
Seven Complete Chloroplast Genomes from Symplocos: Genome Organization and Comparative Analysis


Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection, DNA Extraction, and CP Genome Sequencing
2.2. CP Genome Assembly and Annotation
2.3. Genome Comparison
2.4. Simple-Sequence Repeat (SSR) and Long Repeat Sequence Analysis
2.5. Divergent Hotspot Identification
2.6. Phylogenetic Analysis
3. Results
3.1. General Features of the CP Genomes
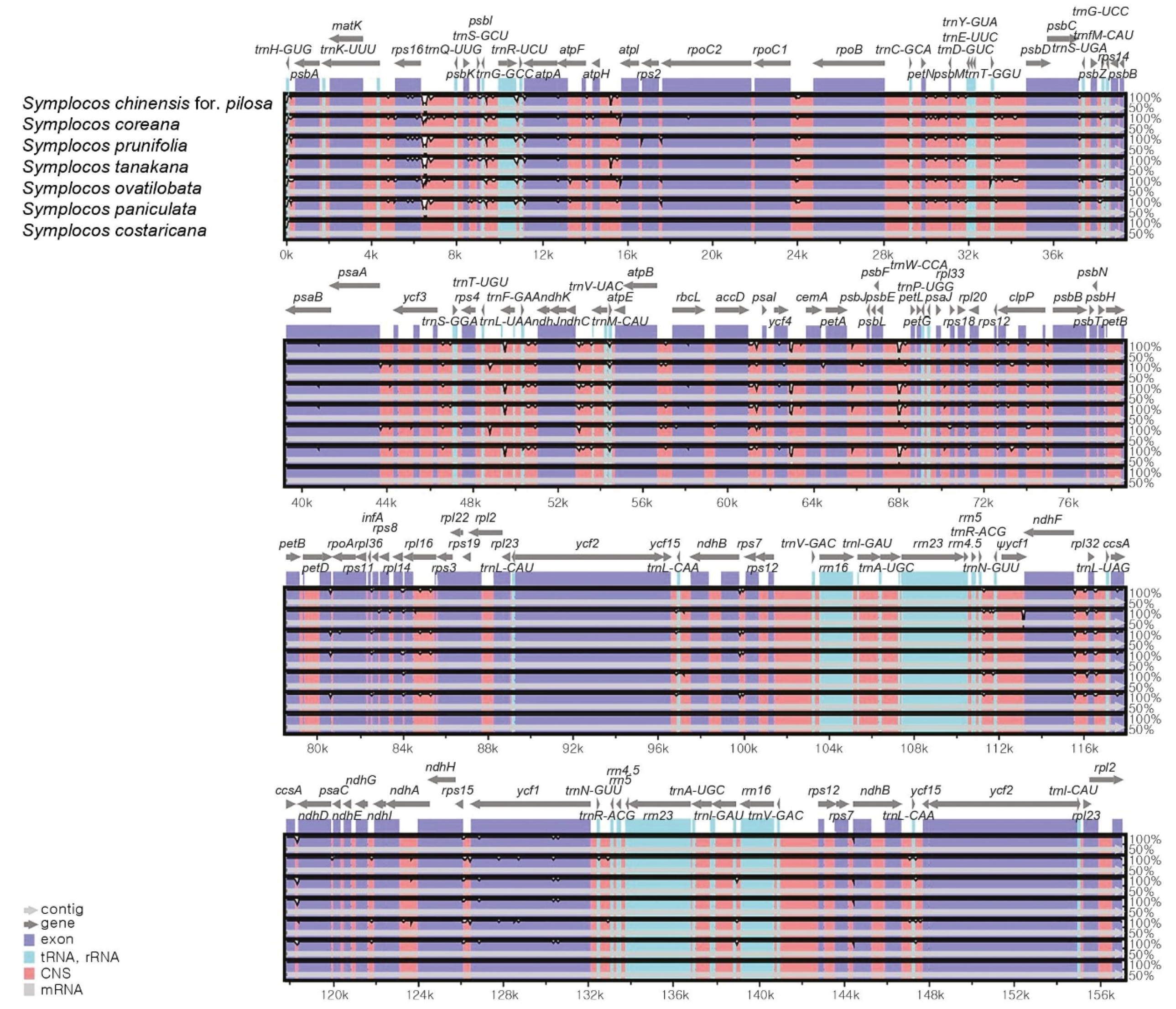
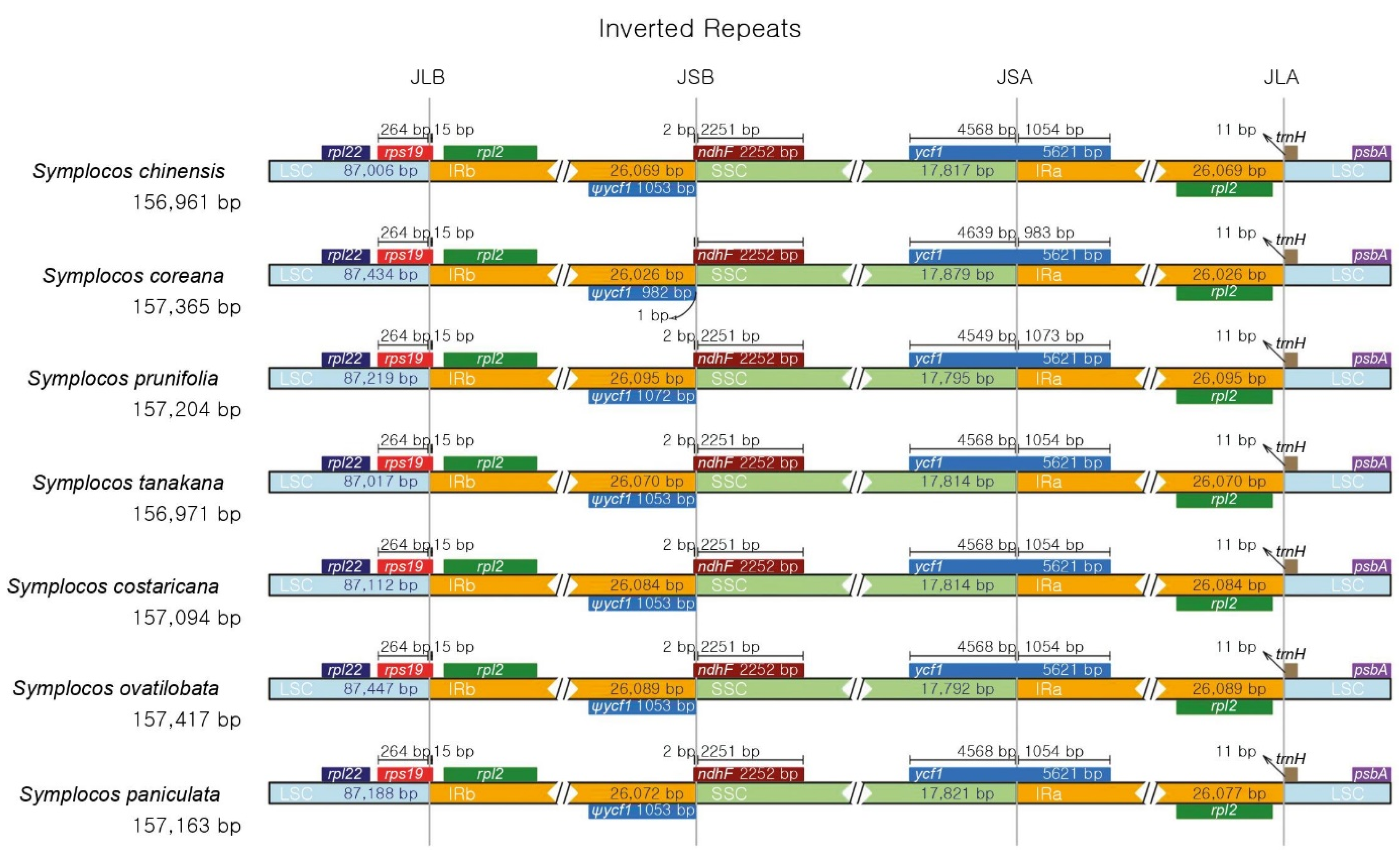
3.2. Comparison of CP Genomes of Seven Symplocos Species
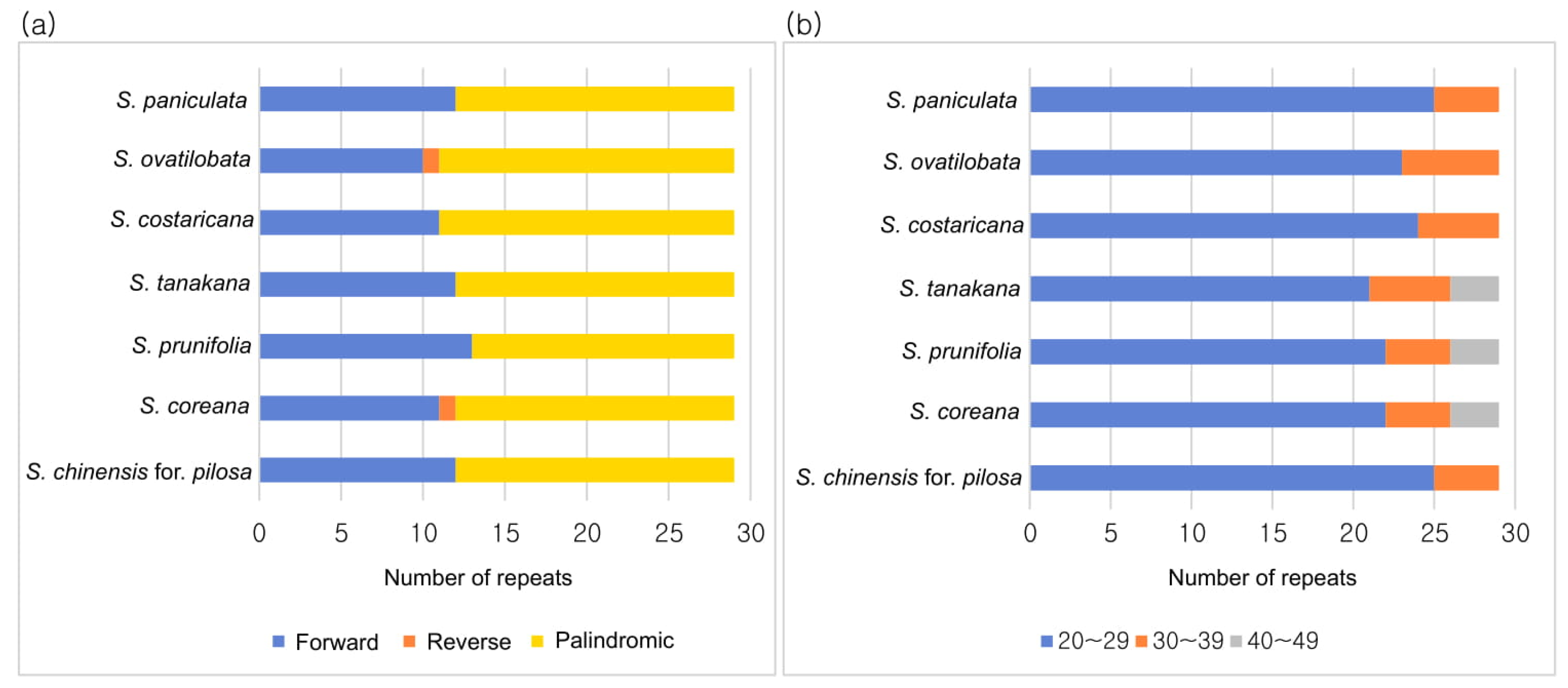
3.3. SSR and Long Repeat Analysis
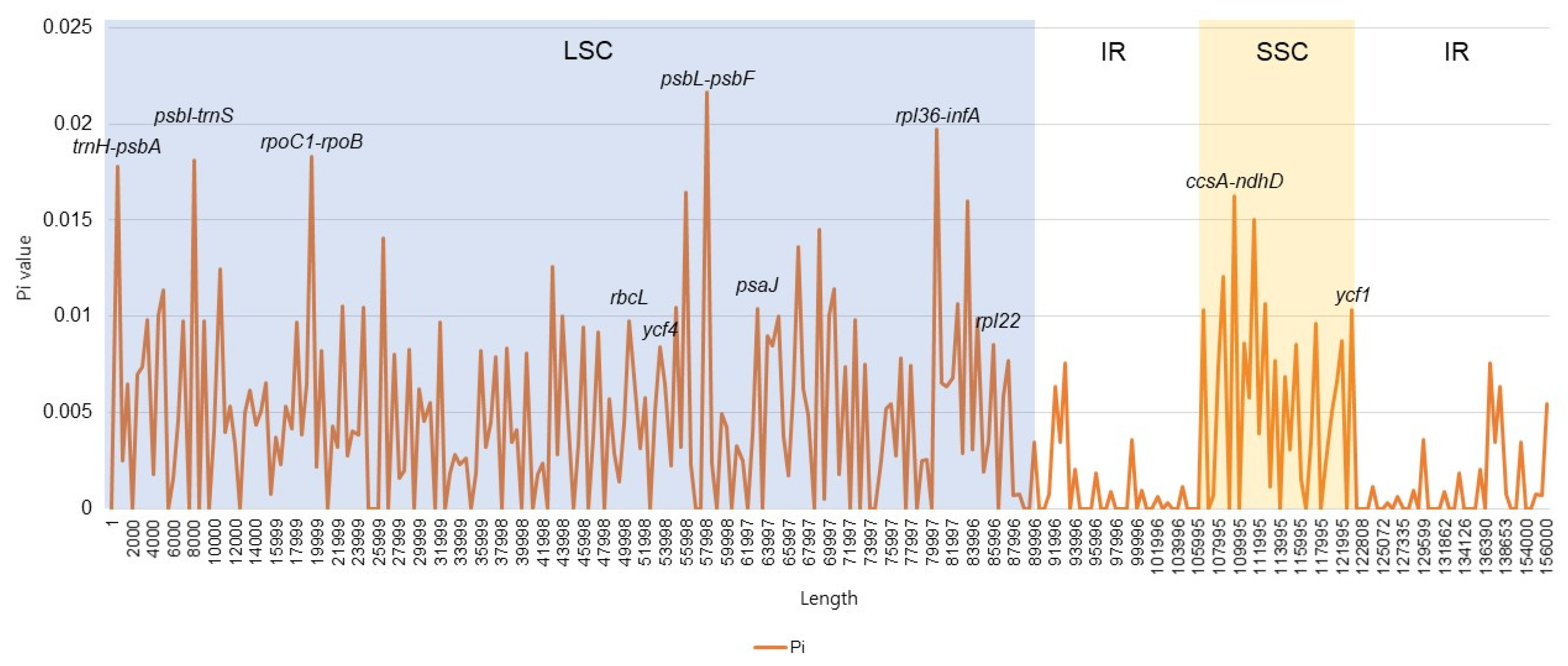
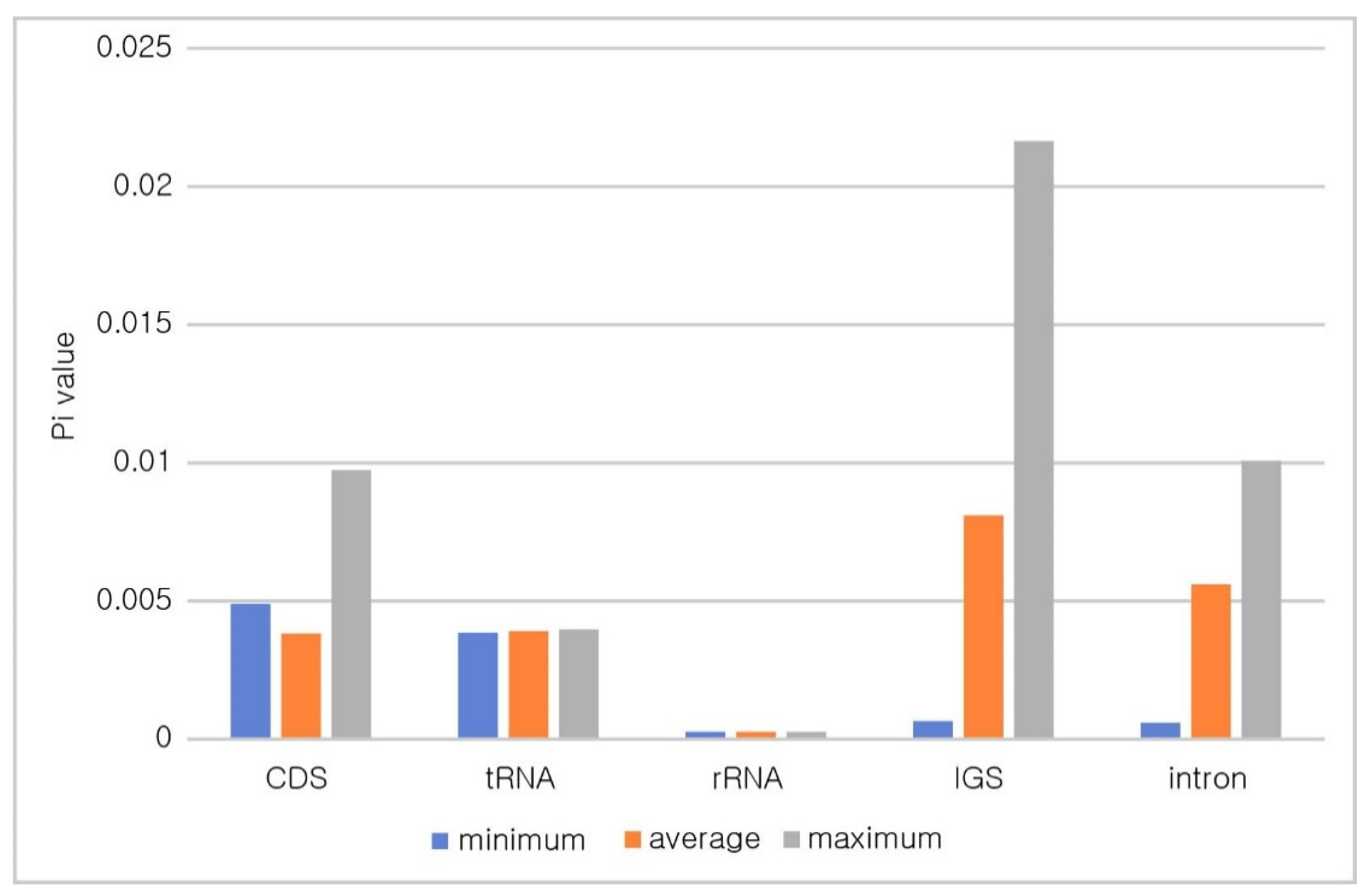
3.4. Divergent Hotspots in the Symplocos CP Genome
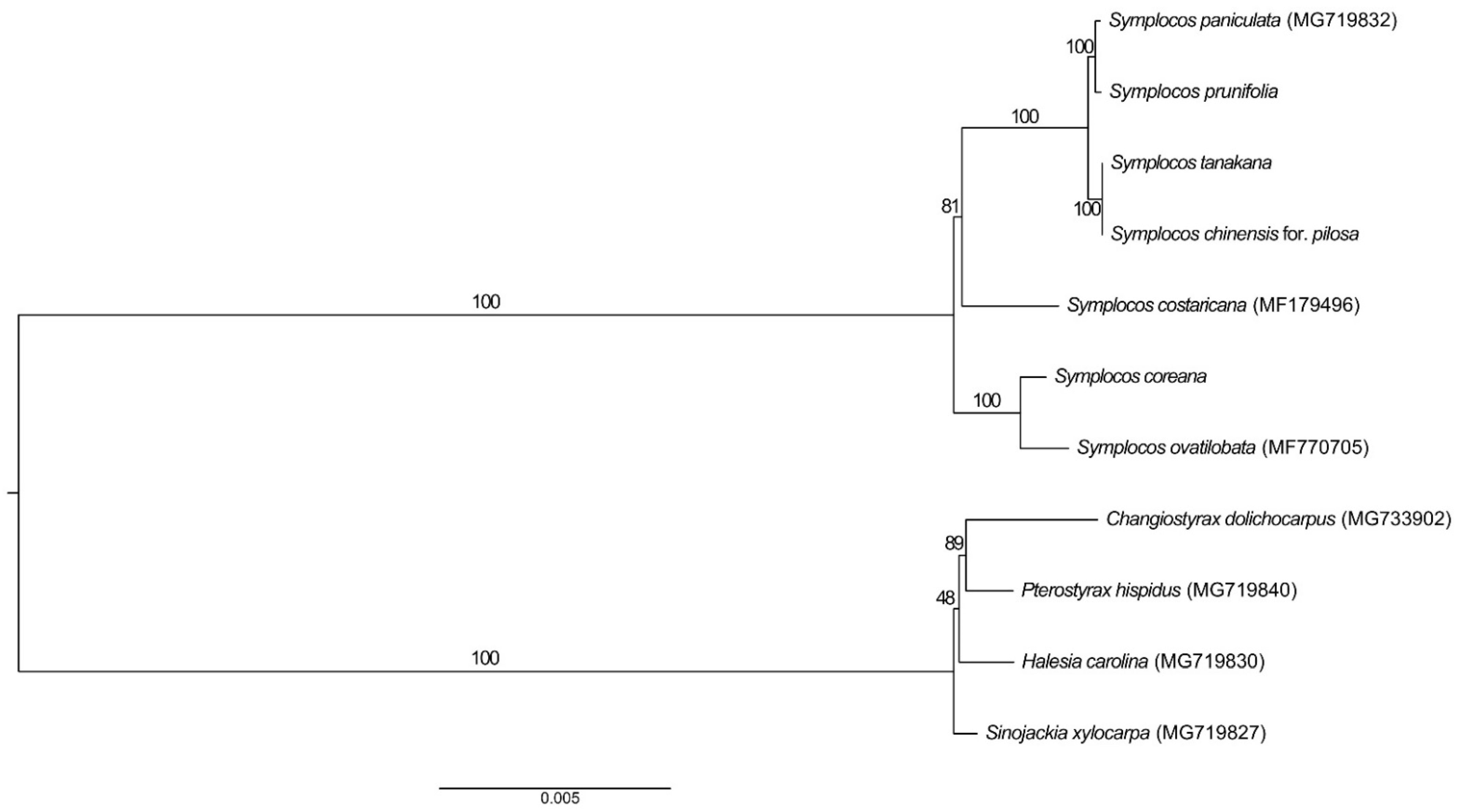
3.5. Phylogenetic Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jansen, R.K.; Raubeson, L.A.; Boore, J.L.; de Pamphilis, C.W.; Chumley, T.W.; Haberle, R.C.; Wyman, S.K.; Alverson, A.J.; Peery, R.; Herman, S.J.; et al. Methods for obtaining and analyzing whole chloroplast genome sequences. Methods Enzymol. 2005, 395, 348–384. [Google Scholar]
