
The Use of “Genotyping-by-Sequencing” to Recover Shared Genealogy in Genetically Diverse Eucalyptus Populations

 , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Materials and Methods
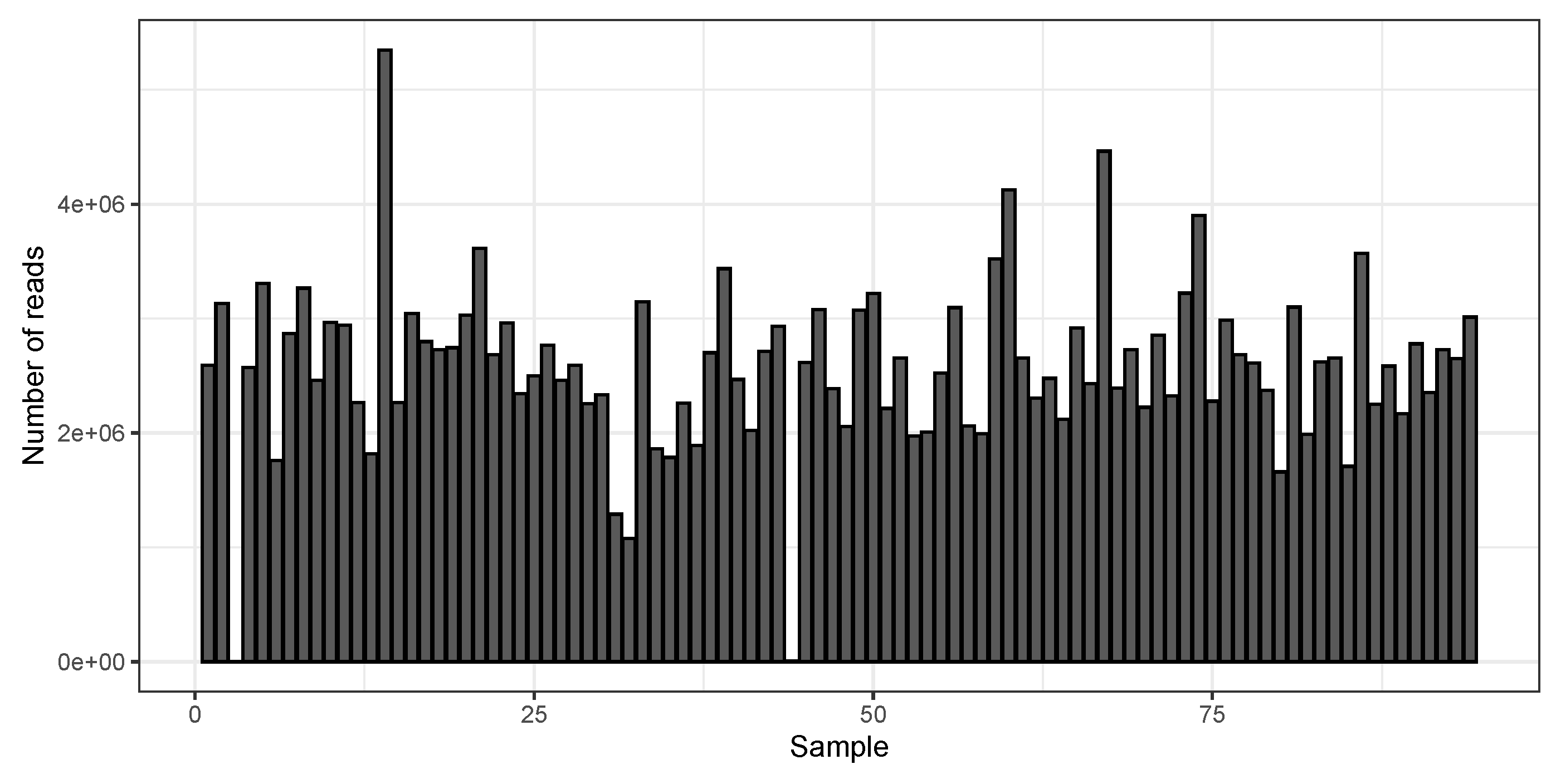
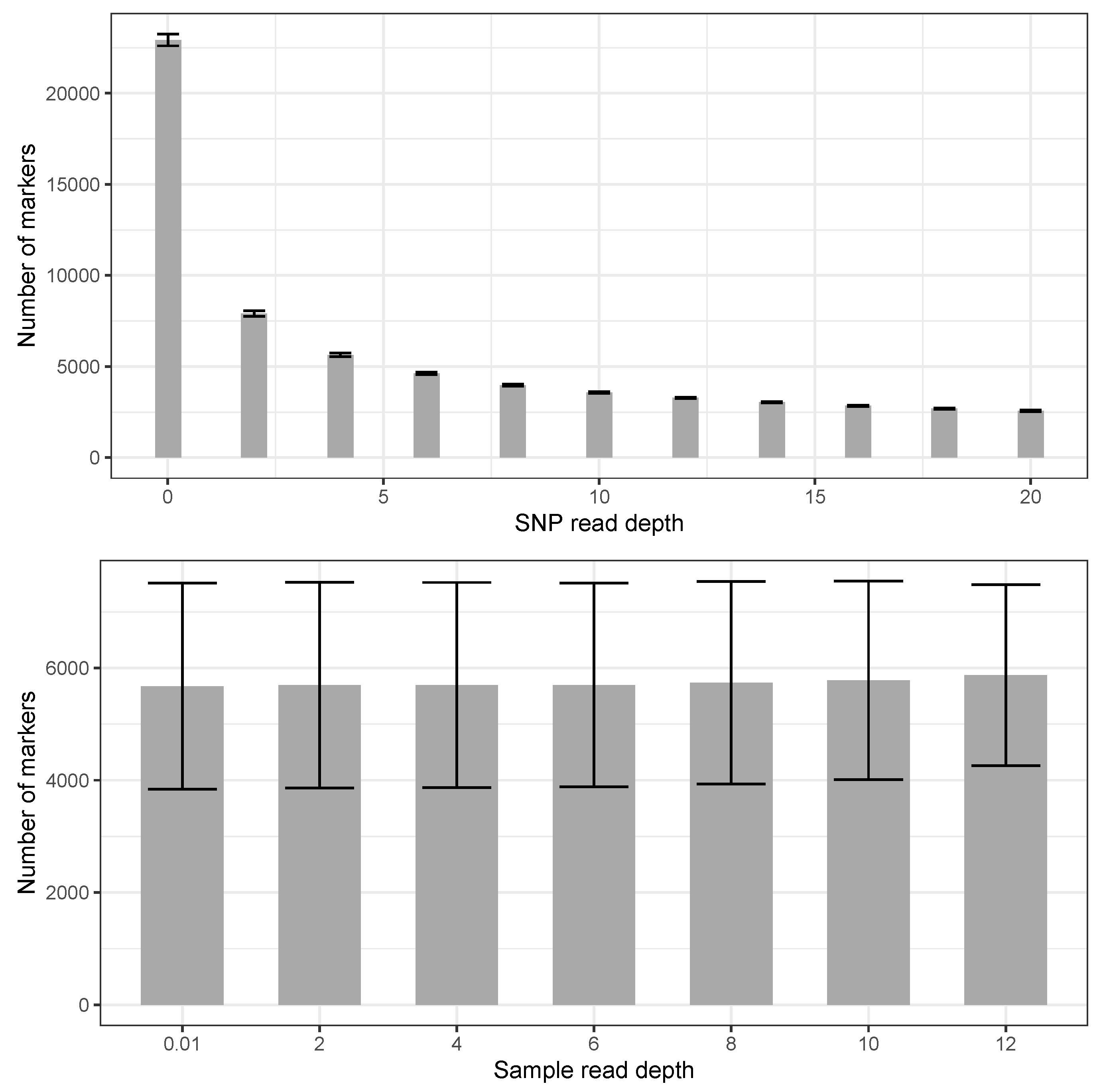
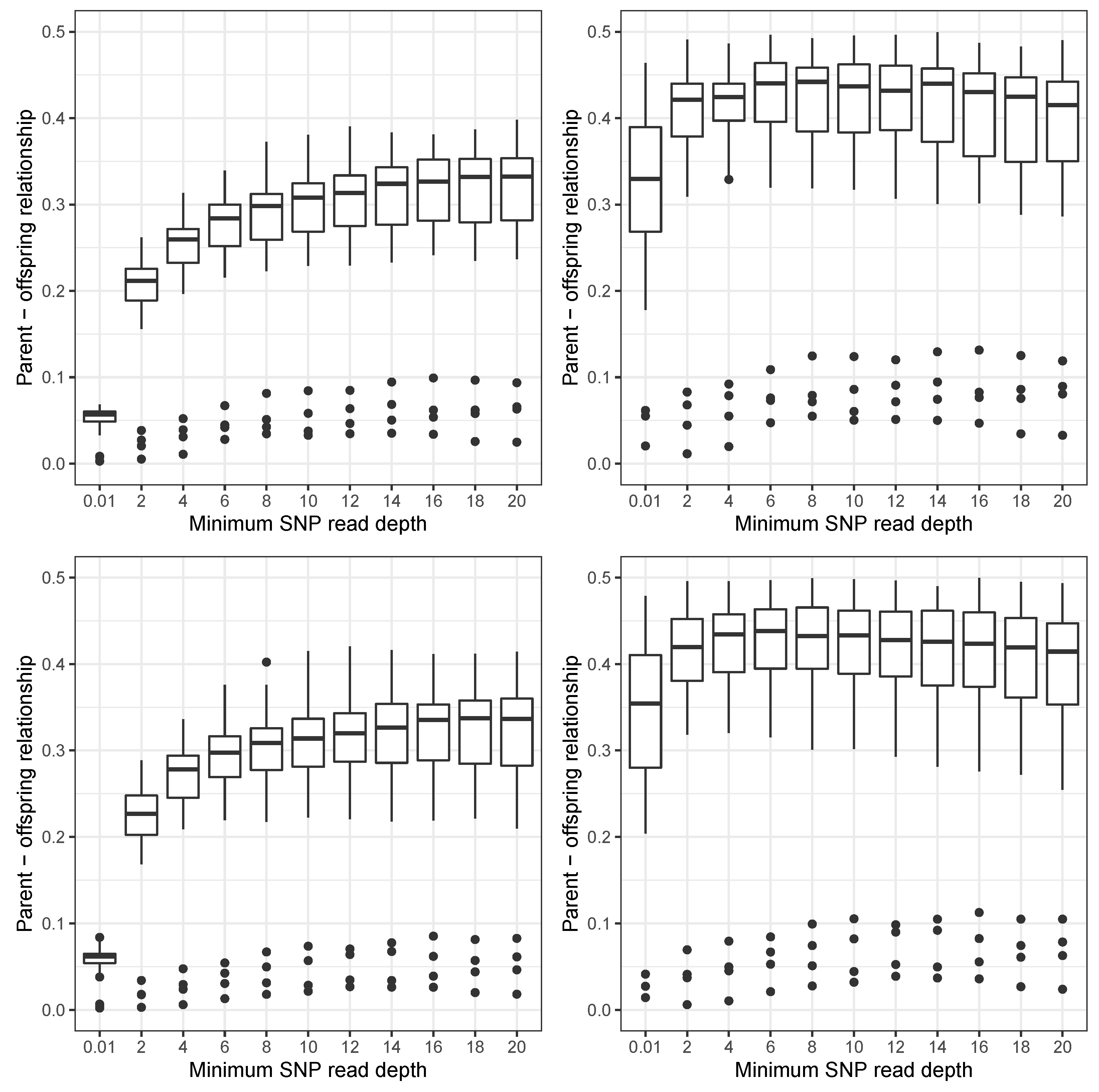
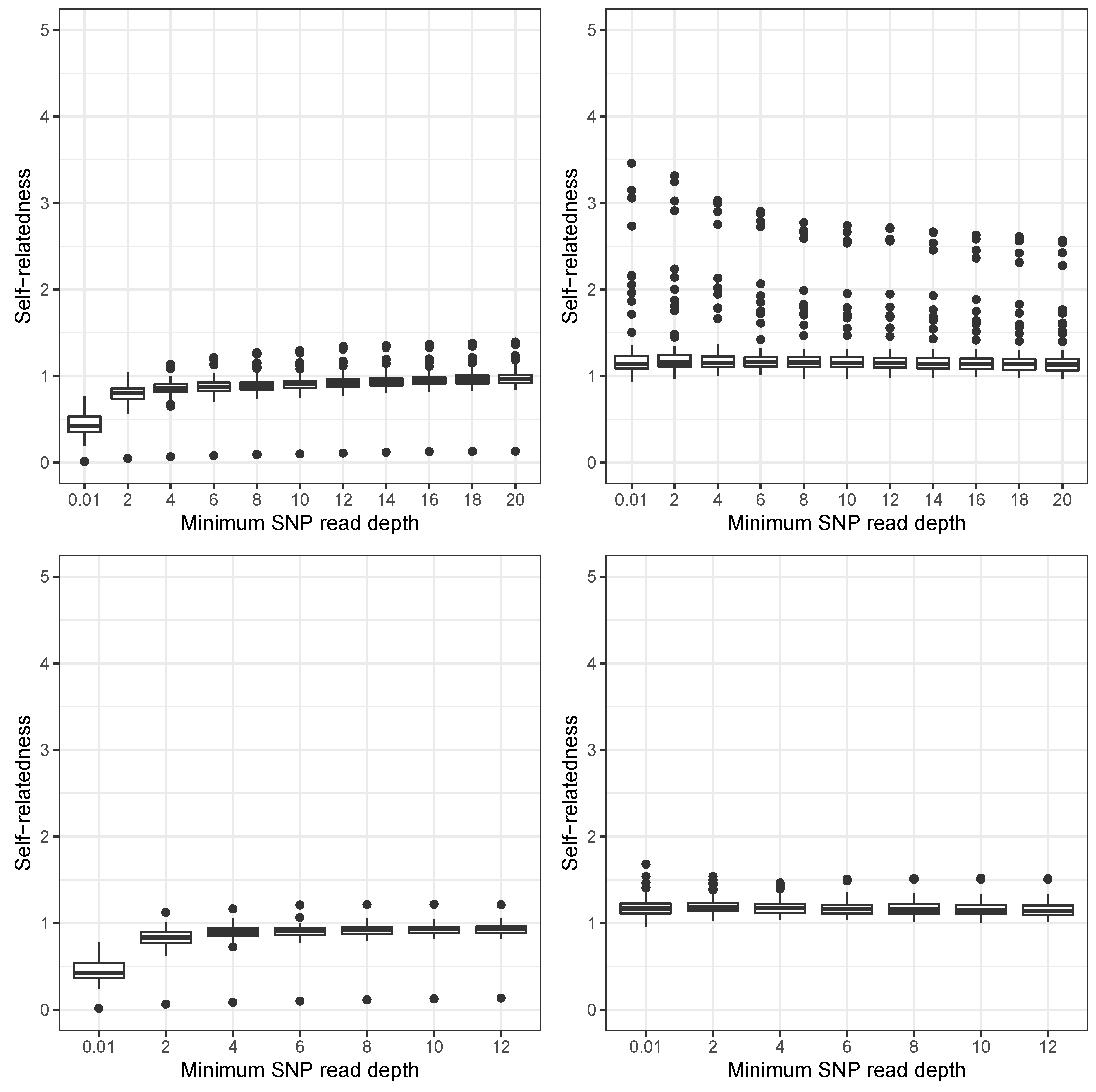
3. Results
4. Discussion
4.1. Challenges to Recover Relatedness in Forest Tree Breeding Populations
4.2. Genomic Resources in Eucalypts and Their Limitations
4.3. Recovery of Genealogy Using GBS
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| GBS | Genotyping by sequencing |
| DNA | Deoxyribonucleic acid |
| IBD | Identity by descent |
| SSR | Simple sequence repeats |
| SNP | Single nucleotide polymorphism |
| QTL | Quantitative trait loci |
| LD | Linkage disequilibrium |
References
- Wright, S. Coefficients of inbreeding and relationship. Am. Nat. 1922, 56, 330–338. [Google Scholar] [CrossRef] [Green Version]
- Malécot, G.; Blaringhem, L.-F. Les Mathématiques de L’hérédité; Masson: Paris, France, 1948. [Google Scholar]
- Mrode, R.A. Linear Models for the Prediction of Animal Breeding Values; CABI: Wallingford, UK, 2014. [Google Scholar]
- Henderson, C.R. Estimation of variances in animal model and reduced animal model for single traits and single records. J. Dairy Sci. 1986, 69, 1394–1402. [Google Scholar] [CrossRef]
- Westell, R.A.; Quaas, R.L.; Van Vleck, L.D. Genetic groups in an animal model. J. Dairy Sci. 1988, 71, 1310–1318. [Google Scholar] [CrossRef]
- Shiotsuki, L.; Cardoso, F.F.; Silva, J.A.V., II; Rosa, G.J.M.; Albuquerque, L.G. Evaluation of an average numerator relationship matrix model and a Bayesian hierarchical model for growth traits in Nellore cattle with uncertain paternity. Livest. Sci. 2012, 144, 89–95. [Google Scholar] [CrossRef] [Green Version]
- Henderson, C.R. Use of an average numerator relationship matrix for multiple-sire joining. J. Anim. Sci. 1988, 66, 1614–1621. [Google Scholar] [CrossRef]
- Lambeth, C.; Lee, B.-C.; O’Malley, D.; Wheeler, N. Polymix breeding with parental analysis of progeny: An alternative to full-sib breeding and testing. Theor. Appl. Genet. 2001, 103, 930–943. [Google Scholar] [CrossRef]
- El-Kassaby, Y.A.; Lstibůrek, M. Breeding without breeding. Genet. Res. 2009, 91, 111–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Kassaby, Y.A.; Cappa, E.P.; Liewlaksaneeyanawin, C.; Klápště, J.; Lstibůrek, M. Breeding without breeding: Is a complete pedigree necessary for efficient breeding? PLoS ONE 2011, 6, e25737. [Google Scholar] [CrossRef] [Green Version]
- Vidal, M.; Plomion, C.; Harvengt, L.; Raffin, A.; Boury, C.; Bouffier, L. Paternity recovery in two maritime pine polycross mating designs and consequences for breeding. Tree Genet. Genomes 2015, 11, 105. [Google Scholar] [CrossRef]
