavino, E.; Du, G.; Frohman, M.A. 5′ end cDNA amplification using classic RACE. Nat. Protoc. 2006, 1, 2555–2562. [Google Scholar] [CrossRef] [PubMed]
- Ochman, H.; Gerber, A.S.; Hartl, D.L. Genetic applications of an inverse polymerase chain reaction. Genetics 1988, 120, 621–623. [Google Scholar] [CrossRef]
- Laney, A.G.; Keller, K.E.; Martin, R.R.; Tzanetakis, I.E. A discovery 70 years in the making: Characterization of the rose rosette virus. J. Gen. Virol. 2011, 92, 1727–1732. [Google Scholar] [CrossRef]
- Kallinen, A.K.; Lindberg, I.L.; Tugume, A.K.; Valkonen, J.P. Detection, distribution, and genetic variability of European mountain ash ringspot-associated virus. Phytopathology 2009, 99, 344–352. [Google Scholar] [CrossRef]
- Führling, M.; Büttner, C. Transmission experiments of viruses to woody seedlings (Quercus robur L. and Sorbus aucuparia L.) by grafting and mechanical inoculation. Eur. J. For. Path. 1995, 25, 129–135. [Google Scholar]
- Liu, H.; Wang, G.; Yang, Z.; Wang, Y.; Zhang, Z.; Li, L.; Waqas, M.; Hong, N.; Liu, H.; Wang, G.; et al. Identification and Characterization of a Pear Chlorotic Leaf Spot-Associated Virus, a Novel Emaravirus Associated with a Severe Disease of Pear Trees in China. Plant Dis. 2021, 104, 2786–2798. [Google Scholar] [CrossRef] [PubMed]
- von Bargen, S.; Bandte, M.; Al Kubrusli, R.; Jalkanen, R.; Büttner, C. First report of European mountain ash ringspot-associated virus in Karpatiosorbus x hybrida in Finland. New Dis. Rep. 2020, 42, 1. [Google Scholar]
- Grimová, L.; Marek, M.; Konrady, M.; Ryšánek, P. Newly identified host range of European mountain ash ringspot-associated virus (EMARaV) and its distribution in the Czech Republic. For. Pathol. 2015, 45, 177–189. [Google Scholar] [CrossRef]
- von Bargen, S.; Tischendorf, M.; Büttner, C. First report of European mountain ash ringspot-associated virus in serviceberry (Amelanchier spp.) in Germany. New Dis. Rep. 2018, 37, 19. [Google Scholar] [CrossRef] [Green Version]
- García-Arenal, F.; Fraile, A.; Malpica, J.M. Variability and genetic structure of plant virus populations. Annu. Rev. Phytopathol. 2001, 39, 157–186. [Google Scholar] [CrossRef]
- von Bargen, S.; Arndt, N.; Robel, J.; Jalkanen, R.; Büttner, C. Detection and genetic variability of European mountain ash ringspot-associated virus (EMARaV) in Sweden. For. Pathol. 2013, 43, 429–432. [Google Scholar] [CrossRef]
- Roßbach, J.; Dieckmann, H.L.; Büttner, T.; Mühlbach, H.P.; von Bargen, S.; Büttner, C. Genetic Variability and Phylogeny of European mountain ash ringspot-associated virus RNA3 and RNA4. Forests 2015, 6, 4072–4087. [Google Scholar] [CrossRef]
- Dong, L.; Lemmetty, A.; Latvala, S.; Samuilova, O.; Valkonen, J.P.T. Occurrence and genetic diversity of Raspberry leaf blotch virus (RLBV) infecting cultivated and wild Rubus species in Finland. Ann. Appl. Biol. 2016, 168, 122–132. [Google Scholar] [CrossRef]