- Jansen, R.K.; Ruhlman, T.A. Plastid genomes of seed plants. In Genomics of Chloroplasts and Mitochondria; Bock, R., Knoop, V., Eds.; Springer: Dutch, The Netherlands, 2012; pp. 103–126. [Google Scholar]
- Palmer, J.D. Comparative organization of chloroplast genomes. Annu. Rev. Genet. 1985, 19, 325–354. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.C.; Lee, J.W. The complete chloroplast genome of Chamaecyparis obtusa (Cupressaceae). Mitochondrial DNA B Resour. 2020, 5, 3278–3279. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.; Kim, S.C.; Hong, K.N.; Kang, H.; Lee, J.W. The complete chloroplast genome of Torreya nucifera (Taxaceae) and phylogenetic analysis. Mitochondrial DNA B Resour. 2019, 4, 2537–2538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- CBOL Plant Working Group. A DNA barcode for land plants. Proc. Natl. Acad. Sci. USA 2009, 106, 12794–12797. [Google Scholar] [CrossRef] [Green Version]
- Cronquist, A.; Takhtadzhian, A.L. An Integrated System of Classification of Flowering Plants; Columbia University Press: New York, NY, USA, 1981. [Google Scholar]
- Kang, H.I.; Lee, H.O.; Lee, I.H.; Kim, I.S.; Lee, S.W.; Yang, T.J.; Shim, D. Complete chloroplast genome of Pinus densiflora Siebold & Zucc. and comparative analysis with five pine trees. Forests 2019, 10, 600. [Google Scholar]
- Kim, S.C.; Lee, J.W.; Lee, M.W.; Baek, S.H.; Hong, K.N. The complete chloroplast genome sequences of Larix kaempferi and Larix olgensis var. koreana (Pinaceae). Mitochondrial DNA B Resour. 2018, 3, 36–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.C.; Kim, J.S.; Kim, J.H. Insight into infrageneric circumscription through complete chloroplast genome sequences of two Trillium species. AoB Plants 2016, 8, plw015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabah, S.O.; Shrestha, B.; Hajrah, N.H.; Sabir, M.J.; Alharby, H.F.; Sabir, M.J.; Alhebshi, A.M.; Sabir, J.S.M.; Gilbert, L.E.; Ruhlman, T.A.; et al. Passiflora plastome sequencing reveals widespread genomic rearrangements. J. Syst. Evol. 2019, 57, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Parks, M.; Cronn, R.; Liston, A. increasing phylogenetic resolution at low taxonomic levels using massively parallel sequencing of chloroplast