- Munoz, P.R.; Resende, M.F.R.; Huber, D.A.; Quesada, T.; Resende, M.D.V.; Neale, D.B.; Wegrzyn, J.L.; Kirst, M.; Peter, G.F. Genomic relationship matrix for correcting pedigree errors in breeding populations: Impact on genetic parameters and genomic selection accuracy. Crop Sci. 2014, 54, 1115–1123. [Google Scholar] [CrossRef] [Green Version]
- Silva-Junior, O.B.; Faria, D.A.; Grattapaglia, D. A flexible multi-species genome-wide 60K SNP chip developed from pooled resequencing of 240 Eucalyptus tree genomes across 12 species. New Phytol. 2015, 206, 1527–1540. [Google Scholar] [CrossRef] [Green Version]
- Geraldes, A.; Difazio, S.P.; Slavov, G.T.; Ranjan, P.; Muchero, W.; Hannemann, J.; Gunter, L.E.; Wymore, A.M.; Grassa, C.J.; Farzaneh, N.; et al. A 34K SNP genotyping array for Populus trichocarpa: Design, application to the study of natural populations and transferability to other Populus species. Mol. Ecol. Resour. 2013, 13, 306–323. [Google Scholar] [CrossRef]
- Silva, P.I.T.; Silva-Junior, O.B.; Resende, L.V.; Sousa, V.A.; Aguiar, A.V.; Grattapaglia, D. A 3K Axiom® SNP array from a transcriptome-wide SNP resource sheds new light on the genetic diversity and structure of the iconic subtropical conifer tree Araucaria angustifolia (Bert.) Kuntze. PLoS ONE 2020, 15, e0230404. [Google Scholar] [CrossRef]
- Howe, G.T.; Yu, J.; Knaus, B.; Cronn, R.; Kolpak, S.; Dolan, P.; Lorenz, W.W.; Dean, J.F.D. A SNP resource for Douglas-fir: de novo transcriptome assembly and SNP detection and validation. BMC Genom. 2013, 14, 137. [Google Scholar] [CrossRef] [Green Version]
- Howe, G.T.; Jayawickrama, K.; Kolpak, S.E.; Kling, J.; Trappe, M.; Hipkins, V.; Ye, T.; Guida, S.; Cronn, R.; Cushman, S.A.; et al. An Axiom SNP genotyping array for Douglas-fir. BMC Genom. 2020, 21, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plomion, C.; Chancerel, E.; Endelman, J.; Lamy, J.-B.; Mandrou, E.; Lesur, I.; Ehrenmann, F.; Isik, F.; Bink, M.C.A.M.; van Heerwaarden, J.; et al. Genome-wide distribution of genetic diversity and linkage disequilibrium in a mass-selected population of maritime pine. BMC Genom. 2014, 15, 171. [Google Scholar] [CrossRef] [PubMed]
- Azaiez, A.; Pavy, N.; Gérardi, S.; Laroche, J.; Boyle, B.; Gagnon, F.; Mottet, M.-J.; Beaulieu, J.; Bousquet, J. A catalog of annotated high-confidence SNPs from exome capture and sequencing reveals highly polymorphic genes in Norway spruce (Picea abies). BMC Genom. 2018, 19, 942. [Google Scholar] [CrossRef] [PubMed]
- Pavy, N.; Gagnon, F.; Rigault, P.; Blais, S.; Deschênes, A.; Boyle, B.; Pelgas, B.; Deslauriers, M.; Clément, S.; Lavigne, P.; et al. Development of high-density SNP genotyping arrays for white spruce (Picea glauca) and transferability to subtropical and nordic congeners. Mol. Ecol. Resour. 2013, 13, 324–336. [Google Scholar] [CrossRef]
- Ukrainetz, N.K.; Mansfield, S.D. Assessing the sensitivities of genomic selection for growth and wood quality traits in lodgepole pine using Bayesian models. Tree Genet. Genomes 2020, 16, 14. [Google Scholar] [CrossRef]