- Walia, J.J.; Willemsen, A.; Elci, E.; Caglayan, K.; Falk, B.W.; Rubio, L. Genetic variation and possible mechanisms driving the evolution of worldwide Fig mosaic virus isolates. Phytopathology 2014, 104, 108–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massart, S.; Candresse, T.; Gil, J.; Lacomme, C.; Predajna, L.; Ravnikar, M.; Reynard, J.-S.; Rumbou, A.; Saldarelli, P.; Škorić, D.; et al. A framework for the evaluation of biosecurity, commercial, regulatory, and scientific impacts of plant viruses and viroids identified by NGS technologies. Front. Microbiol. 2017, 8, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genome Segment | Primer | Sequence 5′-3′ | Amplicon Size (bp) |
|---|---|---|---|
| RNA1 | S104-684F S152-97R S50-1081F S294-146R | GGTGACAACTATACAAGATCAG CTAAATCATTGGCATATACACC GTATGAAAGGGATGATTCAGATC ATAGTCTATTGCTGGTGTGGAG | 429 289 |
| RNA2 | S68-933F S71-187R | CAGCAACTATTTGTGGTTCATG CATTTGTCCAGTGTACACATGG | 243 |
| RNA3 | S55-579F S55-920R | AAGATCTGCTCCTGATCCTGC CTGGTTGTCCCAATATCTCTGG | 342 |
| RNA4 | S65-99F S65-596R | CAATGGCATAGAAAGCATCACT GGTAATGTCTTCCATGATACATC | 498 |
| RNA5 | S164-41F S164-441R | GACAATTAGAGAGGCTCATGA CATGTACAGTTGATACCACAG | 401 |
| RNA Genomic Segment | Scaffold Number | Size (nt) | % Total Reads | Mean Coverage | Best Match in Blastx (Accession Number) | Query Coverage (%) | E Value | Sequence Identity (%) |
|---|---|---|---|---|---|---|---|---|
| Fraxinus excelsior | ||||||||
| RNA1 | S34 | 1455 | 0.37 | 6.54 | RdRP, Actinidia emaravirus 2 (QEE82886.1) | 99 | 0.0 | 73.76 |
| S50 | 1195 | 0.31 | 7.40 | RdRP, Actinidia emaravirus 2 (QEE82886.1) | 99 | 0.0 | 87.12 | |
| S104 | 797 | 0.20 | 5.27 | RdRP, Actinidia emaravirus 2 (QEE82886.1) | 95 | 1 × 10−102 | 63.53 | |
| S152 | 661 | 0.17 | 6.33 | RdRP, Pigeonpea sterility mosaic emaravirus 2 (QBA83607.1) | 94 | 3 × 10−116 | 85.58 | |
| S206 | 548 | 0.14 | 5.83 | RdRP, Actinidia emaravirus 2 (QEE82886.1) | 99 | 6 × 10−90 | 75.41 | |
| S294 | 465 | 0.12 | 7.54 | RdRP, Fig mosaic emaravirus (QBH72678.1) | 99 | 1 × 10−86 | 87.66 | |
| RNA2 | S68 | 1043 | 0.27 | 18.90 | GPP, Pigeonpea sterility mosaic emaravirus 2 (YP_009268865.1) | 90 | 5 × 10−138 | 56.51 |
| S71 | 1024 | 0.26 | 18.45 | GPP, Pigeonpea sterility mosaic emaravirus 2 (YP_009268865.1) | 96 | 7 × 10−153 | 61.49 | |
| RNA3 | S55 | 1177 | 0.30 | 24.46 | NC, Pea-associated emaravirus (QJX15716.1) | 80 | 0.0 | 99.05 |
| RNA4 | S65 | 1070 | 0.27 | 16.79 | MP, Fig mosaic emaravirus (QBI22245.1) | 97 | 0.0 | 77.30 |
| RNA5 | S164 | 635 | 0.16 | 13.04 | P6, Actinidia emaravirus 2 (QEE82891.1) | 78 | 2 × 10−54 | 46.39 |
| Fraxinus ornus | ||||||||
| RNA1 | S10 | 7122 | 0.12 | 57.80 | RdRP, Actinidia emaravirus 2 (QEE82886.1) | 98 | 0.0 | 77.88 |
| RNA2 | S146 | 1929 | 0.03 | 233.64 | GPP, Pigeonpea sterility mosaic emaravirus 2 (YP_009268865.1) | 95 | 0.0 | 59.90 |
| RNA3 | S1644 | 1463 | 0.01 | 267.67 | NC, Pea-associated emaravirus (QJX15716.1) | 64 | 0.0 | 99.05 |
| RNA4 | S81 | 1496 | 0.04 | 121.29 | MP, Fig mosaic emaravirus (QBI22245.1) | 72 | 0.0 | 77.29 |
| RNA5 | S164 | 996 | 0.03 | 70.28 | P6, Actinidia emaravirus 2 (QEE82891.1) | 60 | 1 × 10−65 | 47.50 |
| Virus | RNA1 (RdRp) | RNA2 (GPP) | RNA3 (N) | RNA4 (MP) | RNA5 (P 26–28 kDa) |
|---|---|---|---|---|---|
| AcCRaV | 48.7 | 39.5 | 35.6 | 29.0 | 10.8 (P5, 27 kDa) |
| AcV-2 | 77.9 | 54.7 | 73.1 | 73.6 | 42.4 (P6, 27 kDa) |
| AsMaV | 71.6 | 53.8 | 57.2 | 65.0 | 34.4 (P28, 28 kDa) |
| BLMaV | 68.4 | 50.6 | 59.1 | 56.1 | 28.7 (P5, 26 kDa) |
| CjaV-1 | 25.5 | 19.1 | 16.4 | 10.2 | - |