genomes. BMC Biol. 2009, 7, 84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bock, R. Structure, function, and inheritance of plastid genomes. In Topics in Current Genetics; Springer: Berlin/Heidelberg, Germany, 2007; pp. 29–63. [Google Scholar]
- Wang, Y.; Fritsch, P.W.; Shi, S.; Almeda, F.; Cruz, B.C.; Kelly, L.M. Phylogeny and infrageneric classification of Symplocos (Symplocaceae) inferred from DNA sequence data. Am. J. Bot. 2004, 91, 1901–1914. [Google Scholar] [CrossRef]
- Takhtajan, A.L. Diversity and Classification of Flowering Plants; Columbia University Press: New York, NY, USA, 1997. [Google Scholar]
- Angiosperm Phylogeny Group. An update of the angiosperm phylogeny group classification for the orders and families of flowering plants: APG III. Bot. J. Linn. Soc. 2009, 161, 105–121. [Google Scholar] [CrossRef] [Green Version]
- Angiosperm Phylogeny Group. An update of the angiosperm phylogeny group classification for the orders and families of flowering plants: APG IV. Bot. J. Linn. Soc. 2016, 181, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Fritsch, P.W.; Cruz, B.C.; Almeda, F.; Wang, Y.; Shi, S. Phylogeny of Symplocos based on DNA sequences of the chloroplast trnC–trnD intergenic region. Syst. Bot. 2006, 31, 181–192. [Google Scholar] [CrossRef]