- Perry, A.; Wachowiak, W.; Downing, A.; Talbot, R.; Cavers, S. Development of a single nucleotide polymorphism array for population genomic studies in four European pine species. Mol. Ecol. Resour. 2020, 20, 1697–1705. [Google Scholar] [CrossRef]
- Lepoittevin, C.; Bodénès, C.; Chancerel, E.; Villate, L.; Lang, T.; Lesur, I.; Boury, C.; Ehrenmann, F.; Zelenica, D.; Boland, A.; et al. Single-nucleotide polymorphism discovery and validation in high-density SNP array for genetic analysis in European white oaks. Mol. Ecol. Resour. 2015, 15, 1446–1459. [Google Scholar] [CrossRef]
- Neves, L.G.; Davis, J.M.; Barbazuk, W.B.; Kirst, M. Whole-exome targeted sequencing of the uncharacterized pine genome. Plant J. 2013, 75, 146–156. [Google Scholar] [CrossRef]
- Chen, Z.Q.; Baison, J.; Pan, J.; Karlsson, B.; Andersson, B.; Westin, J.; García-Gil, M.R.; Wu, H.X. Accuracy of genomic selection for growth and wood quality traits in two control-pollinated progeny trials using exome capture as the genotyping platform in Norway spruce. BMC Genom. 2018, 19, 946. [Google Scholar] [CrossRef] [Green Version]
- Thistlethwaite, F.R.; Ratcliffe, B.; Klápště, J.; Porth, I.; Chen, C.; Stoehr, M.U.; El-Kassaby, Y.A. Genomic prediction accuracies in space and time for height and wood density of Douglas-fir using exome capture as the genotyping platform. BMC Genom. 2017, 18, 930. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Mitchell, S.E.; Elshire, R.J.; Buckler, E.S.; El-Kassaby, Y.A. Mining conifers’ mega-genome using rapid and efficient multiplexed high-throughput genotyping-by-sequencing (GBS) SNP discovery platform. Tree Genet. Genomes 2013, 9, 1537–1544. [Google Scholar] [CrossRef]
- Elshire, R.J.; Glaubitz, J.C.; Sun, Q.; Poland, J.A.; Kawamoto, K.; Buckler, E.S.; Mitchell, S.E. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE 2011, 6, e19379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Dien, O.G.; Ratcliffe, B.; Klápště, J.; Chen, C.; Porth, I.; El-Kassaby, Y.A. Prediction accuracies for growth and wood attributes of interior spruce in space using genotyping-by-sequencing. BMC Genom. 2015, 16, 370. [Google Scholar]
- Ratcliffe, B.; El-Dien, O.G.; Klápště, J.; Porth, I.; Chen, C.; Jaquish, B.; El-Kassaby, Y.A. A comparison of genomic selection models across time in interior spruce (Picea engelmannii × glauca) using unordered SNP imputation methods. Heredity 2015, 115, 547–555. [Google Scholar] [CrossRef] [Green Version]
- Parchman, T.L.; Jahner, J.P.; Uckele, K.A.; Gall, L.M.; Eckert, A.J. RADseq approaches and applications for forest tree genetics. Tree Genet. Genomes 2018, 14, 39. [Google Scholar] [CrossRef]
- Aguirre, N.C.; Filippi, C.V.; Zaina, G.; Rivas, J.G.; Acuña, C.V.; Villalba, P.V.; García, M.N.; González, S.; Rivarola, M.; Martínez, M.C.; et al. Optimizing ddRADseq in non-model species: A case study in Eucalyptus dunnii maiden. Agronomy 2019, 9, 484. [Google Scholar] [CrossRef] [Green Version]