| CjaV-2 | 26.2 | 18.3 | 13.1 | 9.4 | - |
| CORaV | 31.1 | 21.7 | 17.6 | 16.4 | - |
| EMARaV | 49.1 | 36.5 | 34.8 | 28.2 1 | 10.6 (P4, 27 kDa) 2 |
| FMV | 73.8 | 55.2 | 69.2 | 76.1 | 31.0 (P6, 26 kDa) |
| HPWMoV | 30.3 | 21.6 | 15.1 | 16.5 | - |
| JYMaV | 31.5 | 21.8 | 20.5 | 15.8 | - |
| LiCRaV | 48.6 | 38.2 | 34.3 | 30.7 | 13.0 (P5, 26 kDa) |
| MaMaV | 67.5 | 50.1 | 47.6 | 62.6 | 32.7 (P6, 27 kDa) |
| PCLSaV | 28.0 | 14.2 | 14.8 | 9.2 | - |
| PerMV | 25.3 | 18.4 | 16.3 | 10.2 | 7.5 (P, 27 kDa) |
| PaEV | - | - | 99.0 | - | - |
| PiVB | 71.0 | 53.6 | 57.7 | 69.8 | 30.7 (P6, 28 kDa) |
| PPSMV-1 | 53.2 | 44.0 | 40.6 | 38.5 | 32.7 (P6, 27 kDa) |
| PPSMV-2 | 73.4 | 58.2 | 71.1 | 73.4 | 32.3 (P6, 27 kDa) |
| PVBV | 31.4 | 19.3 | 15.2 | 16.1 | - |
| RLBV | 31.3 | 21.4 | 18.8 | 17.3 | - |
| RRV | 68.3 | 52.7 | 59.8 | 59.8 | 28.9 (P6b, 27 kDa) |
| RYRSaV | 48.2 | 40.5 | 35.8 | 30.2 | 11.6 (P5, 26 kDa) |
| TiRSaV | 31.0 | 20.8 | 18.5 | 14.0 | - |
| Location | Host Species | Samples | Symptomatic/ASaV Infected | Asymptomatic/ASaV Infected |
|---|---|---|---|---|
| Germany 1 | F. excelsior | 5 | 5/4 | - |
| F. ornus | 44 | 34/34 | 10/1 | |
| Germany 2 | F. ornus | 11 | 9/9 | 2/2 |
| Italy | F. ornus | 2 | 2/1 | - |
| Sweden | F. excelsior | 39 | 29/26 | 10/6 |
| Switzerland | F. excelsior | 8 | 6/6 | 2/0 |
| Total | F. excelsior | 52 | 40/36 (90%) | 12/6 (50%) |
| F. ornus | 57 | 45/44 (98%) | 12/3 (25%) | |
| Total | 109 | 85/80 (94%) | 24/9 (38%) | |
| Host Species and Genome Segment | RNA | n 1 | Nt 2 | Min 3 | Max 3 | Mean | SEM 5 | aa | Min 4 | Max 4 | Mean | SEM 5 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| F. excelsior, F. ornus | 3 | 52 | 297 | 0 | 0.096 | 0.030 | 0.001 | 99 | 0 | 0.063 | 0.012 | 0.001 |
| F. excelsior | 18 | 0 | 0.088 | 0.031 | 0.002 | 0 | 0.063 | 0.011 | 0.002 | |||
| F. ornus | 34 | 0 | 0.089 | 0.025 | 0.001 | 0 | 0.063 | 0.012 | 0.001 | |||
| F. excelsior, F. ornus | 4 | 17 | 453 | 0 | 0.062 | 0.025 | 0.001 | 151 | 0 | 0.034 | 0.004 | 0.001 |
| F. excelsior | 10 | 0 | 0.055 | 0.026 | 0.003 | 0 | 0.034 | 0.007 | 0.002 | |||
| F. ornus | 7 | 0.002 | 0.044 | 0.025 | 0.003 | 0 | 0 | 0 | 0 | |||
| F. excelsior, F. ornus | 5 | 49 | 357 | 0 | 0.086 | 0.039 | 0.001 | 119 | 0 | 0.107 | 0.040 | 0.001 |
| F. excelsior | 17 | 0 | 0.072 | 0.027 | 0.002 | 0 | 0.097 | 0.033 | 0.002 | |||
| F. ornus | 32 | 0 | 0.086 | 0.036 | 0.001 | 0 | 0.106 | 0.033 | 0.002 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gaskin, T.R.; Tischendorf, M.; Günther, I.; Rehanek, M.; Büttner, C.; von Bargen, S. Characterization of a Novel Emaravirus Affecting Ash Species (Fraxinus spp.) in Europe. Forests 2021, 12, 1574. https://doi.org/10.3390/f12111574
Gaskin TR, Tischendorf M, Günther I, Rehanek M, Büttner C, von Bargen S. Characterization of a Novel Emaravirus Affecting Ash Species (Fraxinus spp.) in Europe. Forests. 2021; 12(11):1574. https://doi.org/10.3390/f12111574
Chicago/Turabian StyleGaskin, Thomas R., Max Tischendorf, Ines Günther, Marius Rehanek, Carmen Büttner, and Susanne von Bargen. 2021. "Characterization of a Novel Emaravirus Affecting Ash Species (Fraxinus spp.) in Europe" Forests 12, no. 11: 1574. https://doi.org/10.3390/f12111574