- Soejima, A.; Nagamasu, H. Phylogenetic analysis of Asian Symplocos (Symplocaceae) based on nuclear and chloroplast DNA sequences. J. Plant Res. 2004, 117, 199–207. [Google Scholar] [CrossRef]
- Ghimire, B.; Park, B.K.; Oh, S.; Lee, J.; Son, D.C. Wood anatomy of Korean Symplocos Jacq. (Symplocaceae). Korean J. Pl. Taxon. 2020, 50, 333–342. [Google Scholar] [CrossRef]
- Park, S.H.; Lee, J.K.; Kim, J.H. A morphological study of Symplocaceae in Korea. Korean J. Pl. Taxon 2007, 37, 255–273. [Google Scholar] [CrossRef]
- Park, S.H.; Lee, J.K.; Kim, J.H. A systematic relationship of the Korean Symplocaceae based on RAPD analysis. Korean J. Pl. Taxon. 2007, 37, 225–237. [Google Scholar] [CrossRef]
- Kim, C.S.; Son, S.G.; Tho, J.H.; Kim, J.E.; Hwang, S.I.; Cheong, J.H. Distribution characteristics of woody plants resources in Jeiu, Korea. Korean J. Plant Resour. 2007, 20, 424–436. [Google Scholar]
- Korea National Arboretum. Rare Plants Data Book in Korea; Korea Forest Service: Seoul, Korea, 2008.