- Miki, Y.; Yoshida, K.; Enoki, H.; Komura, S.; Suzuki, K.; Inamori, M.; Nishijima, R.; Takumi, S. GRAS-Di system facilitates high-density genetic map construction and QTL identification in recombinant inbred lines of the wheat progenitor Aegilops tauschii. Sci. Rep. 2020, 21, 21455. [Google Scholar] [CrossRef]
- Ott, A.; Liu, S.; Schnable, J.C.; Yeh, C.T.E.; Wang, K.S.; Schnable, P.S. tGBS® genotyping-by-sequencing enables reliable genotyping of heterozygous loci. Nucleic Acids Res. 2017, 45, e178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuskan, G.A.; Difazio, S.; Jansson, S.; Bohlmann, J.; Grigoriev, I.; Hellsten, U.; Putnam, N.; Ralph, S.; Rombauts, S.; Salamov, A.; et al. The genome of black cottonwood, Populus trichocarpa (Torr. & Gray). Science 2006, 313, 1596–1604. [Google Scholar] [PubMed] [Green Version]
- Nystedt, B.; Street, N.R.; Wetterbom, A.; Zuccolo, A.; Lin, Y.-C.; Scofield, D.G.; Vezzi, F.; Delhomme, N.; Giacomello, S.; Alexeyenko, A.; et al. The Norway spruce genome sequence and conifer genome evolution. Nature 2013, 497, 579–584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myburg, A.A.; Grattapaglia, D.; Tuskan, G.A.; Hellsten, U.; Hayes, R.D.; Grimwood, J.; Jenkins, J.; Lindquist, E.; Tice, H.; Bauer, D.; et al. The genome of Eucalyptus grandis. Nature 2014, 510, 356–362. [Google Scholar] [CrossRef] [Green Version]
- Neale, D.B.; Wegrzyn, J.L.; Stevens, K.A.; Zimin, A.V.; Puiu, D.; Crepeau, M.W.; Cardeno, C.; Koriabine, M.; Holtz-Morris, A.E.; Liechty, J.D.; et al. Decoding the massive genome of loblolly pine using haploid DNA and novel assembly strategies. Genome Biol. 2014, 15, R59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Telfer, E.; Graham, N.; Macdonald, L.; Sturrock, S.; Wilcox, P.; Stanbra, L. Approaches to variant discovery for conifer transcriptome sequencing. PLoS ONE 2018, 13, e0205835. [Google Scholar] [CrossRef] [Green Version]
- Telfer, E.; Graham, N.; Macdonald, L.; Li, Y.; Klápště, J.; Resende, M., Jr.; Neves, L.G.; Dungey, H.; Wilcox, P. A high-density exome capture genotype-by-sequencing panel for forestry breeding in Pinus radiata. PLoS ONE 2019, 14, e0222640. [Google Scholar] [CrossRef] [Green Version]
- Neale, D.B.; Savolainen, O. Association genetics of complex traits in conifers. Trends Plant Sci. 2004, 9, 330–337. [Google Scholar] [CrossRef]
- VanRaden, P.M. Efficient methods to compute genomic predictions. J. Dairy Sci. 2008, 91, 4414–4423. [Google Scholar] [CrossRef] [Green Version]
- Nejati-Javaremi, A.; Smith, C.; Gibson, J.P. Effect of total allelic relationship on accuracy of evaluation and response to selection. J. Anim. Sci.e 1997, 75, 1738–1745. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Pressoir, G.; Briggs, W.H.; Bi, I.V.; Yamasaki, M.; Doebley, J.F.; McMullen, M.D.; Gaut, B.S.; Nielsen, D.M.; Holl, J.B.; et al. A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat. Genet. 2006, 38, 203–208. [Google Scholar] [CrossRef]
- Kang, H.M.; Sul, J.H.; Zaitlen, N.A.; Kong, S.-Y.; Freimer, N.B.; Sabatti, C.; Eskin, E. Variance component model to account for sample structure in genome-wide association studies. Nat. Genet. 2010, 42, 348–354. [Google Scholar] [CrossRef] [Green Version]
- Habier, D.; Fernando, R.L.; Dekkers, J.C.M. The impact of genetic relationship information on genome-assisted breeding values. Genetics 2007, 177, 2389–2397. [Google Scholar] [CrossRef] [Green Version]
- Habier, D.; Fernando, R.L.; Garrick, D.J. Genomic BLUP decoded: A look into the black box of genomic prediction. Genetics 2013, 194, 597–607. [Google Scholar] [CrossRef] [Green Version]
- El-Kassaby, Y.A.; Klápště, J.; Guy, R.D. Breeding without breeding: Selection using the genomic best linear unbiased predictor method (GBLUP). New For. 2012, 43, 631–637. [Google Scholar] [CrossRef]
- Yang, J.; Benyamin, B.; McEvoy, B.P.; Gordon, S.; Henders, A.K.; Nyholt, D.R.; Madden, P.A.; Heath, A.C.; Martin, N.G.; Montgomery, G.W.; et al. Common SNPs explain a large proportion of the heritability for human height. Nat. Genet. 2010, 42, 565–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porth, I.; Klápště, J.; Skyba, O.; Lai, B.S.K.; Geraldes, A.; Muchero, W.; Tuskan, G.A.; Douglas, C.J.; El-Kassaby, Y.A.; Mansfield, S.D. Populus trichocarpa cell wall chemistry and ultrastructure trait variation, genetic control and genetic correlations. New Phytol. 2013, 197, 777–790. [Google Scholar] [CrossRef]
- El-Dien, O.G.; Ratcliffe, B.; Klápště, J.; Porth, I.; Chen, C.; El-Kassaby, Y.A. Implementation of the realized genomic relationship matrix to open-pollinated white spruce family testing for disentangling additive from nonadditive genetic effects. G3 Genes Genomes Genet. 2016, 6, 743–753. [Google Scholar]
- Nazarian, A.; Gezan, S.A. Integrating nonadditive genomic relationship matrices into the study of genetic architecture of complex traits. J. Hered. 2015, 107, 153–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Dungey, H.S. Expected benefit of genomic selection over forward selection in conifer breeding and deployment. PLoS ONE 2018, 13, e0208232. [Google Scholar] [CrossRef] [Green Version]
- Resende, M.F.R.; Munoz, P.; Acosta, J.J.; Peter, G.F.; Davis, J.M.; Grattapaglia, D.; Resende, M.D.V.; Kirst, M. Accelerating the domestication of trees using genomic selection: Accuracy of prediction models across ages and environments. New Phytol. 2012, 193, 617–624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beaulieu, J.; Doerksen, T.; Clément, S.; MacKay, J.; Bousquet, J. Accuracy of genomic selection models in a large population of open-pollinated families in white spruce. Heredity 2014, 113, 343–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguirre, N.C.; Filippi, C.V.; Zaina, G.; Rivas, J.G.; Acuña, C.; Villalba, P.V.; García, M.N.; Scaglione, D.; Morgante, M.; González, S.; et al. Development of a genotyping by sequencing strategy for assisted breeding of Eucalyptus dunnii. In Proceedings of the VII Reunión Genética y Mejoramiento Forestal, San Miguel de Tucumán, Argentina, 22–26 August 2016. [Google Scholar]