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [Green Version]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Wyman, S.K.; Jansen, R.K.; Boore, J.L. Automatic annotation of organellar genomes with DOGMA. Bioinformatics 2004, 20, 3252–3255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lowe, T.M.; Chan, P.P. TRNAscan-SE On-Line: Integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 2016, 44, W54–W57. [Google Scholar] [CrossRef] [PubMed]
- Lohse, M.; Drechsel, O.; Bock, R. OrganellarGenomeDRAW (OGDRAW): A tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr. Genet. 2007, 52, 267–274. [Google Scholar] [CrossRef]
- Katoh, K.; Kuma, K.I.; Toh, H.; Miyata, T. MAFFT Version 5: Improvement in accuracy of multiple sequence alignment. Nucleic Acids Res. 2005, 33, 511–518. [Google Scholar] [CrossRef]
- Mayor, C.; Brudno, M.; Schwartz, J.R.; Poliakov, A.; Rubin, E.M.; Frazer, K.A.; Pachter, L.S.; Dubchak, I. VISTA: Visualizing global DNA sequence alignments of arbitrary length. Bioinformatics 2000, 16, 1046–1047. [Google Scholar] [CrossRef] [Green Version]
- Amiryousefi, A.; Hyvönen, J.; Poczai, P. IRscope: An online program to visualize the junction sites of chloroplast genomes. Bioinformatics 2018, 34, 3030–3031. [Google Scholar] [CrossRef]
- Thiel, T.; Michalek, W.; Varshney, R.K.; Graner, A. Exploiting EST databases for the development and characterization of gene-derived SSR-Markers in barley (Hordeum vulgare L.). Theor. Appl. Genet. 2003, 106, 411–422. [Google Scholar] [CrossRef]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef] [Green Version]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Santorum, J.M.; Darriba, D.; Taboada, G.L.; Posada, D. Jmodeltest.org: Selection of nucleotide substitution models on the cloud. Bioinformatics 2014, 30, 1310–1311. [Google Scholar] [CrossRef] [Green Version]
- Stamatakis, A.; Hoover, P.; Rougemont, J. A rapid bootstrap algorithm for the RAxML web servers. Syst. Biol. 2008, 57, 758–771. [Google Scholar] [CrossRef] [PubMed]
- Acharya, N.; Acharya, S.; Shah, U.; Shah, R.; Hingorani, L. A comprehensive analysis on Symplocos racemosa Roxb.: Traditional uses, botany, phytochemistry and pharmacological activities. J. Ethnopharmacol. 2016, 181, 236–251. [Google Scholar] [CrossRef]
- Im, H.T.; Hong, H.H.; Son, H.D.; Park, M.S.; Nam, B.M.; Kwon, B.K.; Lee, C.H.; Chung, G.Y. The usage of regional folk plants in Gyeongsangnam-do. Korean J. Plant Resour. 2011, 24, 419–429. [Google Scholar] [CrossRef]
- Zhu, Z.X.; Wang, J.H.; Cai, Y.C.; Zhao, K.K.; Zhou, R.C.; Wang, H.F. Characterization of the complete chloroplast genome sequence of Symplocos ovatilobata (Symplocaceae). Conserv. Genet. Resour. 2018, 10, 503–506. [Google Scholar] [CrossRef]
- Grassi, F.; Labra, M.; Scienza, A.; Imazio, S. Chloroplast SSR markers to assess DNA diversity in wild and cultivated grapevines. Vitis 2002, 41, 157–158. [Google Scholar]
- Xue, J.; Wang, S.; Zhou, S.L. Polymorphic chloroplast microsatellite loci in Nelumbo (Nelumbonaceae). Am. J. Bot. 2012, 99, e240–e244. [Google Scholar] [CrossRef]
- Mariette, S.; Le Corre, V.; Austerlitz, F.; Kremer, A. Sampling within the genome for measuring within-population diversity: Trade-offs between markers. Mol. Ecol. 2002, 11, 1145–1156. [Google Scholar] [CrossRef] [Green Version]
- Torokeldiev, N.; Ziehe, M.; Gailing, O.; Finkeldey, R. Genetic diversity and structure of natural Juglans regia L. populations in the southern Kyrgyz Republic revealed by nuclear SSR and EST-SSR markers. Tree Genet. Genomes 2019, 15, 1–12. [Google Scholar] [CrossRef]
- Goulding, S.E.; Wolfe, K.H.; Olmstead, R.G.; Morden, C.W. Ebb and flow of the chloroplast inverted repeat. Mol. Gen. Genet. 1996, 252, 195–206. [Google Scholar] [CrossRef]