- Klápště, J.; Suontama, M.; Telfer, E.; Graham, N.; Low, C.; Stovold, T.; McKinley, R.; Dungey, H. Exploration of genetic architecture through sib-ship reconstruction in advanced breeding population of Eucalyptus nitens. PLoS ONE 2017, 12, e0185137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burdon, R.D.; Shelbourne, C.J.A. Breeding populations for recurrent selection: Conflicts and possible solutions. N. Z. J. For. Sci. 1971, 1, 174–193. [Google Scholar]
- Dodds, K.G.; McEwan, J.C.; Brauning, R.; Anderson, R.M.; Stijn, T.C.; Kristjánsson, T.; Clarke, S.M. Construction of relatedness matrices using genotyping-by-sequencing data. BMC Genom. 2015, 16, 1047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Telfer, E.J.; Stovold, G.T.; Li, Y.; Silva-Junior, O.B.; Grattapaglia, D.G.; Dungey, H.S. Parentage reconstruction in Eucalyptus nitens using SNPs and microsatellite markers: A comparative analysis of marker data power and robustness. PLoS ONE 2015, 10, e0130601. [Google Scholar] [CrossRef]
- Telfer, E.; Graham, N.; Stanbra, L.; Manley, T.; Wilcox, P. Extraction of high purity genomic DNA from pine for use in a high-throughput genotyping platform. N. Z. J. For. Sci. 2013, 43, 3. [Google Scholar] [CrossRef] [Green Version]
- Lu, F.; Lipka, A.E.; Glaubitz, J.; Elshire, R.; Cherney, J.H.; Casler, M.D.; Buckler, E.S.; Costich, D.E. Switchgrass genomic diversity, ploidy, and evolution: Novel insights from a network-based SNP discovery protocol. PLoS Genet. 2013, 9, e1003215. [Google Scholar] [CrossRef] [Green Version]
- Bradbury, P.J.; Zhang, Z.; Kroon, D.E.; Casstevens, T.M.; Ramdoss, Y.; Buckler, E.S. TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics 2007, 23, 2633–2635. [Google Scholar] [CrossRef]
- Lopes, M.S.; Silva, F.F.; Harlizius, B.; Duijvesteijn, N.; Lopes, P.S.; Guimaraes, S.E.F.; Knol, E.F. Improved estimation of inbreeding and kinship in pigs using optimized SNP panels. BMC Genet. 2013, 14, 92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dodds, K.G.; McEwan, J.C.; Brauning, R.; van Stijn, T.C.; Rowe, S.J.; McEwan, K.M.; Clarke, S.M. Exclusion and genomic relatedness methods for assignment of parentage using genotyping-by-sequencing data. G3 Genes Genomes Genet. 2019, 9, 3239–3247. [Google Scholar] [CrossRef] [Green Version]
- Whalen, A.; Gorjanc, G.; Hickey, J.M. Parentage assignment with genotyping-by-sequencing data. J. Anim. Breed. Genet. 2019, 136, 102–112. [Google Scholar] [CrossRef] [Green Version]
- Grattapaglia, D.; Ribeiro, V.J.; Rezende, G.D.S.P. Retrospective selection of elite parent trees using paternity testing with microsatellite markers: An alternative short term breeding tactic for Eucalyptus. Theor. Appl. Genet. 2004, 109, 192–199. [Google Scholar] [CrossRef]
- Lstibůrek, M.; Ivanková, K.; Kadlec, J.; Kobliha, J.; Klápště, J.; El-Kassaby, Y.A. Breeding without breeding: Minimum fingerprinting effort with respect to the effective population size. Tree Genet. Genomes 2011, 7, 1069–1078. [Google Scholar] [CrossRef]