- Wang, R.J.; Cheng, C.L.; Chang, C.C.; Wu, C.L.; Su, T.M.; Chaw, S.M. Dynamics and evolution of the inverted repeat-large single copy junctions in the chloroplast genomes of monocots. BMC Evol. Biol. 2008, 8, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, K.S.; Ha, Y.H.; Gil, H.Y.; Choi, K.; Kim, D.K.; Oh, S.H. Two Korean endemic Clematis chloroplast genomes: Inversion, reposition, expansion of the inverted repeat region, phylogenetic analysis, and nucleotide substitution rates. Plants 2021, 10, 397. [Google Scholar] [CrossRef]
- Kim, S.C.; Lee, J.W.; Baek, S.H.; Lee, M.W.; Hong, K.N. The complete chloroplast genome of Fraxinus chiisanensis (Oleaceae). Mitochondrial DNA B Resour. 2017, 2, 823–824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Tan, W.; Sun, J.; Du, J.; Zheng, C.; Tian, X.; Zheng, M.; Xiang, B.; Wang, Y. Comparison of four complete chloroplast genomes of medicinal and ornamental Meconopsis species: Genome organization and species discrimination. Sci. Rep. 2019, 9, 10567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Xu, W.; Zou, W.; Jiang, D.; Liu, X. Complete chloroplast genome sequences of two endangered Phoebe (Lauraceae) species. Bot. Stud. 2017, 58, 37. [Google Scholar] [CrossRef]
- Kim, S.C.; Baek, S.H.; Lee, J.W.; Hyun, H.J. Complete chloroplast genome of Vaccinium oldhamii and phylogenetic analysis. Mitochondrial DNA B Resour. 2019, 4, 902–903. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Chen, T.; Zhang, Y.; Li, Y.; Gong, J.; Yi, Y. The complete chloroplast genome of Rhododendron delavayi (Ericaceae). Mitochondrial DNA B Resour. 2020, 5, 37–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez-Alberola, F.; Del Campo, E.M.; Lázaro-Gimeno, D.; Mezquita-Claramonte, S.; Molins, A.; Mateu-Andrés, I.; Pedrola-Monfort, J.; Casano, L.M.; Barreno, E. Balanced gene losses, duplications and intensive rearrangements led to an unusual regularly sized genome in Arbutus unedo chloroplasts. PLoS ONE 2013, 8, e79685. [Google Scholar]
- Särkinen, T.; George, M. Predicting plastid marker variation: Can complete plastid genomes from closely related species help? PLoS ONE 2013, 8, e82266. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Shen, X.; Liao, B.; Xu, J.; Hou, D. Comparing and phylogenetic analysis chloroplast genome of three Achyranthes species. Sci. Rep. 2020, 10, 10818. [Google Scholar] [CrossRef]
- Luo, J.; Hou, B.W.; Niu, Z.T.; Liu, W.; Xue, Q.Y.; Ding, X.Y. Comparative chloroplast genomes of photosynthetic orchids: Insights into evolution of the Orchidaceae and development of molecular markers for phylogenetic applications. PLoS ONE 2014, 9, e99016. [Google Scholar] [CrossRef] [PubMed]
- Moore, M.J.; Soltis, P.S.; Bell, C.D.; Burleigh, J.G.; Soltis, D.E. Phylogenetic analysis of 83 plastid genes further resolves the early diversification of eudicots. Proc. Natl. Acad. Sci. USA 2010, 107, 4623–4628. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | S. chinensis for. pilosa | S. coreana | S. prunifolia | S. tanakana |
|---|---|---|---|---|
| Specimen number | WTFRC10032701 | WTFRC10031678 | WTFRC10032813 | WTFRC10031670 |
| Accession number | MW307951 | MW307952 | MW307953 | MW307954 |
| Total bases (GB) | 7.24 | 11 | 9.57 | 12.8 |
| Total reads | 42,998,603 | 64,043,107 | 56,918,749 | 77,435,124 |
| Read length (bp) | 177 | 180 | 175 | 171 |
| Genome size [GC (%)] | 156,961 [37.5] | 157,365 [37.5] | 157,204 [37.5] | 156,971 [37.5] |
| LSC [GC (%)] | 87,006 [35.5] | 87,434 [35.5] | 87,219 [35.4] | 87,017 [35.5] |
| SSC [GC (%)] | 17,817 [31.0] | 17,879 [31.0] | 17,795 [31.0] | 17,814 [31.0] |
| IR [GC (%)] | 26,069 [43.1] | 26,026 [43.1] | 26,095 [43.1] | 26,070 [43.1] |
| Gene Category | Gene Group | Gene Names |
|---|---|---|
| Self-replication | Large subunit ribosomal protein | rpl2(×2) *, rpl14, rpl16 *, rpl20, rpl22, rpl23(×2), rpl32, rpl33, rpl36 |