- Lstibůrek, M.; Klápště, J.; Kobliha, J.; El-Kassaby, Y.A. Breeding without Breeding: Effect of gene flow on fingerprinting effort. Tree Genet. Genomes 2012, 8, 873–877. [Google Scholar] [CrossRef]
- Bush, D.; Kain, D.; Kanowski, P.; Matheson, C. Genetic parameter estimates informed by a marker-based pedigree: A case study with Eucalyptus cladocalyx in southern Australia. Tree Genet. Genomes 2015, 11, 798. [Google Scholar] [CrossRef]
- Hill, W.G.; Weir, B.S. Variation in actual relationship as a consequence of Mendelian sampling and linkage. Genet. Res. 2011, 93, 47–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J. Pedigrees or markers: Which are better in estimating relatedness and inbreeding coefficient? Theor. Popul. Biol. 2016, 107, 4–13. [Google Scholar] [CrossRef]
- Goddard, M. Genomic selection: Prediction of accuracy and maximisation of long term response. Genetica 2009, 136, 245–257. [Google Scholar] [CrossRef]
- Lippert, C.; Quon, G.; Kang, E.Y.; Kadie, C.M.; Listgarten, J.; Heckerman, D. The benefits of selecting phenotype-specific variants for applications of mixed models in genomics. Sci. Rep. 2013, 3, 1815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grattapaglia, D.; Kirst, M. Eucalyptus applied genomics: From gene sequences to breeding tools. New Phytol. 2008, 179, 911–929. [Google Scholar] [CrossRef]
- Müller, B.S.F.; Neves, L.G.; de Almeida Filho, J.E.; Resende, M.F.R.; Munoz, P.R.; dos Santos, P.E.T.; Filho, E.P.; Kirst, M.; Grattapaglia, D. Genomic prediction in contrast to a genome-wide association study in explaining heritable variation of complex growth traits in breeding populations of Eucalyptus. BMC Genom. 2017, 18, 524. [Google Scholar] [CrossRef] [Green Version]
- Durán, R.; Isik, F.; Zapata-Valenzuela, J.; Balocchi, C.; Valenzuela, S. Genomic predictions of breeding values in a cloned Eucalyptus globulus population in Chile. Tree Genet. Genomes 2017, 13, 74. [Google Scholar] [CrossRef]
- Tan, B.; Grattapaglia, D.; Martins, G.S.; Ferreira, K.Z.; Sundberg, B.; Ingvarsson, P.K. Evaluating the accuracy of genomic prediction of growth and wood traits in two Eucalyptus species and their F1 hybrids. BMC Plant Biol. 2017, 17, 110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suontama, M.; Klápště, J.; Telfer, E.; Graham, N.; Stovold, T.; Low, C.; McKinley, R.; Dungey, H. Efficiency of genomic prediction across two Eucalyptus nitens seed orchards with different selection histories. Heredity 2019, 122, 370–379. [Google Scholar] [CrossRef] [Green Version]
- Grattapaglia, D.; Resende, M.D.V. Genomic selection in forest tree breeding. Tree Genet. Genomes 2011, 7, 241–255. [Google Scholar] [CrossRef]
- Gorjanc, G.; Clevel, M.A.; Houston, R.D.; Hickey, J.M. Potential of genotyping-by-sequencing for genomic selection in livestock populations. Genet. Sel. Evol. 2015, 47, 12. [Google Scholar] [CrossRef] [PubMed]