| DNA dependent RNA polymerase | rpoA, rpoB, rpoC1 *, rpoC2 | |
| Small subunit ribosomal protein | rps2, rps3, rps4, rps7(×2), rps8, rps11, rps12(×2) **, rps14, rps15, rps16 *, rps18, rps19 | |
| rRNA | rrn4.5S(×2), rrn5S(×2), rrn16S(× 2), rrn23S(×2) | |
| tRNA | trnA-UGC(×2) *, trnC-GCA, trnD-GUC, trnE-UUC, trnF-GAA, trnfM-CAU, trnG-GC C *, trnG-UCC, trnH-GUG, trnI-GAU(×2) *, trnI-CAU(×2), trnK-UUU *, trnL-CAA(×2), trnL-UAA *, trnL-UAG, trnM-CAU, trnN-GUU(×2), trnP-UGG, trnQ-UUG, trnR-ACG(×2), trnR-UCU, trnS-GCU, trnS-GGA, trnS-UGA, trnT-GGU, trnT-UGU, trnV-GAC(×2), trnV-UAC *, trnW-CCA, trnY-GUA | |
| Photosynthesis | ATP synthase subunit | atpA, atpB, atpE, atpF *, atpH, atpI |
| NADH-dehydrogenase subunit | ndhA *, ndhB(×2) *, ndhC, ndhD, ndhE, ndhF, ndhG, ndhH, ndhI, ndhJ, ndhK | |
| Cytochrome b/f complex subunit | petA, petB *, petD, petG, petL, petN | |
| Photosystem I subunit | psaA, psaB, psaC, psaI, psaJ, ycf4 | |
| Photosystem II subunit | psbA, psbB, psbC, psbD, psbE, psbF, psbH, psbI, psbJ, psbK, psbL, psbM, psbN, psbT, psbZ | |
| Rubisco subunit | rbcL | |
| Other genes | Acetyl-CoA-carboxylase subunit | accD |
| C-type cytochrome synthesis gene | ccsA | |
| Envelope membrane protein | cemA | |
| ATP-dependent protease subunit P | clpP ** | |
| Translational initiation factor | infA | |
| Maturase | matK | |
| Unknown function | Conserved open reading frame | ycf1, ycf2(×2), ycf3 **, ycf15(×2) |
| SSR Type | Repeat Unit | S. chinensis for. pilosa | S. coreana | S. prunifolia | S. tanakana | S. costaricana | S. ovatilobata | S. paniculata | Total |
|---|---|---|---|---|---|---|---|---|---|
| Mono- | A/T | 27 | 29 | 38 | 31 | 37 | 28 | 36 | 230 |
| C/G | 1 | 1 | 0 | 0 | 1 | 0 | 1 | ||
| Di- | AT/AT | 6 | 3 | 6 | 6 | 5 | 4 | 6 | 36 |
| Tri- | AAT/ATT | 1 | 1 | 1 | 1 | 2 | 1 | 1 | 9 |
| AGC/CTG | 0 | 1 | 0 | 0 | 0 | 0 | 0 | ||
| Tetra- | AAAG/CTTT | 3 | 2 | 3 | 3 | 2 | 2 | 3 | 79 |
| AAAT/ATTT | 3 | 2 | 3 | 3 | 2 | 2 | 3 | ||
| AACC/GGTT | 0 | 0 | 0 | 0 | 1 | 1 | 0 | ||
| AAGG/CCTT | 1 | 0 | 1 | 1 | 2 | 0 | 1 | ||
| AATC/ATTG | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ||
| AATT/AATT | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ||
| ACAG/CTGT | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ||
| AGAT/ATCT | 1 | 0 | 1 | 1 | 1 | 1 | 1 | ||
| ATCC/ATGG | 2 | 0 | 2 | 2 | 0 | 0 | 2 | ||
| Penta- | AACTT/AAGTT | 0 | 1 | 0 | 0 | 1 | 1 | 0 | 8 |
| AAGGT/ACCTT | 0 | 0 | 0 | 0 | 0 | 1 | 0 | ||
| AATAT/ATATT | 0 | 0 | 0 | 0 | 1 | 0 | 0 | ||
| ACTAT/AGTAT | 0 | 1 | 0 | 0 | 1 | 1 | 0 | ||
| Total | 48 | 44 | 58 | 51 | 59 | 45 | 57 | 362 |
| Repeat Type | S. chinensis for. pilosa | S. coreana | S. prunifolia | S. tanakana | S. costaricana | S. ovatilobata | S. paniculata |
|---|---|---|---|---|---|---|---|
| Forward | 12 | 11 | 13 | 12 | 11 | 10 | 12 |
| Reverse | 0 | 1 | 0 | 0 | 0 | 1 | 0 |
| Palindromic | 17 | 17 | 16 | 17 | 18 | 18 | 17 |
| Total | 29 | 29 | 29 | 29 | 29 | 29 | 29 |
| Repeat length (bp) | |||||||
| 20–29 | 25 | 22 | 22 | 21 | 24 | 23 | 25 |
| 30–39 | 4 | 4 | 4 | 4 | 5 | 6 | 4 |
| 40–49 | 0 | 3 | 3 | 3 | 0 | 0 | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, S.-C.; Lee, J.-W.; Choi, B.-K. Seven Complete Chloroplast Genomes from Symplocos: Genome Organization and Comparative Analysis. Forests 2021, 12, 608. https://doi.org/10.3390/f12050608
Kim S-C, Lee J-W, Choi B-K. Seven Complete Chloroplast Genomes from Symplocos: Genome Organization and Comparative Analysis. Forests. 2021; 12(5):608. https://doi.org/10.3390/f12050608
Chicago/Turabian StyleKim, Sang-Chul, Jei-Wan Lee, and Byoung-Ki Choi. 2021. "Seven Complete Chloroplast Genomes from Symplocos: Genome Organization and Comparative Analysis" Forests 12, no. 5: 608. https://doi.org/10.3390/f12050608