- Ahuja, M.R.; Neale, D.B. Evolution of genome size in conifers. Silvae Genet. 2005, 54, 126–137. [Google Scholar] [CrossRef] [Green Version]
- Moghaddar, N.; Gore, K.P.; Daetwyler, H.D.; Hayes, B.J.; Werf, J.H.J. Accuracy of genotype imputation based on random and selected reference sets in purebred and crossbred sheep populations and its effect on accuracy of genomic prediction. Genet. Sel. Evol. 2015, 47, 97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jarquín, D.; Kocak, K.; Posadas, L.; Hyma, K.; Jedlicka, J.; Graef, G.; Lorenz, A. Genotyping by sequencing for genomic prediction in a soybean breeding population. BMC Genom. 2014, 15, 740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savolainen, O.; Pyhäjärvi, T. Genomic diversity in forest trees. Curr. Opin. Plant Biol. 2007, 10, 162–167. [Google Scholar] [CrossRef] [PubMed]
- Meirmans, P.G.; Gros-Louis, M.-C.; Lamothe, M.; Perron, M.; Bousquet, J.; Isabel, N. Rates of spontaneous hybridization and hybrid recruitment in co-existing exotic and native mature larch populations. Tree Genet. Genomes 2014, 10, 965–975. [Google Scholar] [CrossRef] [Green Version]
- Meirmans, P.G.; Lamothe, M.; Gros-Louis, M.-C.; Khasa, D.; Périnet, P.; Bousquet, J.; Isabel, N. Complex patterns of hybridization between exotic and native North American poplar species. Am. J. Bot. 2010, 97, 1688–1697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Visscher, P.M.; Hill, W.G.; Wray, N.R. Heritability in the genomics era–concepts and misconceptions. Nat. Rev. Genet. 2008, 9, 255–266. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Seed Orchard | Nr. of Parents | Nr. of Offspring |
|---|---|---|---|
| E. nitens | Alexandra | 13 | 9 |
| E. nitens | Drumfern | 0 | 9 |
| E. nitens | Tinkers | 30 | 18 |
| E. grandis x E. nitens | NA | 4 | 0 |
| E. quadrangulata | NA | 1 | 0 |
| E. regnans | NA | 1 | 0 |
| E. saligna | NA | 1 | 0 |
| E. cladocalyx | NA | 1 | 0 |
| E. camaldensis | NA | 1 | 0 |
| E. bosistoana | NA | 1 | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Klápště, J.; Ashby, R.L.; Telfer, E.J.; Graham, N.J.; Dungey, H.S.; Brauning, R.; Clarke, S.M.; Dodds, K.G. The Use of “Genotyping-by-Sequencing” to Recover Shared Genealogy in Genetically Diverse Eucalyptus Populations. Forests 2021, 12, 904. https://doi.org/10.3390/f12070904
Klápště J, Ashby RL, Telfer EJ, Graham NJ, Dungey HS, Brauning R, Clarke SM, Dodds KG. The Use of “Genotyping-by-Sequencing” to Recover Shared Genealogy in Genetically Diverse Eucalyptus Populations. Forests. 2021; 12(7):904. https://doi.org/10.3390/f12070904
Chicago/Turabian StyleKlápště, Jaroslav, Rachael L. Ashby, Emily J. Telfer, Natalie J. Graham, Heidi S. Dungey, Rudiger Brauning, Shannon M. Clarke, and Ken G. Dodds. 2021. "The Use of “Genotyping-by-Sequencing” to Recover Shared Genealogy in Genetically Diverse Eucalyptus Populations" Forests 12, no. 7: 904. https://doi.org/10.3390/f12070904